

Abstract
There is a growing appreciation of the extent of transcriptome variation across individual cells of the same cell type. While expression variation may be a byproduct of, for example, dynamic or homeostatic processes, here we consider whether single‐cell molecular variation per se might be crucial for population‐level function. Under this hypothesis, molecular variation indicates a diversity of hidden functional capacities within an ensemble of “identical” cells, and this functional diversity facilitates collective behavior that would be inaccessible to a homogenous population. In reviewing this topic, we explore possible functions that might be carried by a heterogeneous ensemble of cells; however, this question has proven difficult to test, both because methods to manipulate molecular variation are limited and because it is complicated to define, and measure, population‐level function. We consider several possible methods to further pursue the hypothesis that “variation is function” through the use of comparative analysis and novel experimental techniques.
Keywords: bet‐hedging, evolution of variation, fractional response, functional variation, single cell transcriptome, single cell variation
Introduction
Cells in multi‐cellular organisms are frequently treated as fundamental units of function. While cell differentiation generates classes of cells with unique phenotypic identity, ensembles of cells within a cell type have been seen as (nearly) identical building blocks. However, recent technological advancements have enabled increasingly high‐resolution measurements of gene expression in single cells 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, resulting in a growing appreciation for the extent of individual expression variability 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33. This variability has been examined from many vantage points: as an indicator of the vast diversity of cell types 9, 17, 18, 19, as a byproduct of redundancy in regulatory networks 21, 22, as a temporal snapshot of asynchronous dynamic processes 23, 24, 25, 26, 27, 28, 29, 30, or as evidence that RNA abundance may be irrelevant for cell phenotype 31, 32. (For further discussion, see 34.) An alternative perspective is to consider whether single cell transcriptome, proteome, and other molecular variability might be part of what establishes tissue/population‐level function. Are individual cells in a multi‐cellular organism like individual organisms in a cooperative community, where each cell's behavior contributes to a higher‐level functional ecology?
Before we begin our exposition, we briefly consider some preliminary concepts. When we use the term “single cell variability” or “single cell heterogeneity,” it is not meant to refer to diversity of cell types that are clearly distinct and already recognized. Rather, we use the terms to describe diversity within an ensemble that has been previously defined as being generally homogeneous (e.g. pyramidal neurons from the CA3 area of the hippocampus). Of course, no two cells are exactly identical, so there is an implicit quantitative degree of variation implied in when single cell variation is discussed. “Function” is an elusive term spanning different scales from proximate function (e.g. biochemical action) to distal evolutionary fitness function. It is not our goal to precisely define function or create an ontology of single cell function. Our main focus is to ask how variation among individual cells might interact to causally generate higher‐level function, regardless of how function is defined. We leave out the discussion of certain kinds of variation that might be more of a homeostatic adaptation or a consequence of robustness promoting mechanisms rather than the result of each cell carrying out a distinct action that leads to aggregate function.
We note that variation or variability of a trait refers to the entire characteristics of the distribution of the trait over the ensemble of cells. This is distinct from a particular statistic like “variance,” which is a number computed from the distribution (average squared deviation from the mean). Two different single cell measurements can have the same variance but a different variation (Fig. 1). And, if there are threshold effects (e.g. resistance to chemotherapy agent), the two different populations may have very different numerical responses (Fig. 1). Because variation is a multifaceted trait, when comparing different studies or carrying out analyses, it is important to be precise about which aspect of variation is being analyzed and its model expectations (see below). An important concept is the idea that a trait's single cell variation may be changed or modulated, perhaps through genetic mechanisms. For example, a gene may be “ON” in 50% of individual cells under one genetic background while “ON” in 25% of the cells under another genetic background. Modulation of single cell variability may involve changing the variance of a trait or some other aspect of the distribution. From this perspective, the term “stochastic expression” does not imply that expression is not regulated – gene expression may have randomness, but the characteristics of the resulting variation may be regulated (see 35, 36).
Figure 1.
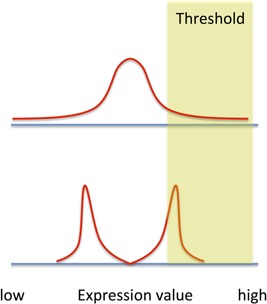
A schematic diagram of two different single cell distributions with identical variance value. If there is a threshold value that triggers some cellular response, the two populations will respond very differently.
In this essay, we review some possible scenarios in which cell‐to‐cell heterogeneity may be important for higher‐level function, and discuss possible ways of testing such a “variation is function” hypothesis (see Table 1). In particular, we note that asking whether variability is needed for certain functions is distinct and complementary to the idea that biological mechanisms exist to “allow function despite variability.” There is no doubt that robustness, homeostasis, and canalization are important phenomena in organisms, and a contributing factor to single cell variability. Here, we focus on the idea that variation, in and of itself, is required for function.
Table 1.
Scenarios where aggregate function may depend on single cell variation
| Hypothesis | Description |
|---|---|
| Bet hedging | A pre‐existing diversity of cell states allows rapid population adaptation to an unpredictable environmental change |
| Generalized bet hedging | Extensive randomized phenotypic diversity allows population adaptation of vast diversity of environments |
| Response distribution | Cell‐to‐cell variation in binary decisions allows a fractional or dose‐dependent population response |
| Fate plasticity and priming | Uncorrelated, sub‐threshold fluctuations in regulators of cell fates create subpopulations of cells primed for multiple fate decisions |
| Information coding and transfer | A diverse ensemble of individuals enables the population to encode and transmit complex information |
| Crowd control | Rare individuals with capacity to respond to perturbation emit local signals that coordinate population behavior |
Bet hedging: A pre‐existing diversity of cell states allows rapid population adaptation to a new environment
In fluctuating, unpredictable environments, a population may benefit by maintaining a diversity of cell phenotypes, each advantageous in a distinct context. Unlike a strategy where individual cells sense and respond to the environment, maintenance of a standing diversity may be preferred when a rapid response of at least a subpopulation is advantageous and there is insufficient time for signal transduction 37. Because this maintenance of diversity protects against a future crisis, this behavior has been termed “bet hedging” and has been extensively studied in single‐celled organisms 37, 38, 39, 40, 41, 42. In a classic example, E. coli populations maintain a subset of cells in persistence, a quiescent phenotypic state 38, 39. Though the presence of persistent cells reduces population growth in nutrient‐rich environments, it allows the population to survive unexpected antibiotic agents that target rapidly proliferating cells. To generate the standing population diversity in a uniform environment, individual E. coli cells stochastically switch into and out of persistence. Phenotype switching has been observed broadly, suggesting that this single cell behavior provides a fitness advantage in certain contexts 39. Experimental evolution of Pseudomonas fluorescens demonstrated that, under a fluctuating selection regime, stochastic phenotype switching could evolve 41. The rate of bi‐stable state switching can be a function of the gene regulatory network, and can affect fitness, with an optimal switching rate dependent on the rate of environmental fluctuations 36, 37
We know of no cases of bet hedging in healthy mammalian tissues, perhaps because of the interdependence of cells in multicellular organisms 39 or lack of experiments assessing individual cell turnover dynamics. However, it may be that mammalian cancers exhibit this behavior 43, 44, 45. As in the E. coli example, cancer populations may survive chemotherapies that target proliferating cells by switching into and out of a proliferative states 43, 44. Phenotype switching has also been hypothesized to play a role in cancer metastasis. Lee et al. characterized a regulatory network that may be capable of producing coexisting noninvasive and pro‐metastatic expression states within a triple‐negative breast cancer population 45. Models suggested that transient perturbations could trigger a cancer cell to switch into a malignant state and that pro‐metastatic cells may relax back into a noninvasive state. The implication for functional relevance is only speculative; however, one may imagine that state switching between noninvasive and metastatic states may be akin to whole organisms' ecological life history decisions on migration and colonization 46. The key question is whether normal cells might employ such bet‐hedging strategies. One obvious possibility is with tissues such as skin that directly interact with unpredictable external environment or unpredictable changes in whole organism physiology (e.g. injury response). A more speculative possibility is in developmental contexts where cell proliferation and death in response to patterning gradients is part of morphogenesis. J. J. Kupiec has proposed the novel idea that variation and selection of specific cellular phenotypes (“Darwinian cell theory”) may be an intrinsic mechanism in multi‐cellular development 47.
Generalized bet hedging: Random phenotype generation enables population response to novel environments
If the diversity of environments that may be encountered is vast, it may be of use for a population of cells to contain as broad a range of phenotypes as possible – to have individuals extensively sample phenotypic space, potentially through use of random mechanisms such as highly variable transcription, errors in transcription or DNA replication, or random genomic rearrangements 48, 49, 50, 51. We may consider this as a more generalized form of bet hedging. Though under this strategy individual phenotypes may not be reproducible, it may be that the population benefits substantially by containing at least one successful phenotype. Archetypal examples include the adaptive immune system 48, 49, and stress, where the generation of diversity through increased molecular error rates may produce an individual who survives 52. The benefits of such extensive diversity may also be relevant in disease. Cancer populations are highly heterogeneous, molecularly and phenotypically, and this population heterogeneity has been associated with resistance to drug treatment and patient survival 52, 53, 54, 55. Roux et al. show that fluctuations in protein levels can lead to recurring sub‐populations of cells that are more resistant to ligand‐induced apoptosis 56, 57.
Response distribution: Variation across single cells may allow a graded population response
Tissues rely on binary decisions made by individual cells, such as whether to enter the cell cycle or apoptosis. Uniformity across cells in binary decisions would produce switch‐like population behavior, and in many cases this would be undesirable. Instead, fractional quantitative responses can be achieved by integrating expression fluctuations in decision‐making, fluctuations that may be generated by stochastic gene expression. The role of stochastic fluctuations 58, 59, 60 and quantitatively distributed population states in the regulation of population abundance within ecological communities has been noted. Given predator‐prey dynamics, if all predators acted homogeneously, all prey would be eaten simultaneously at carrying capacity, and the population would collapse to extinction (e.g. 61). Incorporating heterogeneity to desynchronize populations can generate fractional or dose‐dependent population responses. Recent studies have suggested this type of heterogeneity‐dependent population behavior in contexts such as fractional population death in response to chemotherapy 54, maintenance of adult adipose tissue size by fractional differentiation of pre‐adipocytes 58, fractional apotoptic response to ligands 57, 59, and graded response to growth factors in the decision to enter the cell cycle in mammary epithelial cells 60. Graded or fractional population response mediated by individual variation may be an important general mechanism bridging the discrete outputs of a cell and the need for quantitative responses (e.g. neuronal activity).
Priming and fate plasticity: Gene expression variation endows cells and populations with fate plasticity
For some tissues, function depends on cell fate plasticity or the ability of its members to take on a diversity of cell states, as in stem cell populations. Fate plasticity has been associated with expression variation, such as stochastic, semi‐indiscriminant gene activation 62, 63, which has also been called “promiscuous gene expression” 64. Numerous studies of stem and progenitor cells have reported variable expression of developmental regulators 2, 62, 63, 65, 66 and have associated this with heterogeneity in differentiation potential 62, 63. Recently, Kumar et al. reported extensive variation across mouse pluripotent stem cells (PSCs) in the activation of stem‐ and cell‐fate regulators, as well as genes that sense and respond to environmental cues. They also reported that the extent of variation was associated with the rate of differentiation 63. In an environment containing cues for both differentiation and self‐renewal, the authors found multiple subpopulations of PSCs: one subpopulation demonstrated relatively homogenous expression and a bias towards self‐renewal; a second showed variable activation of cell‐fate regulators and higher rates of spontaneous differentiation.
In many cases, genes critical to cell fate decisions are involved in regulatory networks with switch‐like behavior. As gene expression levels approach the network's switching threshold, the probability that induction by external cues will trigger threshold crossing is increased. A cell with expression level near a threshold level for phenotypic switching might be considered to be “primed” for a cell‐fate decision 62, 63. If the population contains a set of cells at variable distance from the threshold, a subset of cells might be ever ready to cross the threshold immediately. This expression variability across cells may be generated by stochastic gene activation, which – in a process akin to signal amplification by white noise 67 – assists cells in crossing thresholds. If the expression state of any individual fluctuates over time, as seen in populations of pluripotent stem cells 62, 63, then, even as cells differentiate, the population may maintain a characteristic diversity of primed cells 62.
Information propagation: Population diversity may enable information coding and transfer
There is an association of high variation with high information content (i.e. high entropy). Single cell variation can represent both high information content and, if cells are processing information, the capacity to transfer high information content. For example, medullary thymic epithelial cells (mTEC) stochastically transcribe tissue‐restricted genes in the mTEC population so that collectively the population exposes thousands of self‐antigens to developing T cells. This diversity plays an instructive role in T cell differentiation, so that only T cells with low self‐affinity are directed to an effector fate 68. In the brain, extensive phenotypic diversity may broaden the extent of possible neural circuitry, and so enhance the brain's capacity for information transfer 50, 69, 70, 71. Increased rates of Line1 (L1) retrotranposition, a source of somatic genetic diversity, have been found during neurogenesis 69, speculatively supporting a functional advantage to heightened diversity in the brain.
Crowd control: Rare cells respond rapidly to perturbations and coordinate population behavior
Several recent single‐cell studies of anti‐viral 72, 73, 74 or inflammatory 73, 74, 75 response and cell fate choice 76 have reported cases where a rare subset of cells in a population responded rapidly to perturbation and emitted signals that coordinated population behavior 72, 73, 74, 75, 76. Described as sentinels 72, first responders 73, precocious cells 74 or pioneers 76, these cells uniquely express 72, 73, 74, 76 and secrete 75 key cytokines in response to the stimulus. By contrast, the majority of the population was incapable of responding in kind to the same stimulus 72, 73, 74, 75, even over extended periods of time. This two‐tiered signaling mechanism coordinated population behaviors, eliciting a uniform response 72, 73, 74, 75, 76 or more complex behavior, such as modulating phenotype heterogeneity spatially or over time 72, 73, 74, 75. The concept of sentinel or first responder cells is that a subset of cells in a signal responding state can dynamically reprogram the greater cell population, and this helps balance competing needs of the physiological dynamics. For example, the immune system requires a balance between rapid response to assault and avoidance of self‐toxicity 73, 74. Recently, Patil et al. reported that when human dendritic cells were infected with Newcastle disease virus, a small fraction of cells activated Ifnb1 promptly 73. Paracrine signals emitted by these early responders activated Ifnb1 expression in the majority of cells, but in a manner that elicited large variation across cells in time‐to‐activation. Dynamic coordinated population behavior activated through single cell variation may also be critical in other contexts, such as tissue morphogenesis. A recent study provided suggestive evidence, reporting that a subpopulation of rare cells was essential in normal breast epithelial cell morphogenesis in 3D culture for enforcement of quiescence 77.
Evolutionary comparisons to test the “variation is function” hypothesis
The examples discussed in the above sections suggest that cell‐to‐cell variations of molecular states in seemingly homogeneous population of cells may have functional rationale in terms of population/tissue level function. Here, we discuss possible approaches to test the hypothesis that “variation is function” and the challenges associated with such tests.
Leveraging the predictions of neutral theory of molecular evolution 78, one approach to test the idea that variation is functionally important is to assess whether single cell variability of particular genes is an evolutionarily conserved trait. Suppose we choose variance as the appropriate statistic for measuring variation (see Introduction about difference between “variation” and “variance”), we can compute the single cell variance of a gene's expression in mice and ask whether the variance is essentially the same in rats. But, if single cell variance of MAPK is 10 (normalized read units) in mouse and it is 12 in rats, how do we know that this difference is significantly different than expected under neutral evolution? When similar inferences are made with sequence analysis, a standard method is to compare rates of divergence against the divergence of sequences whose functional effects are a priori assumed to be neutral (e.g. synonymous positions of codons). In the case of gene expression, it is difficult to directly measure such expectations because it is difficult to point to some gene's expression that can be a priori assumed to be neutral in function. It is especially difficult to measure for single cell variation when we currently have no a priori theory of its function.
One possible approach is to compare the conservation of the particular degree of variance (or some similar statistic) associated with a particular gene in relation to other genes in the genome. That is, we might hypothesize that if gene A has greater cell‐to‐cell variance than gene B in mouse, it might also have greater variance in rat if the variation is functionally important enough to affect fitness. Thus, we can compute a correlation‐based test of genes' variances amongst homologous genes in mouse and rat. In Dueck et al., we applied one such test for a small dataset and found significant correlations between the gene‐variance of mouse and rat 33.
Problems to overcome for comparative analysis
There are two important problems to overcome with the comparative approach to assessing whether single cell variation is functionally important. The first is a technical problem. A statistic that measures variability such as variance can be correlated with other features of the transcriptome. Sampling each cell's mRNA into RNAseq counts generates a mathematical relationship (nearly linear) between expression intensity and expression variance by multinomial sampling theory. Therefore, if we find a significant correlation of single cell variance of homologous genes between mouse and rat, it may be due to the correlation of expression intensity amongst homologous genes rather than due to biological functions of cell‐to‐cell variation (Fig. 2A and B). Therefore, any comparative correlation test needs to correct for other covariance factors; e.g. by computing partial correlations as was done in Dueck et al. (Fig. 2C). A difficulty is that other additional factors besides sampling may also affect the observed variation. For example, certain mRNA might be membrane‐bound and harder to recover, certain mRNAs' sequence features (e.g. GC percent, length, 5′ and 3′ motifs, etc.) may affect cDNA synthesis or amplification; mRNA of very low abundance may be especially difficult to capture in the first cDNA synthesis, etc. All of these factors can modulate measured single cell variance. These ancillary features of transcripts may also be conserved due to conservation of genes' sequences and cellular functions. Thus, the hypothesis test becomes compounded with the effects of evolutionary forces governing these other traits. The features that complicate the measurement of single cell variation – such as membrane association – may be exactly the feature that is important for the functional mechanism underlying cell‐to‐cell variation. In sum, for both evolutionary hypothesis testing and for general inference of single cell variation, it is critical to develop a robust measurement theory of single cell variation 79. We need a model of expected variance associated with each gene and each measurement protocol that can be used to “correct” the observed variance (see Dueck et al. 33).
Figure 2.
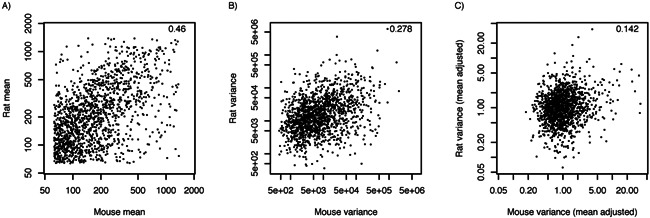
Computing the correlation of gene expression variance between two species can be influenced by other correlated factors. A: Correlation of expression level of between homologous genes of rat and mouse neurons. B: Correlation of single cell variance between homologous genes of rat and mouse neurons. C: Partial correlation conditioned on expression level, correcting for the conservation of expression levels.
The second problem for comparing single cell variability amongst genes across species is biological. If we construct a test based on conservation of relative levels of variance amongst genes across species (e.g. correlation of variance), a significant correlation pattern may emerge due to conserved functional importance of either the high‐variability genes or the low variability genes (or both). A gene may have high single cell variability vis‐à‐vis the rest of the transcriptome that is conserved between species due to the various possible higher‐level functions discussed above. Or, a gene may have conserved low single cell variability because the precision of its expression is important 80, 81, 82, which is a different hypothesis from the idea of “variance is function.” One possible approach to distinguish between these two hypotheses is to examine the (corrected) correlation of variance across multiple cell types. Suppose we were to find significant inter‐species correlation of genome‐wide single cell variances in, say, both cardiomyocytes and neurons, respectively. Overlap in the identity of the high variability genes between the two different cell types would suggest consistent function of the high variability genes. Unfortunately, the lack of such overlap does not necessarily imply lack of functional rationale for single cell variability. Different genes may be highly variable in different cell types because the higher‐level function of different tissues might require variability of different genes, as in many of the scenarios listed above. This might be resolvable with the inclusion of many different cell types in a cross‐species comparison and identifying finite number of conserved high‐variability gene clusters.
Prospects for directly testing “variance is function”
What are the prospects for directly testing whether cell‐to‐cell variation in gene expression has functional significance? Classic studies such as canalization mechanisms show that a variety of mechanisms exist to control the variability of molecular processes 83. There is an increasing number of studies showing that the distribution of gene expression across cells can be modulated by regulation 14, 58, 63, 84, 85, 86, 87, 88, through mechanisms involving promoter accessibility 86, transcript degradation rate 33, 63, gene copy number 14, or regulatory network structure 58, 63. Additionally, multiple studies have suggested separate control of expression mean and variance 85, 86, 87. Recently, Lagha et al. showed that paused Pol II decreased temporal variation in gene activation in response to Dpp signaling during Drosophila development 84. Benayoun et al. provided evidence that genes broadly covered with H3K4me3 histone modifications demonstrate low expression variation, uncorrelated with expression level 85. Interestingly, Vinuelas et al. 89 report that chromatin modifying reagents can induce significant effects on the stochastic expression variation while Dar et al. 87 report almost hundred different compounds that modulate HIV gene expression variation. Examples listed suggest that there might be accessible control points to manipulate single cell variability. But complex manipulation of single‐cell variation is not a simple matter. It is possible that the absolute amounts of multiple RNAs may be important along with single cell variation 90. This suggests that the both the amounts of variable RNAs and their relative abundances need to be controlled within the biological constraints of the cell, where approximately 200,000–400,000 mRNA molecules are thought to be the normal complement of mRNAs. Another complication is that over‐expression of particular RNAs might cause a non‐specific change in cellular dynamics related to titration of RNA polymerases or translational machinery not related to the variability itself, and so altered expression profiles need to be carefully controlled.
General perturbation of single cell variability
If the functionally relevant single‐cell expression variation is non‐specific, such as may be the case in the generalized bet hedging scenario, one possible approach to experimentally manipulate variation is to use non‐specific effects of siRNA. While siRNA is usually utilized to target specific complementary RNA, it is well known that there are significant off‐target effects with estimates of 100‐1,000 RNAs changing as a result of the RNA manipulation 91, 92. These effects are generally thought to be non‐specific and include both increases and decreases in RNA abundances encoded by many genes 93, 94. If indeed the off‐target action of siRNA is non‐specific, then there is little reason to believe that these off‐target effects would selectively modify individual biological systems. Generation of an siRNA with primarily off‐target action, for example an siRNA of random sequence not present in the mammalian genome, would permit a test of the role of non‐specific alterations in transcriptome variation across single cells. Ideally siRNA would be introduced into cells using lentivirus where siRNA expression is under regulated promoter control (e.g. tet – on promoter) 95. In this paradigm, cells would be transfected, baseline population phenotypic measures quantified, and then the siRNA turned on by addition of tetracycline. At various times after activation of the siRNA, the population would be high‐content screened for phenotype alterations followed by transcriptome analysis to assess the extent of siRNA‐induced transcriptome variability between cells. If the siRNA promoter can also be turned off (removal of Tet) then, presuming that the siRNA effect is non‐specific, any phenotypic changes that have occurred at a population level would be expected to return to baseline unless the cells have responded permanently. Further, when the promoter is turned back on a new single‐cell variation will be generated (because of different off‐target effects than those initially produced by siRNA activation), facilitating an examination of reproducibility.
Targeted perturbation of sub‐systems
If functionally relevant cell‐to‐cell variability is specific to mRNAs that comprise a particular regulatory system, a different approach will be required. To assess the role of gene expression variability of specific genes in the production of population function, we need approaches to modulate multiple specific RNAs simultaneously in many cells. We discuss several possible approaches including, (1) transfection of selected in vitro transcribed RNAs in user specified amounts and ratios; (2) transfection of transcription factor (TF) encoding RNAs into cells; and, (3) use of a miRNA that anneals to multiple RNAs to modulate their translation or stability.
In 96 we created a method to quantitatively transfect multiple RNAs into cells called Transcriptome Induced Phenotype Remodeling (TIPeR). TIPeR transfects a predefined library of mRNA, either native or synthetic, into cells (usually using lipid mediated, electroporation or phototransfection procedures). Ideally, a well‐defined pool of mRNA might be transfected into individual cells and directly control cell‐to‐cell variation. This most direct approach, isolated transfection of mRNAs into individual cells, is not trivial and currently its throughput is limited so only a limited numbers of cells could be assessed. Furthermore, it would be difficult to apply to naturally interacting in vivo cell populations. The transfection of transcription factor (TF) encoding RNAs into cells permits modulation of any of the cell‐specific target genes of that specific transcription factor. The change in TF levels will add to the existing single cell variation to change the distribution. The key will be to place the TFs under regulatable promoter control, preferably one that allows the careful titration of transcription so that small or large amounts of RNA are made in the cell 97. Fortunately such promoters exist, including those regulated by the cumate operon 98, 99 which can be easily used in cell culture and intact animals through lentivirus infection. Targeting multiple components of some pathway may require transfecting multiple TFs. The use of miRNAs as modulators of multiple RNA expression is particularly intriguing as there is evidence to suggest that many RNAs within a regulatory pathway have the same predicted miRNA binding sites 100, 101. Schmiedel et al. 102 has recently shown miRNA modulation of protein expression variation. This suggests that variability of RNA expression for particular systems may be more easily manipulated using miRNAs. Tight regulation of abundances can be achieved, again using the cumate operon 95, 99. Performing time courses by turning the miRNAs on and off within the same cells and quantifying changes in population phenotype and transcriptome variability promises to highlight functional components of cellular transcriptome variability. TIPeR, TF transfection, or miRNA, all of the experiments will require measurable definition of higher‐level function enabled by single cell variation. Evolutionary comparisons may help narrow down some of the possible functions listed in Table 1 and help design the right experiments. Ideal future experiments might include ex vivo reconstruction of 3D tissues with coupled modulation of individual cell variability as pioneered in 103, 104
Conclusions and outlook
In this essay, we have explored the idea that single cell variation may, at least in part, be required for higher‐level system function. Higher‐level group properties that arise from heterogeneous ensembles are often seen in ecological communities. For example, the nutrient cycles, food webs, social groups, etc., all involve an ensemble of individuals with differentiated roles. (Many such assemblies are not selected for the group property but rather the group property arises out of the interaction of the participants). Similar dynamics amongst individual cells might be an important component of organismal physiology, and the pursuit of this topic may improve our understanding of both healthy and diseased tissues.
When considering differentiated roles, it may seem that the main question is that of classification of previously unrealized subtypes (e.g. 105). Classification of types is a classic systematics problem, and even in whole organisms, systematicists occasionally find cryptic subtypes. It may be that, if a theory or principle of cell phenotype emerges, similar to the biological species concept 106, much of the single cell variation might indicate multiple cryptic subtypes; however, it is also likely that much of the single cell variation is plastic and context dependent. In a more speculative model, J. J. Kupiec 47 as well as A. Paldi 107 suggest that variation may be part of a kind of Darwinian mechanism for driving developmental decisions, where stochastic variation generates possible different cell fates and subsequent mechanisms apply a kind of “natural selection” for proper differentiation. This is an extremely intriguing speculation and more broadly, we speculate that cell variation could be a mechanism for incorporating environmental information (through aforementioned Darwinian mechanism) into organismal information. In sum, we propose that understanding the mechanism and higher‐level function of single cell variation will be the key to understanding multi‐cellular systems.
Acknowledgments
This work has been supported in part by Ellison Medical Foundation Aging Award, NIMH 5U01MH098953, and Health Research Formula Funds from the Commonwealth of Pennsylvania. The funding bodies had no role in study design, in collection, analysis, interpretation of data, or in the writing of the manuscript.
The authors have declared no conflicts of interest.
References
- 1. Tang F, Barbacioru C, Wang Y, Nordman E, et al. 2009. mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods 6: 377–82. [DOI] [PubMed] [Google Scholar]
- 2. Islam S, Kjällquist U, Moliner A, Zajac P, et al. 2011. Characterization of the single‐cell transcriptional landscape by highly multiplex RNA‐seq. Genome Res 21: 1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hashimshony T, Wagner F, Sher N, Yanai I. 2012. CEL‐seq: single‐cell rna‐seq by multiplexed linear amplification. Cell Rep 2: 666–73. [DOI] [PubMed] [Google Scholar]
- 4. Ramsköld D, Luo S, Wang Y‐C, Li R, et al. 2012. Full‐length mRNA‐Seq from single‐cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30: 777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Picelli S, Björklund ÅK, Faridani OR, Sagasser S, et al. 2013. Smart‐seq2 for sensitive full‐length transcriptome profiling in single cells. Nat Methods 10: 1096–8. [DOI] [PubMed] [Google Scholar]
- 6. Sasagawa Y, Nikaido I, Hayashi T, Danno H, et al. 2013. Quartz‐Seq: a highly reproducible and sensitive single‐cell RNA sequencing method, reveals non‐genetic gene‐expression heterogeneity. Genome Biol 14: R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Islam S, Zeisel A, Joost S, La Manno G, et al. 2014. Quantitative single‐cell RNA‐seq with unique molecular identifiers. Nat Methods 11: 163–6. [DOI] [PubMed] [Google Scholar]
- 8. Lovatt D, Ruble BK, Lee J, Dueck H, et al. 2014. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods 11: 190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jaitin DA, Kenigsberg E, Keren‐Shaul H, Elefant N, et al. 2014. Massively parallel single‐cell RNA‐Seq for marker‐free decomposition of tissues into cell types. Science 343: 776–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JH, Daugharthy ER, Scheiman J, Kalhor R, et al. 2014. Highly multiplexed subcellular RNA sequencing in situ. Science 343: 1360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smallwood SA, Lee HJ, Angermueller C, Krueger F, et al. 2014. Single‐cell genome‐wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11: 817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, et al. 2015. Localization and abundance analysis of human lncRNAs at single‐cell and single‐molecule resolution. Genome Biol 16: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen KH, Boettiger AN, Moffitt JR, Wang S, et al. 2015. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348: 6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dey SS, Kester L, Spanjaard B, Bienko M, et al. 2015. Integrated genome and transcriptome sequencing of the same cell. Nat Biotech 33: 285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Satija R, Farrell JA, Gennert D, Schier AF, et al. 2015. Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol 33: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson NK, Kent DG, Buettner F, Shehata M, et al. 2015. Combined single‐cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell 16: 712–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poulin J‐F., Zou J, Drouin‐Ouellet J, Kim K‐YA, et al. 2014. Defining midbrain dopaminergic neuron diversity by single‐cell gene expression profiling. Cell Rep 9: 930–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chiu IM, Barrett LB, Williams EK, Strochlic DE, et al. 2014. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. eLife 3: e04660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Usoskin D, Furlan A, Islam S, Abdo H, et al. 2015. Unbiased classification of sensory neuron types by large‐scale single‐cell RNA sequencing. Nat Neurosci 18: 145–53. [DOI] [PubMed] [Google Scholar]
- 20. Park J, Brureau A, Kernan K, Starks A, et al. 2014. Inputs drive cell phenotype variability. Genome Res 24: 930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marder E, Goaillard J‐M. 2006. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 7: 563–74. [DOI] [PubMed] [Google Scholar]
- 22. Schulz DJ, Goaillard J‐M., Marder EE. 2007. Quantitative expression profiling of identified neurons reveals cell‐specific constraints on highly variable levels of gene expression. Proc Natl Acad Sci USA 104: 13187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Durruthy‐Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, et al. 2014. Reconstruction of the mouse otocyst and early neuroblast lineage at single‐cell resolution. Cell 157: 964–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bieler J, Cannavo R, Gustafson K, Gobet C, et al. 2014. Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Mol Syst Biol 10: 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deng Q, Ramskold D, Reinius B, Sandberg R. 2014. Single‐cell RNA‐seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343: 193–6. [DOI] [PubMed] [Google Scholar]
- 26. McDavid A, Dennis L, Danaher P, Finak G, et al. 2014. Modeling bi‐modality improves characterization of cell cycle on gene expression in single cells. PLoS Comput Biol 10: e1003696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Piras V, Tomita M, Selvarajoo K. 2014. Transcriptome‐wide variability in single embryonic development cells. Sci Rep 4: 7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Treutlein B, Brownfield DG, Wu AR, Neff NF, et al. 2014. Reconstructing lineage hierarchies of the distal lung epithelium using single‐cell RNA‐seq. Nature 509: 371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, et al. 2015. Decoding the regulatory network of early blood development from single‐cell gene expression measurements. Nat Biotech 33: 269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buettner F, Natarajan KN, Casale FP, Proserpio V, et al. 2015. Computational analysis of cell‐to‐cell heterogeneity in single‐cell. Nat Biotechnol 33: 155–60. [DOI] [PubMed] [Google Scholar]
- 31. Piras V, Selvarajoo K. 2015. The reduction of gene expression variability from single cells to populations follows simple statistical laws. Genomics 105: 137–44. [DOI] [PubMed] [Google Scholar]
- 32. Eckersley‐Maslin MA, Thybert D, Bergmann JH, Marioni JC, et al. 2014. Random monoallelic gene expression increases upon embryonic stem cell differentiation. Dev Cell 28: 351–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dueck H, Khaladkar M, Kim TK, Spaethling JM, et al. 2015. Deep sequencing reveals cell‐type‐specific patterns of single‐cell transcriptome variation. Genome Biol 16: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eberwine J, Kim J. 2015. Cellular deconstruction: finding meaning in individual cell variation. Trends Cell Biol 25: 569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yvert G. 2014. “ Particle genetics”: treating every cell as unique. Trends Genet 30: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cagatay T, Turcotte M, Elowitz MB, Garcia‐Ojalvo J, et al. 2009. Architecture‐dependent noise discriminates functionally analogous differentiation circuits. Cell 139: 512–22. [DOI] [PubMed] [Google Scholar]
- 37. Acar M, Mettetal JT, van Oudenaarden A. 2008. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet 40: 471–5. [DOI] [PubMed] [Google Scholar]
- 38. Eldar A, Elowitz MB. 2010. Functional roles for noise in genetic circuits. Nature 467: 167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Losick R, Desplan C. 2008. Stochasticity and cell fate. Science 320: 65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martins BM, Locke JC. 2015. Microbial individuality: how single‐cell heterogeneity enables population level strategies. Cell Regul 24: 104–12. [DOI] [PubMed] [Google Scholar]
- 41. Beaumont HJE, Gallie J, Kost C, Ferguson GC, et al. 2009. Experimental evolution of bet hedging. Nature 462: 90–3. [DOI] [PubMed] [Google Scholar]
- 42. Mineta K, Matsumoto T, Osada N, Araki H. 2015. Population genetics of non‐genetic traits: Evolutionary roles of stochasticity in gene expression. Gene 562: 16–21. [DOI] [PubMed] [Google Scholar]
- 43. Gupta PB, Fillmore CM, Jiang G, Shapira SD, et al. 2011. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146: 633–44. [DOI] [PubMed] [Google Scholar]
- 44. Zhou D, Wang Y, Wu B. 2014. A multi‐phenotypic cancer model with cell plasticity. J Theor Biol 357: 35–45. [DOI] [PubMed] [Google Scholar]
- 45. Lee J, Lee J, Farquhar KS, Yun J, et al. 2014. Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc Natl Acad Sci USA 111: E364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stearns SC. 1976. Life‐history tactics: a review of the ideas. Q Rev Biol 51: 3–47. [DOI] [PubMed] [Google Scholar]
- 47. Kupiec JJ. 1997. A Darwinian theory for the origin of cellular differentiation. Mol Gen Genet 255: 201–8. [DOI] [PubMed] [Google Scholar]
- 48. Briney BS, Jr JEC. 2013. Secondary mechanisms of diversification in the human antibody repertoire. Front Immunol 4: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaeger S, Fernandez B, Ferrier P. 2013. Epigenetic aspects of lymphocyte antigen receptor gene rearrangement or “when stochasticity completes randomness”. Immunology 139: 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Erwin JA, Marchetto MC, Gage FH. 2014. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci 15: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Woods HA. 2014. Mosaic physiology from developmental noise: within‐organism physiological diversity as an alternative to phenotypic plasticity and phenotypic flexibility. J Exp Biol 217: 35–45. [DOI] [PubMed] [Google Scholar]
- 52. Lee M‐CW, Lopez‐Diaz FJ, Khan SY, Tariq MA, et al. 2014. Single‐cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci USA 111: E4726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, et al. 2014. Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim K‐T., Lee HW, Lee H‐O., Kim SC, et al. 2015. Single‐cell mRNA sequencing identifies subclonal heterogeneity in anti‐cancer drug responses of lung adenocarcinoma cells. Genome Biol 16: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh DK, Ku C‐J., Wichaidit C, Steininger RJ 3rd, et al. 2010. Patterns of basal signaling heterogeneity can distinguish cellular populations with different drug sensitivities. Mol Syst Biol 6: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roux J, Hafner M, Bandara S, Sims JJ, et al. 2015. Fractional killing arises from cell‐to‐cell variability in overcoming a caspase activity threshold. Mol Syst Biol 11: 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bertaux F, Stoma S, Drasdo D, Batt G. 2014. Modeling dynamics of cell‐to‐cell variability in TRAIL‐induced apoptosis explains fractional killing and predicts reversible resistance. PLoS Comput Biol 10: e1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ahrends R, Ota A, Kovary KM, Kudo T, et al. 2014. Controlling low rates of cell differentiation through noise and ultrahigh feedback. Science 344: 1384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spencer SL, Gaudet S, Albeck JG, Burke JM, et al. 2009. Non‐genetic origins of cell‐to‐cell variability in TRAIL‐induced apoptosis. Nature 459: 428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Overton KW, Spencer SL, Noderer WL, Meyer T, et al. 2014. Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proc Natl Acad Sci USA 111: E4386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hanski I. 1981. Coexistence of competitors in patchy environment with and without predation. Oikos 37: 306–12. [Google Scholar]
- 62. Chang HH, Hemberg M, Barahona M, Ingber DE, et al. 2008. Transcriptome‐wide noise controls lineage choice in mammalian progenitor cells. Nature 453: 544–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kumar RM, Cahan P, Shalek AK, Satija R, et al. 2014. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature 516: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sansom SN, Shikama‐Dorn N, Zhanybekova S, Nusspaumer G, et al. 2014. Population and single‐cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self‐antigen expression in thymic epithelia. Genome Res 24: 1918–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ohnishi Y, Huber W, Tsumura A, Kang M, et al. 2014. Cell‐to‐cell expression variability followed by signal reinforcement progressively segregates early mouse lineages. Nat Cell Biol 16: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Singer ZS, Yong J, Tischler J, Hackett JA, et al. 2014. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 55: 319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Badzey RL, Mohanty P. 2005. Coherent signal amplification in bistable nanomechanical oscillators by stochastic resonance. Nature 437: 995–8. [DOI] [PubMed] [Google Scholar]
- 68. Hogquist KA, Baldwin TA, Jameson SC. 2005. Central tolerance: learning self‐control in the thymus. Nat Rev Immunol 5: 772–82. [DOI] [PubMed] [Google Scholar]
- 69. Muotri AR, Chu VT, Marchetto MCN, Deng W, et al. 2005. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435: 903–10. [DOI] [PubMed] [Google Scholar]
- 70. Muotri AR, Marchetto MCN, Coufal NG, Oefner R, et al. 2010. L1 retrotransposition in neurons is modulated by Me CP2. Nature 468: 443–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. O'Rourke NA, Weiler NC, Micheva KD, Smith SJ. 2012. Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat Rev Neurosci 13: 365–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rand U, Rinas M, Schwerk J, Nohren G, et al. 2012. Multi‐layered stochasticity and paracrine signal propagation shape the type‐I interferon response. Mol Syst Biol 8: 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Patil S, Fribourg M, Ge Y, Batish M, et al. 2015. Single‐cell analysis shows that paracrine signaling by first responder cells shapes the interferon‐beta response to viral infection. Sci Signal 8: ra6. [DOI] [PubMed] [Google Scholar]
- 74. Shalek AK, Satija R, Shuga J, Trombetta JJ, et al. 2014. Single‐cell RNA‐seq reveals dynamic paracrine control of cellular variation. Nature 510: 363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xue Q, Lu Y, Eisele MR, Sulistijo ES, et al. 2015. Analysis of single‐cell cytokine secretion reveals a role for paracrine signaling in coordinating macrophage responses to TLR4 stimulation. Sci Signal 8: ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fang M, Xie H, Dougan SK, Ploegh H, et al. 2013. Stochastic cytokine expression induces mixed T helper cell states. PLoS Biol 11: e1001618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bajikar SS, Fuchs C, Roller A, Theis FJ, et al. 2014. Parameterizing cell‐to‐cell regulatory heterogeneities via stochastic transcriptional profiles. Proc Natl Acad Sci USA 111: E626–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kimura M. 1983. The Neutral Theory of Molecular Evolution. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 79. Grun D, Kester L, van Oudenaarden A. 2014. Validation of noise models for single‐cell transcriptomics. Nat Methods 11: 637–40. [DOI] [PubMed] [Google Scholar]
- 80. Morris KV, Mattick JS. 2014. The rise of regulatory RNA. Nat Rev Genet 15: 423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Panning B, Segal E. 2015. Editorial overview: Genome architecture and expression. Curr Opin Genet Dev 31: 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sharon E, van Dijk D, Kalma Y, Keren L, et al. 2014. Probing the effect of promoters on noise in gene expression using thousands of designed sequences. Genome Res 24: 1698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stearns SC. 2002. Progress on canalization. Proc Natl Acad Sci USA 99: 10229–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lagha M, Bothma JP, Esposito E, Ng S, et al. 2013. Paused Pol II coordinates tissue morphogenesis in the Drosophila embryo. Cell 153: 976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, et al. 2014. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158: 673–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dey SS, Foley JE, Limsirichai P, Schaffer DV, et al. 2015. Orthogonal control of expression mean and variance by epigenetic features at different genomic loci. Mol Syst Biol 11: 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dar RD, Hosmane NN, Arkin MR, Siliciano RF, et al. 2014. Screening for noise in gene expression identifies drug synergies. Science 344: 1392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Senecal A, Munsky B, Proux F, Ly N, et al. 2014. Transcription factors modulate c‐Fos transcriptional bursts. Cell Rep 8: 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vinuelas J, Kaneko G, Coulon A, Vallin E, et al. 2013. Quantifying the contribution of chromatin dynamics to stochastic gene expression reveals long, locus‐dependent periods between transcriptional bursts. BMC Biol 11: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kim J, Eberwine J. 2010. RNA: state memory and mediator of cellular phenotype. Trends Cell Biol 20: 311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lin X, Ruan X, Anderson MG, McDowell JA, et al. 2005. siRNA‐mediated off‐target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res 33: 4527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Persengiev SP, Zhu X, Green MR. 2004. Nonspecific, concentration‐dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA 10: 12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Scacheri PC, Rozenblatt‐Rosen O, Caplen NJ, Wolfsberg TG, et al. 2004. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA 101: 1892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jackson AL, Bartz SR, Schelter J, Kobayashi SV, et al. 2003. Expression profiling reveals off‐target gene regulation by RNAi. Nat Biotechnol 21: 635–7. [DOI] [PubMed] [Google Scholar]
- 95. Weidenfeld I. 2012. Inducible microRNA‐mediated knockdown of the endogenous human lamin A/C gene. Methods Mol Biol 815: 289–305. [DOI] [PubMed] [Google Scholar]
- 96. Sul J‐Y, Wu C‐w.K, Zeng F, Jochems J, et al. 2009. Transcriptome transfer produces a predictable cellular phenotype. Proc Natl Acad Sci USA 106: 7624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tenen DG, Hromas R, Licht JD, Zhang DE. 1997. Transcription factors, normal myeloid development, and leukemia. Blood 90: 489–519. [PubMed] [Google Scholar]
- 98. Schulze W, Hayata‐Takano A, Kamo T, Nakazawa T, et al. 2015. Simultaneous neuron‐ and astrocyte‐specific fluorescent marking. Biochem Biophys Res Commun 459: 81–6. [DOI] [PubMed] [Google Scholar]
- 99. Liu G, Sprenger C, Wu P‐J., Sun S, et al. 2015. MED1 mediates androgen receptor splice variant induced gene expression in the absence of ligand. Oncotarget 6: 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ambros V. 2003. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell 113: 673–6. [DOI] [PubMed] [Google Scholar]
- 101. Buhagiar A, Ayers D. 2015. Chemoresistance, cancer stem cells, and miRNA influences: the case for neuroblastoma. Anal Cell Pathol Amst 2015: 150634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schmiedel JM, Klemm SL, Zheng Y, Sahay A, et al. 2015. Gene expression. MicroRNA control of protein expression noise. Science 348: 128–32. [DOI] [PubMed] [Google Scholar]
- 103. Todhunter ME, Jee NY, Hughes AJ, Coyle MC, et al. 2015. Programmed synthesis of three‐dimensional tissues. Nat Methods 12: 975–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu JS, Farlow JT, Paulson AK, Labarge MA, et al. 2012. Programmed cell‐to‐cell variability in Ras activity triggers emergent behaviors during mammary epithelial morphogenesis. Cell Rep 2: 1461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Grun D, Lyubimova A, Kester L, Wiebrands K, et al. 2015. Single‐cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525: 251–5. [DOI] [PubMed] [Google Scholar]
- 106. Mayr E. 1942. Systematics and the Origin of Species, from the Viewpoint of a Zoologist. Cambridge, MA: Harvard University Press. [Google Scholar]
- 107. Paldi A. 2003. Stochastic gene expression during cell differentiation: order from disorder? Cell Mol Life Sci 60: 1775–8. [DOI] [PMC free article] [PubMed] [Google Scholar]