

Abstract
An N,N′‐dioxide/scandium(III) complex catalyzed, highly efficient conjugate addition of malonic esters to enynes is described. A range of trisubstituted 1,2‐allenyl ketones were obtained in high yields (up to 99 %) with good d.r. (up to 95/5) and excellent ee values (97 %–99 %). Moreover, the products could be easily transformed into chiral furan and 5‐hydroxypyrazoline derivatives, both of which are important skeletons of many biologically active compounds and pharmacologicals.
Keywords: 1,2-allenyl ketones; 5-hydroxypyrazoline; asymmetric catalysis; enyne addition; furans
Chiral allenes are valuable compounds for their importance in organic synthesis and pharmaceuticals.1, 2 Over the past two decades, substantial efforts have been devoted to their asymmetric synthesis.3 Early successful examples were limited to the use of stoichiometric amounts of chiral auxiliaries/promoters or enantioenriched substrates.4 Until now, some ingenious methods for catalytic asymmetric synthesis of chiral allenes have been reported. They include the isomerization of 3‐alkynes,5 kinetic resolution of racemic allenes,6 β‐hydride elimination of enol triflates,7 desymmetrization of meso‐allenes8 or alkyenes,9 rearrangements of alkynes,10 C−H insertion of α‐diazoesters into 1‐alkynes,11 functionalization of racemic allenes,12 and addition to enynes.13 Among them, addition to enynes is a simple but very efficient route to obtain chiral allenes. For instance, Hayashi has pioneered a metal‐catalyzed enyne addition for synthesis of chiral allenes (Scheme 1 a).13a–13c Subsequently, Tang has provided an intramolecular enyne addition for the highly diastereo‐ and enantioselective synthesis of bromoallenes in the presence of chiral ureas (Scheme 1 b).13d Zhang has developed an intermolecular enyne addition in the assistance of chiral thioureas (Scheme 1 c).13e Although a series of 2,3‐allenoates were obtained with up to 98 % ee value, there was only one example containing the axial and carbon center chirality with 4:1 d.r. Furthermore, the reaction also gave the byproduct alkynoate as a result of direct conjugate addition (Scheme 1 c). Zhang has also reported two racemic enyne addtion to construct contiguous axial and carbon center,14 but the catalytic asymmetric manner is still unrealized. In short, the highly diastereoselective and enantioselective construction of continuous axial and carbon center chirality by intermolecular enyne addition is a challenge. Stimulated by the successful application of our unique catalysts in conjugate addition,15 herein, we report a highly efficient conjugate addition of malonic esters to enynes catalyzed by an N,N′‐dioxide/scandium(III) complex, affording a range of trisubstituted 1,2‐allenyl ketones. It should be mentioned that the direct conjugate addition product alkynones 4 were not detected during the reaction course (Scheme 1).
Scheme 1.
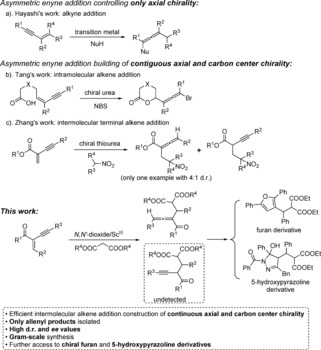
Comparison of previous catalytic asymmetric enyne addition for synthesis of chiral allenes with current work.
Our investigation began with the addition of diethyl malonate (2 b) to enyne (1 a) as the model reaction to optimize the reaction conditions. Initially, chiral ligands were evaluated (Table 1, entries 1–3). The results showed that by complexing with Sc(OTf)3, l‐proline‐derived l ‐PrEt2Me, l‐ramipril‐derived l‐RaEt2Me, and l‐pipecolic acid‐derived l‐PiEt2Me, all of the complexes could promote the reaction smoothly in the presence of 4‐dimethylaminopyridine (DMAP) at 30 °C. However, the diastereoselectivity was low even though the enantioselectivity was moderate to excellent. To improve the diastereoselectivity, other bases were investigated (Table 1, entries 4–6). To our delight, nBu3N gave the highest 88:12 d.r. (Table 1, entry 6). Gratifyingly, when the reaction temperature was decreased to 0 °C, the diastereoselectivity could be further enhanced to 92:8 (Table 1, entry 7). Unfortunately, the d.r. value could not be improved significantly (93:7) by continuing to decrease the reaction temperature to −5 °C (Table 1, entry 8). Thus, the optimized reaction conditions were established as Sc(OTf)3/l‐PiEt2Me (10 mol %), nBu3N (10 mol %), 1 a:2 b=1:4 in CH2Cl2 at 0 °C.
Table 1.
Optimization of the reaction conditions.[a]
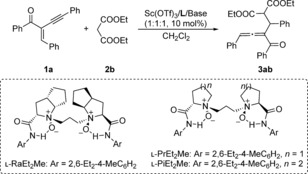
| Entry | L | Base | Yield [%][b] | ee [%][c] | d.r.[d] |
|---|---|---|---|---|---|
| 1 | l‐PrEt2Me | DMAP | 98 | 76:76 | 67:33 |
| 2 | l‐RaEt2Me | DMAP | 98 | 99:99 | 64:36 |
| 3 | l‐PiEt2Me | DMAP | 98 | 99:99 | 68:32 |
| 4 | l‐PiEt2Me | Na2CO3 | 93 | 98:99 | 84:16 |
| 5 | l‐PiEt2Me | iPr2EtN | 89 | 95:95 | 86:14 |
| 6 | l‐PiEt2Me | nBu3N | 88 | 98:98 | 88:12 |
| 7[e] | l‐PiEt2Me | nBu3N | 85 | 99:99 | 92:8 |
| 8[f] | l‐PiEt2Me | nBu3N | 66 | 99:99 | 93:7 |
[a] Unless otherwise noted, reactions were carried out with L (10 mol %), Sc(OTf)3 (10 mol %), base (10 mol %), 1 a (0.2 mmol), 2 b (0.8 mmol) in CH2Cl2 (0.8 mL) at 30 °C for 24 h. [b] Yield of isolated product. [c] Determined by chiral HPLC analysis. [d] Determined by 1H NMR analysis. [e] Reactions were carried out at 0 °C. [f] Reactions were carried out at −5 °C.
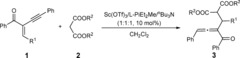
With the optimized conditions in hand, the reaction scope was next examined. As shown in Table 2, enynes tethering aryl or alkyl substituents (R1) on the carbonyl group are suitable substrates, providing the desired trisubstituted 1,2‐allenyl ketones in high yields with high d.r. and excellent ee values (Table 2, entries 1–4). Regardless of the electron‐withdrawing or electron‐donating groups on the 4‐position of aromatic rings on the double bond (R2), there was little influence on the reaction. The corresponding products were obtained in high yields (88–94 %) with excellent d.r. (92:8–95:5) and ee values (99 %) (Table 2, entries 6–9). When the R2 group was changed to 2‐chlorophenyl, it just gave a moderate yield (77 %) and d.r. (78:22; Table 2, entries 5). Various substituents (R3) on the triple bond were also examined. The aryl groups bearing an electron‐donating substituent furnished higher diastereoselectivities than electron‐withdrawing groups (Table 2, entries 15–17 vs. entries 10–14). The enyne 1 r containing a 3‐thienyl group afforded the desired product 3 rb in 90 % yield with 97 % ee and 94:6 d.r. by decreasing the reaction temperature to −5 °C (Table 2, entry 18). The n‐butyl‐substituted 1 s was also a competent substrate, providing the allene 3 sb with good results (Table 2, entry 19).
Table 2.
Substrate scope for enynes.[a,b]

| Entry | R1/R2/R3 | Yield [%][c] | ee [%][d] | d.r.[e] |
|---|---|---|---|---|
| 1 | Ph/Ph/Ph | 85 (3 ab) | 99 | 92:8 |
| 2 | 3‐MeC6H4/Ph/Ph | 84 (3 bb) | 99 | 91:9 |
| 3[f] | 3‐ClC6H4/Ph/Ph | 98 (3 cb) | 99 | 91:9 |
| 4 | CH3/Ph/Ph | 92 (3 db) | 98 | 89:11 |
| 5 | Ph/2‐ClC6H4/Ph | 77 (3 eb) | 97 | 78:22 |
| 6[f] | Ph/4‐ClC6H4/Ph | 90 (3 fb) | 99 | 92:8 |
| 7[f] | Ph/4‐BrC6H4/Ph | 94 (3 gb) | 99 | 93:7 |
| 8[f] | Ph/4‐MeC6H4/Ph | 90 (3 hb) | 99 | 94:6 |
| 9 | Ph/4‐MeOC6H4/Ph | 88 (3 ib) | 99 | 95:5 |
| 10[f] | Ph/Ph/2‐FC6H4 | 94 (3 jb) | 99 | 91:9 |
| 11[f] | Ph/Ph/3‐FC6H4 | 91 (3 kb) | 99 | 92:8 |
| 12 | Ph/Ph/4‐FC6H4 | 90 (3 lb) | 99 | 88:12 |
| 13 | Ph/Ph/2‐ClC6H4 | 93 (3 mb) | 99 | 89:11 |
| 14[f] | Ph/Ph/4‐ClC6H4 | 96 (3 nb) | 99 | 89:11 |
| 15[f] | Ph/Ph/3‐MeC6H4 | 75 (3 ob) | 99 | 94:6 |
| 16 | Ph/Ph/4‐MeC6H4 | 91 (3 pb) | 99 | 93:7 |
| 17 | Ph/Ph/4‐MeOC6H4 | 59 (3 qb) | 99 | 93:7 |
| 18[f] | Ph/Ph/3‐Thienyl | 90 (3 rb) | 97 | 94:6 |
| 19 | Ph/Ph/nBu | 73 (3 sb) | 99 | 89:11 |
[a] Unless otherwise noted, reactions were carried out with l‐PiEt2Me (10 mol %), Sc(OTf)3 (10 mol %), nBu3N (10 mol %), 1 (0.2 mmol), 2 b (0.8 mmol) in CH2Cl2 (0.8 mL) at 0 °C for 24 h. [b] Enyne 1 t (R1=OEt, R2=H, and R3=Ph) was also examined. For results, see the Supporting Information. [c] Yield of isolated product. [d] Determined by chiral HPLC analysis. [e] Determined by 1H NMR analysis. [f] Reactions were carried out at −5 °C.
Several malonic esters 2 were also tested (Table 3). Although the reactivity and diastereoselectivity markedly decreased with the increased steric hindrance of ester alkyl group (R2), the ee values were maintained. To determine the absolute configuration of products, 3 cd was synthesized (Table 3). After recrystallized from CH2Cl2, 3 cd with >99:1 d.r. and >99 % ee could be obtained. The absolute configuration of 3 cd was determined by X‐ray crystallography to be (R,Sa).16
Table 3.
Substrate scope for malonic esters.[a]
|
|---|
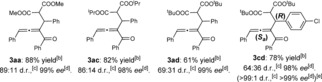
|
[a] Reactions were carried out with l‐PiEt2Me (10 mol %), Sc(OTf)3 (10 mol %), nBu3N (10 mol %), 1 (0.2 mmol), 2 (0.8 mmol) in CH2Cl2 (0.8 mL) at 0 °C (for 3 aa) or 30 °C (for 3 ac, 3 ad, and 3 cd) for 24 h. [b] Yield of isolated product. [c] Determined by 1H NMR analysis. [d] Determined by chiral HPLC analysis. [e] After recrystallization from CH2Cl2, the absolute configuration of 3 cd was determined by X‐ray crystallography.
To show the synthetic potential of this strategy, a gram scale synthesis of 3 ab was carried out (Scheme 2 a). Under the optimized condition, 3 mmol of 1 a reacted smoothly with 4 equivalents of 2 b to provide 1.27 g 3 ab (91 % yield) with 91:9 d.r. and 99 % ee. Furthermore, when treated with 1.2 equiv of 4 a at 30 °C, 3 ab could be transformed to the tetrasustituted 5‐hydroxypyrazoline 5 a in 62 % yield with 54:46 d.r. (99 % ee each).17 Importantly, the family of 5 a has been reported to possess antimicrobial,18 anti‐PDE3,19 anti‐inflammatory,20 antimalarial,21 hypolipidemic,22 and analgesic23 activities (Scheme 2 b). Additionally, 3 ab could also be transformed to trisustituted furan 5 b under treatment by AuCl3 in 79 % yield with 99 % ee,24, 25 which are important skeletons for many biologically active compounds and pharmacologicals (Scheme 2 b).
Scheme 2.

a) Gram‐scale version of the reaction. b) Transformation of 1,2‐allenyl ketone 3 ab.
To gain insight into the function of nBu3N in our system, some control experiments were carried out (Scheme 3). First, when the reaction of 1 a and 2 b was performed in the absence of nBu3N (Scheme 3 a), it gave a trace amount of 3 ab. This suggested that nBu3N served as an effective promoter for the formation of enolate from 2. Second, when 3 ab (91:9 d.r. and 99 % ee) was dissolved in CH2Cl2 and stirred at room temperature for 40 h, there was no influence on its d.r. and ee values. In contrast, when nBu3N was added to the mixture, the d.r. value rapidly decreased to 62:38 (Scheme 3 b). Clearly, nBu3N was the cause of the reducion in the d.r. value of 3 ab. An additional derivation of the recovered 3 ab (62:38 d.r.) was undertaken, and the produced furan was obtained with 99 % ee. This demonstrated that the low d.r. was attributed to the partial racemization of axial chirality of the allene (Scheme 3 b). In other words, nBu3N might not participate in the allenes production, but instead serves as an effective promoter for the formation of enolates from 2 in the catalytic system.
Scheme 3.

Control experiments.
Based on the determination of the absolute configuration of product 3 cd,16 control experiments, and our previous study,26 a possible catalytic cycle with a transition‐state model was proposed (Figure 1). First, the N‐oxides and amide oxygen atoms of l‐PiEt2Me coordinate to Sc3+ in a tetradentate manner to form two six‐membered chelate rings. Then, enynes 1 attach to Sc3+ at the favorable equatorial position to give intermediate T1. Additionally, the enolates are generated from malonates 2 assisted by nBu3N. The Re face of enynes 1 are strongly shielded by the nearby benzyl ring. Therefore, the incoming enolates prefers to attack enynes 1 from the Si face (T2). Finally, the desired products 3 dissociat after a protonation of the favored intermediate T3, and the catalyst is regenerated to accomplish one catalytic cycle.
Figure 1.

Proposed catalytic cycle.
In summary, we have developed an efficient N,N′‐dioxide/scandium complex catalyst for the intermolecular addition of malonic esters to enynes. A range of trisubstituted 1,2‐allenyl ketones were obtained in good to excellent yields (up to 99 %) with high d.r. (up to 95:5) and excellent ee values (up to 99 %). The products could be easily transformed into furan and 5‐hydroxypyrazoline derivatives, which are important skeletons in many biologically active compound and pharmacologicals. Moreover, based on the experiments and our previous work, a possible catalytic cycle was proposed.
Experimental Section
A dry reaction tube was charged with l‐PiEt2Me (0.02 mmol) and Sc(OTf)3 (0.02 mmol), enyne 1 a (0.2 mmol) under an N2 atmosphere, malonic ester 2 b (0.8 mmol), and CH2Cl2 (0.8 mL) were added and the mixture was stirred at 0 °C for 15 min. Then nBu3N (0.02 mmol) was added and the mixture was stirred at 0 °C for 24 h. The reaction mixture was purified by column chromatography on silica gel (ethyl acetate/petroleum ether 1:50‐1:4) to afford the desired product 3 ab (85 % yield, 99 % ee, 92:8 d.r.).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We appreciate the National Natural Science Foundation of China (Nos. 21372162, 21432006, and 21321061) for financial support.
Q. Yao, Y. Liao, L. Lin, X. Lin, J. Ji, X. Liu, X. Feng, Angew. Chem. Int. Ed. 2016, 55, 1859.
Contributor Information
Prof. Dr. Xiaohua Liu, Email: liuxh@scu.edu.cn
Prof. Dr. Xiaoming Feng, Email: xmfeng@scu.edu.cn
References
- 1.For recent reviews on allenes in organic synthesis, see:
- 1a. Ma S., Acc. Chem. Res. 2003, 36, 701; [DOI] [PubMed] [Google Scholar]
- 1b. Ma S., Chem. Rev. 2005, 105, 2829; [DOI] [PubMed] [Google Scholar]
- 1c. Cowen B. J., Miller S. J., Chem. Soc. Rev. 2009, 38, 3102; [DOI] [PubMed] [Google Scholar]
- 1d. Aubert C., Fensterbank L., Garcia P., Malacria M., Simonneau A., Chem. Rev. 2011, 111, 1954; [DOI] [PubMed] [Google Scholar]
- 1e. Yu S., Ma S., Angew. Chem. Int. Ed. 2012, 51, 3074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3128; [Google Scholar]
- 1f. Ye J., Ma S., Acc. Chem. Res. 2014, 47, 989. [DOI] [PubMed] [Google Scholar]
- 2. Hoffmann-Röder A., Krause N., Angew. Chem. Int. Ed. 2004, 43, 1196; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 1216. [Google Scholar]
- 3.For recent reviews on asymmetric synthesis of allenes, see:
- 3a. Ogasawara M., Tetrahedron: Asymmetry 2009, 20, 259; [Google Scholar]
- 3b. Neff R. K., Frantz D. E., ACS Catal. 2014, 4, 519. [Google Scholar]
- 4.For selected early examples of asymmetric synthesis of allenes using stoichiometric amounts of chiral auxiliaries/promoters or enantioenriched substrates, see:
- 4a. Nishibayashi Y., Singh J. D., Fukuzawa S.-i., Uemura S., J. Org. Chem. 1995, 60, 4114; [Google Scholar]
- 4b. Marshall J. A., Wolf M. A., J. Org. Chem. 1996, 61, 3238; [Google Scholar]
- 4c. Naruse Y., Watanabe H., Ishiyama Y., Yoshida T., J. Org. Chem. 1997, 62, 3862; [Google Scholar]
- 4d. Mikami K., Yoshida A., Angew. Chem. Int. Ed. Engl. 1997, 36, 858; [Google Scholar]; Angew. Chem. 1997, 109, 892. [Google Scholar]
- 5. Liu H., Leow D., Huang K.-W., Tan C.-H., J. Am. Chem. Soc. 2009, 131, 7212. [DOI] [PubMed] [Google Scholar]
- 6. Yu J., Chen W.-J., Gong L.-Z., Org. Lett. 2010, 12, 4050. [DOI] [PubMed] [Google Scholar]
- 7. Crouch I. T., Neff R. K., Frantz D. E., J. Am. Chem. Soc. 2013, 135, 4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manzuna Sapu C., Bäckvall J. E., Deska J., Angew. Chem. Int. Ed. 2011, 50, 9731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9905. [Google Scholar]
- 9. Li H., Müller D., Guénée L., Alexakis A., Org. Lett. 2012, 14, 5880. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Li Z., Boyarskikh V., Hansen J. H., Autschbach J., Musaev D., Davies H. M. L., J. Am. Chem. Soc. 2012, 134, 15497; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Wang Y., Zhang W., Ma S., J. Am. Chem. Soc. 2013, 135, 11517; [DOI] [PubMed] [Google Scholar]
- 10c. Liu Y. B., Hu H. P., Zheng H. F., Xia Y., Liu X. H., Lin L. L., Feng X. M., Angew. Chem. Int. Ed. 2014, 53, 11579; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11763. [Google Scholar]
- 11. Tang Y., Chen Q. G., Liu X. H., G, Wang , Lin L. L., Feng X. M., Angew. Chem. Int. Ed. 2015, 54, 9512; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9648. [Google Scholar]
- 12.
- 12a. Imada Y., Nishida M., Kutsuwa K., Murahashi S.-I., Naota T., Org. Lett. 2005, 7, 5837; [DOI] [PubMed] [Google Scholar]
- 12b. Boutier A., Kammerer-Pentier C., Krause N., Prestat G., Poli G., Chem. Eur. J. 2012, 18, 3840; [DOI] [PubMed] [Google Scholar]
- 12c. Hashimoto T., Sakata K., Tamakuni F., Dutton M. J., Maruoka K., Nat. Chem. 2013, 5, 240; [DOI] [PubMed] [Google Scholar]
- 12d. Wan B., Ma S., Angew. Chem. Int. Ed. 2013, 52, 441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 459; [Google Scholar]
- 12e. Mbofana C. T., Miller S. J., J. Am. Chem. Soc. 2014, 136, 3285. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Han J. W., Tokunaga N., Hayashi T., J. Am. Chem. Soc. 2001, 123, 12915; [DOI] [PubMed] [Google Scholar]
- 13b. Hayashi T., Tokunaga N., Inoue K., Org. Lett. 2004, 6, 305; [DOI] [PubMed] [Google Scholar]
- 13c. Nishimura T., Makino H., Nagaosa M., Hayashi T., J. Am. Chem. Soc. 2010, 132, 12865; [DOI] [PubMed] [Google Scholar]
- 13d. Zhang W., Zheng S. Q., Liu N., Werness J. B., Guzei I. A., Tang W. P., J. Am. Chem. Soc. 2010, 132, 3664; [DOI] [PubMed] [Google Scholar]
- 13e. Qian H., Yu X., Zhang J., Sun J., J. Am. Chem. Soc. 2013, 135, 18020. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Yu X., Ren H., Xiao Y., Zhang J., Chem. Eur. J. 2008, 14, 8481; [DOI] [PubMed] [Google Scholar]
- 14b. Yu X., Zhang J., Adv. Synth. Catal. 2011, 353, 1265. [Google Scholar]
- 15.For reviews on chiral N-oxides in asymmetric catalysis, see:
- 15a. Malkov A. V., Kočovsky P., Curr. Org. Chem. 2003, 7, 1737; [Google Scholar]
- 15b. Chelucci G., Murineddu G., Pinna G. A., Tetrahedron: Asymmetry 2004, 15, 1373; [Google Scholar]
- 15c. Malkov A. V., Kočovsky P., Eur. J. Org. Chem. 2007, 29; [Google Scholar]
- 15d. Feng X. M., Liu X. H., in Scandium: Compounds, Productions and Applicatons (Ed: V. A. Greene), Nova Science, New York, 2011, pp. 1–48; [Google Scholar]
- 15e. Liu X. H., Lin L. L., Feng X. M., Acc. Chem. Res. 2011, 44, 574; [DOI] [PubMed] [Google Scholar]
- 15f. Shen K., Liu X. H., Lin L. L., Feng X. M., Chem. Sci. 2012, 3, 327; [Google Scholar]
- 15g. Liu X. H., Lin L. L., Feng X. M., Org. Chem. Front. 2014, 1, 298; For recent examples on asymmetric conjugate addition, see: [Google Scholar]
- 15h. Wang Z., Zhang Z. L., Yao Q., Liu X. H., Cai Y. F., Lin L. L., Feng X. M., Chem. Eur. J. 2013, 19, 8591; [DOI] [PubMed] [Google Scholar]
- 15i. Wang Z., Yao Q., Kang T. F., Feng J. H., Liu X. H., Lin L. L., Feng X. M., Chem. Commun. 2014, 50, 4918; [DOI] [PubMed] [Google Scholar]
- 15j. Yao Q., Wang Z., Zhang Y. H., Liu X. H., Lin L. L., Feng X. M., J. Org. Chem. 2015, 80, 5704. [DOI] [PubMed] [Google Scholar]
- 16.CCDC 1044535 (3 cd).
- 17. Guo S., Wang J., Guo D., Zhang X., Fan X., Tetrahedron 2012, 68, 7768. [Google Scholar]
- 18.
- 18a. Zhao Y., Bacher A., Illarionov B., Fischer M., Georg G., Ye Q.-Z., Fanwick P. E., Franzblau S. G., Wan B., Cushman M., J. Org. Chem. 2009, 74, 5297; [DOI] [PubMed] [Google Scholar]
- 18b. Karthikeyan M. S., Holla B. S., Kumari N. S., Eur. J. Med. Chem. 2007, 42, 30; [DOI] [PubMed] [Google Scholar]
- 18c. Bonacorso H. G., Wentz A. P., Lourega R. V., Cechinel C. A., Moraes T. S., Coelho H. S., Zanatta N., Martins M. A. P., Höerner M., Alves S. H., J. Fluorine Chem. 2006, 127, 1066. [Google Scholar]
- 19. Kim K. Y., Lee H., Yoo S.-E., Kim S. H., Kang N. S., Bioorg. Med. Chem. Lett. 2011, 21, 1617. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Sauzem P. D., Machado P., Rubin M. A., Sant′Anna G. S., Faber H. B., de Souza A. H., Mello C. F., Beck P., Burrow R. A., Bonacorso H. G., Zanatta N., Martins M. A. P., Eur. J. Med. Chem. 2008, 43, 1237; [DOI] [PubMed] [Google Scholar]
- 20b. de Souza F. R., Fighera M. R., Lima T. T. F., de Bastiani J., Barcellos I. B., Almeida C. E., Oliveira M. R., Bonacorso H. G., Flores A. E., Pharmacol. Biochem. Behav. 2001, 68, 525. [DOI] [PubMed] [Google Scholar]
- 21. Cunico W., Cechinel C. A., Bonacorso H. G., Martins M. A. P., Zanatta N., de Souza M. V. N., Freitas I. O., Soares R. P. P., Krettli A. U., Bioorg. Med. Chem. Lett. 2006, 16, 649. [DOI] [PubMed] [Google Scholar]
- 22. Idrees G. A., Aly O. M., Abuo-Rahma G. E.-D. A. A., Radwan M. F., Eur. J. Med. Chem. 2009, 44, 3973. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Machado P., Rosa F. A., Rossatto M., Sant'Anna G. S., Sauzem P. D., Siqueira da Silva R. M., Rubin M. A., Ferreira J., Bonacorso H. G., Zanatta N., Martins M. A. P., ARKIVOC 2007, 281; [Google Scholar]
- 23b. Machado P., Campos P. T., Lima G. R., Rosa F. A., Flores A. F. C., Bonacorso H. G., Zanatta N., Martins M. A. P., J. Mol. Struct. 2009, 917, 176. [Google Scholar]
- 24.
- 24a. Dudnik A. S., Sromek A. W., Rubina M., Kim J. T., Kel'in A. V., Gevorgyan V., J. Am. Chem. Soc. 2008, 130, 1440; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Hashmi A. S. K., Schwarz L., Choi J.-H., Frost T. M., Angew. Chem. Int. Ed. 2000, 39, 2285; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2382. [Google Scholar]
- 25.
- 25a. Nicolaou K. C., Sorensen E. J., Classics in Total Synthesis, VCH, Weinheim, 1996; [Google Scholar]
- 25b. Pozharskii A. F., Soldatenkov A. T., Katritzky A. R., in Heterocycles in Life and Society, Wiley, Chichester, 1997; [Google Scholar]
- 25c. Lee H.-K., Chan K.-F., Hui C.-W., Yim H.-K., Wu X.-W., Wong H. N. C., Pure Appl. Chem. 2005, 77, 139; [Google Scholar]
- 25d. Ash M., Ash I., in Handbook of Flavors and Fragrances, Synapse Information Resources, New York, 2006. [Google Scholar]
- 26. Wang Z., Chen D. H., Yang Z. Y., Bai S., Liu X. H., Lin L. L., Feng X. M., Chem. Eur. J. 2010, 16, 10130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary