

Abstract
Apolipoprotein E4 (apoE4) is the most prevalent genetic risk factor for Alzheimer's disease. We utilized apoE4‐targeted replacement mice (approved by the Tel Aviv University Animal Care Committee) to investigate whether cholinergic dysfunction, which increases during aging and is a hallmark of Alzheimer's disease, is accentuated by apoE4. This revealed that levels of the pre‐synaptic cholinergic marker, vesicular acetylcholine transporter in the hippocampus and the corresponding electrically evoked release of acetylcholine, are similar in 4‐month‐old apoE4 and apolipoprotein E3 (apoE3) mice. Both parameters decrease with age. This decrease is, however, significantly more pronounced in the apoE4 mice. The levels of cholinacetyltransferase (ChAT), acetylcholinesterase (AChE), and butyrylcholinesterase (BuChE) were similar in the hippocampus of young apoE4 and apoE3 mice and decreased during aging. For ChAT, this decrease was similar in the apoE4 and apoE3 mice, whereas it was more pronounced in the apoE4 mice, regarding their corresponding AChE and BuChE levels. The level of muscarinic receptors was higher in the apoE4 than in the apoE3 mice at 4 months and increased to similar levels with age. However, the relative representation of the M1 receptor subtype decreased during aging in apoE4 mice. These results demonstrate impairment of the evoked release of acetylcholine in hippocampus by apoE4 in 12‐month‐old mice but not in 4‐month‐old mice. The levels of ChAT and the extent of the M2 receptor‐mediated autoregulation of ACh release were similar in the adult mice, suggesting that the apoE4‐related inhibition of hippocampal ACh release in these mice is not driven by these parameters.
Evoked ACh release from hippocampal and cortical slices is similar in 4‐month‐old apoE4 and apoE3 mice but is specifically and significantly reduced in hippocampus, but not cortex, of 12‐month‐old apoE4 mice. This effect is accompanied by decreased VAChT levels. These findings show that the hipocampal cholinergic nerve terminals are specifically affected by apoE4 and that this effect is age dependent.
Keywords: acetylcholine release, Alzheimer's disease (AD), apolipoprotein E4 (apoE4), hippocampus
Abbreviations used
- [3H]‐NMS
[3H]‐N‐methylscopolamine
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AD
Alzheimer's disease
- apoE3
apolipoprotein E3
- apoE4
apolipoprotein E4
- BuChE
butyrylcholinesterase
- ChAT
choline acetyltransferase
- GTP‐γ35S
guanosine 5′‐(γ35‐thio)‐triphosphate
- LTP
long‐term potentiation
- TR
targeted replacement
- VAChT
vesicular acetylcholine transporter
Cholinergic impairments are a hallmark of Alzheimer's disease (AD) (Craig et al. 2011). Genetic studies revealed that the allele ε4 of apolipoprotein (APOE4 gene, apoE4 protein) is the most prevalent genetic risk factor for AD (Corder et al. 1993; Saunders et al. 1993). ApoE4 in AD and normal subjects is associated with biochemical cholinergic impairments (Reinvang et al. 2013). Furthermore, the response of apoE4‐carrying AD patients (Braga et al. 2015) and normal controls (Pomara et al. 2004) to pharmacological cholinergic activation is impaired relative to corresponding subjects who express apolipoprotein E3 (apoE3), the AD benign isoform of this molecule. However, the extent to which apoE4 affects the release of acetylcholine (ACh) has not yet been determined.
Transgenic mice have been prepared that express apoE4 or apoE3 under the regulation of either astrocytic or neuronal promoters, in addition to apoE‐targeted replacement (TR) mice in which the endogenous mouse apoE was replaced by human apoE4 and apoE3 (Sullivan et al. 2011). Of these mice, the TR apoE mice have become the standard model in the field. Measurement of long‐term potentiation (LTP) revealed a lower response in apoE4 TR mice (Yun et al. 2005). This effect is replicated in control mice by cholinergic antagonists (Yun et al. 2005), suggesting that the apoE4‐driven decrease in LTP is related to cholinergic impairment. ApoE4 in the TR mice is also associated with lower recovery of hippocampal synapses following entorhinal lesion (Yun et al. 2005). Cholinergic impairments were also observed in transgenic mice that express apoE4 under regulation of human transgenic constructs (Chapman et al. 2000). These experiments clearly show that the expression of cholinergic molecules is specifically affected by apoE4. However, previous to our study, the extent to which ACh release and the mechanisms underlying it are affected by apoE4 had not been studied. Here, we investigate this by utilizing hippocampal and cortical brain slices of young (4‐month‐old) and adult (12‐month‐old) apoE4 and apoE3 TR mice.
Methods
Animals
ApoE3‐ and apoE4‐targeted replacement mice were created by gene targeting, as previously described (Sullivan et al., 1997). The mice used were purchased from Taconic (Germantown, NY, USA). They were back‐crossed to wild‐type C57BL/6J mice (Harlan 2BL/610) for ten generations and were homozygous for the apoE3 (3/3) or apoE4 (4/4) alleles. These mice are referred to in the text as apoE3 and apoE4 mice, respectively. The experiments were performed on age‐matched 4‐, 8‐, and 12‐ to 14‐month‐old male mice (the latter will be referred to as ‘12‐month‐old’ mice). The experiments were approved by the Tel Aviv University Animal Care Committee and were conducted according to the Council of Europe (Directive 86/609) in accordance with the Declaration of Helsinki. Every effort was made to reduce animal stress and to minimize animal usage.
Immunocytochemistry
Mice were anesthetized with ketamine and xylazine and perfused transcardially with saline and then with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Immunohistochemistry and confocal microscopy were performed as previously described (Belinson and Michaelson 2009) utilizing anti‐VAChT antibodies (Synaptic Systems, dilution 1 : 200).
Acetylcholine release
ACh release from superfused hippocampal or cerebral cortex slices was measured after prelabeling with [3H]choline essentially as previously described (Lazareno et al. 2004; Machova et al. 2010). The superfusion medium contained 10 μM hemicholinium‐3 to prevent re‐uptake of labeled choline derived from released [3H]ACh. The release of [3H]ACh was evoked by two successive electrical stimulations (2‐msec rectangular pulses, current strength 25 mA, 600 pulses at 5 Hz) separated by a time interval of 24 min. The first stimulation (S1) was applied in the presence of 10 μM carbachol (non‐hydrolyzable analog of ACh) to maximally activate pre‐synaptic autoreceptors, whereas the second stimulation (S2) was performed in the presence of 1 μM atropine to block pre‐synaptic autoinhibition of ACh release.
Biochemical determinations
Preparation of homogenates and membranes, measurement of total muscarinic receptors and enzyme activities, and muscarinic receptor‐mediated activation of G‐proteins were carried out as described previously (Machova et al. 2008). Total concentration of muscarinic receptors was determined as the specific binding of the non‐selective muscarinic receptor antagonist [3H]‐N‐methylscopolamine ([3H]‐NMS; 2 nM, specific radioactivity 148 dpm/fmol, American Radiolabeled Chemicals, Saint Louis, USA) in homogenates (10–30 μg of protein). Non‐specific binding was measured in the presence of 10 μM atropine. G‐protein activation was determined as the increase in carbachol‐activated (0.3–100 μM) guanosine 5′‐(γ35‐thio)‐triphosphate (GTP‐γ35S; 500 pM, ARC) binding to membranes (5–15 μg protein). Non‐specific binding was determined in the presence of 10 μM unlabeled GTP‐γS. Incubations were terminated by rapid vacuum filtration. Bound radioactivity was measured using solid scintillator Meltilex A in Wallac Microbeta counter (Perkin Elmer, Turku, Finland). Activity of choline acetyltransferase (ChAT) was measured radioenzymatically in homogenates (30–80 μg of protein) at 37°C. Activities of acetylcholinesterase (AChE) (3–9 μg of protein) and BuChE (15–45 μg of protein) were measured colorimetrically at 405 nm using chromogenic substrate acetylthiocholine (1 mM) and butyrylthiocholine (5 mM), respectively. Kinetics of reactions at 30°C were measured in plate reader Victor™ (Perkin Elmer, Turku, Finland).
Western blots and immunodetection
Western blots and immunodetection were performed as described (Janickova et al. 2013). Results were normalized relative to the house‐keeping protein Na‐K‐ATPase, whose levels were not affected by the apoE genotype.
Processing of data and statistical evaluation
Data presented are mean ± SEM. Enzyme activities were calculated from kinetic data using linear regression. A sigmoidal concentration–response curve was fitted to GTP‐γ35S binding data to calculate the parameters of carbachol‐stimulated binding. Statistical significance between age‐matched apoE3 and apoE4 transgenic mice was assessed by unpaired two‐tailed t‐test. Variations between apoE3 and apoE4 transgenic mice and among tested ages were analyzed by two‐way anova. Curve fitting and statistical evaluation of data were performed using Prism 6 (GraphPad Software, Inc., San Diego, CA, USA).
Results
VAChT immunohistochemistry and ACh release
Immunohistochemical determination of the pre‐synaptic vesicular acetylcholine transporter (VAChT) revealed similar levels of VAChT in the CA1, CA3, and dentate gyrus of young, 4‐month‐old apoE4 and apoE3 mice, which decreased with age significantly more in the apoE4 than in the apoE3 mice (P < 0.001 by two‐tailed t‐test for the differences between VAChT levels in CA1 and CA3 of the 12‐month‐old apoE4 and apoE3 mice and P < 0.005 for the corresponding VAChT levels in the dentate gyrus; Fig. 1). The effects of the apoE genotype and aging on hippocampal ACh release were measured utilizing hippocampal and cortical slices. Labeling of hippocampal slices of 4‐month‐old apoE3 and apoE4 mice with [3H]choline for 30 min resulted in accumulation of 193 ± 26 and 232 ± 22 cpm/μg protein, respectively, whereas similar treatment of hippocampal slices of 12‐month‐old apoE3 and apoE4 mice resulted in the accumulation of 187 ± 15 and 176 ± 13 cpm/μg protein, respectively. [3H]Choline uptake by the corresponding cortical slices was 151 ± 20 and 130 ± 20 cpm/μg protein in 4‐month‐old apoE3 and apoE4 mice and 128 ± 14 and 128 ± 14 cpm/μg protein in 12‐month‐old mice, respectively. Values are mean ± SEM of averaged triplicate samples from five and three 4‐month‐old and 12‐month‐old mice, respectively. T‐test analysis revealed no effects of the apoE genotype in either the hippocampus or cortex of both young and adult mice. The evoked release of prelabeled [3H]ACh (Fig. 2) was first measured in the presence of 10 μM carbachol (S1) to maximally activate pre‐synaptic M2 autoreceptors and then after blockade of autoreceptors by atropine (S2). Maximal [3H]ACh release (S2) was similar in hippocampal slices from 4‐month‐old apoE4 and apoE3 mice and decreased significantly with age in the apoE4 but not in the apoE3 mice (Fig. 2c). Two‐way anova showed age effect (P < 0.0001), genotype effect (P < 0.03), and their interaction (P < 0.002). Similar results were obtained following electrical stimulation of hippocampal slices after activation of pre‐synaptic receptors with carbachol (S1; Fig. 2b). Consistent with these results, the S2/S1 ratio of ACh release in hippocampus, which reflects the functionality of the M2 receptor‐mediated autoregulation, was not affected by either the apoE genotype or age. Age‐dependent decreases in the evoked [3H]ACh release during S1 and S2 with no changes in their ratio were also observed in cortical slices except that in this case the apoE genotype did not affect the release in both age groups (Fig. 2e–h).
Figure 1.
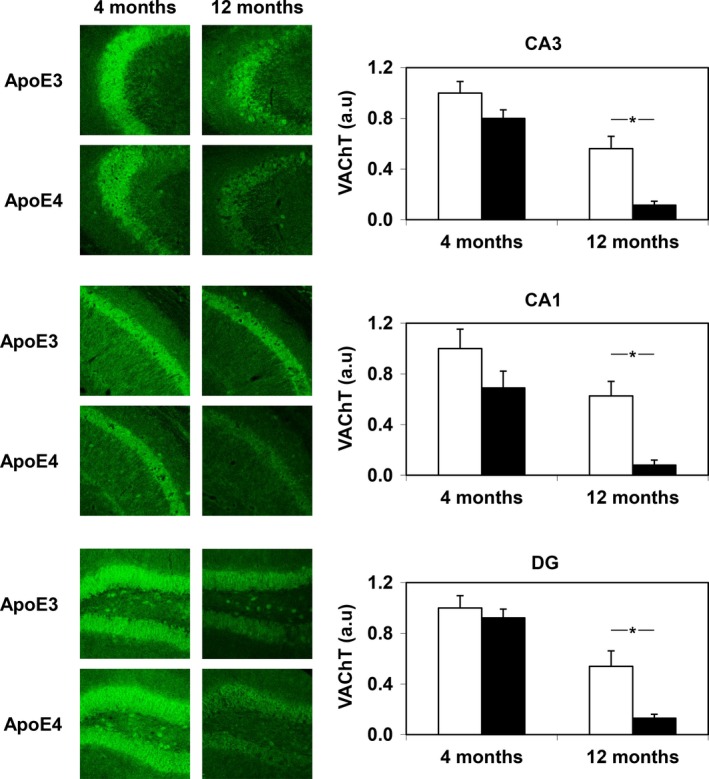
Immunohistochemical determination of vesicular acetylcholine transporter (VAChT) expression in the CA1, CA3, and dentate gyrus (DG) subregions of the hippocampus in young and adult apolipoprotein E3 (apoE3) and apoE4 mice. Representative images (20× magnification) of the indicated hippocampal subregions of young (4‐month‐old) and adult (12‐month‐old) male mice are shown on the panels on the left. Quantification of the results of the apoE3 (white columns) and apoE4 (black columns) mice was performed as described in Methods and is shown on the right. *P < 0.001 for comparison by t‐test for the differences between VAChT levels in CA3 and CA1 of the 12‐month‐old apoE4 and apoE3 mice and *P < 0.005 for the corresponding VAChT levels in the dentate gyrus (DG).
Figure 2.
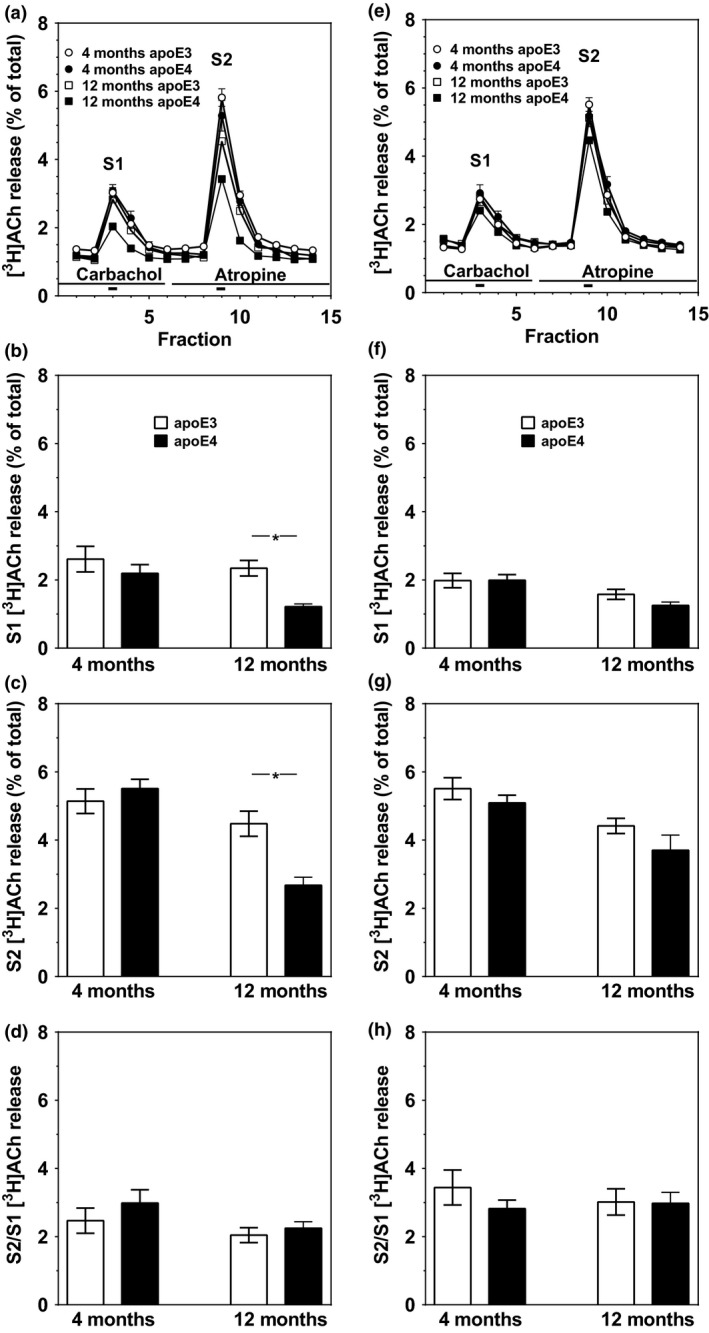
Electrical stimulation‐evoked release of [3H]ACh from the hippocampus (a–d) and cerebral cortex (e–h) of apoE4 and apolipoprotein E3 (apoE3) mice. The release of radioactivity from hippocampal (a) and cerebral cortical (e) slices evoked by electrical stimulation (thick bars under traces) is expressed as fractional release (ordinates). Superfusate was collected in 4‐min fractions (abscissa). Application of drugs during superfusion and description of symbols is shown in the (b and f) evoked [3H]ACh release under conditions of full activation of muscarinic autoreceptors. (c and g) Evoked [3H]ACh release devoid of muscarinic autoinhibition. (d and h) The extent of muscarinic autoregulation of the evoked [3H]ACh release (the ratio of the evoked release of ACh during S2 divided by the evoked release during S1). Description of symbols and columns is shown in panels. Age of mice is indicated under columns. Results are mean ± SEM of 8–15 slices prepared from 3 to 5 mice. *P < 0.001; significant difference, by t‐test, between age‐matched apoE3 and apoE4 mice.
Biochemical measurements of muscarinic receptors and cholinergic enzymes
Levels of muscarinic receptors in hippocampus were higher in the 4‐month‐old apoE4 mice compared with apoE3 mice (Table 1; P < 0.03 by t‐test). In the 8‐month‐old mice, the levels of muscarinic receptors were significantly elevated to a similar extent in the apoE4 and apoE3 mice. Two‐way anova indicated a significant age‐dependent increase in total muscarinic receptor density (P < 0.0001) and interaction between age and genotype (P < 0.02). The relative values of the M1 receptors determined by western blots did not differ between the 4‐month‐old apoE3 and apoE4 mice (1.03 ± 0.05 and 0.98 ± 0.06; n = 6), whereas 8‐month‐old apoE4 mice displayed a significant decrease in the apoE4 mice compared with apoE3 mice (0.88 ± 0.07 vs. 1.13 ± 0.05; n = 6; P < 0.01 by t‐test). Relative density of the M2 receptors was slightly higher in 4‐month‐old apoE4 than in apoE3 mice (1.06 ± 0.06 vs. 0.96 ± 0.04; n = 6; P < 0.01 by t‐test) but not in the corresponding 8‐month‐old mice (1.06 ± 0.08 and 1.00 ± 0.10; n = 6). The potency and efficacy of muscarinic receptor‐mediated activation of G‐proteins in the hippocampus revealed no effect of either the apoE genotype or age (Table 1). Measurements of ChAT and AChE (Table 1) did not detect differences between 4‐month‐old apoE3 and apoE4 mice but revealed age‐dependent decreases in enzyme activities (P < 0.0001 and P < 0.005 for the effects of age, by two‐way anova, on the ChAT and AChE, respectively), which in the case of AChE, but not ChAT, tended to be more pronounced in apoE4 mice. BuChE levels were similar in the apoE4 and apoE3 4‐month‐old mice and decreased with age markedly more in the apoE4 mice than in the apoE3 mice (Table 1; P < 0.0001 for the effect of age and P < 0.02 for the interactions between age and genotype by two‐way anova). Cortical BuChE levels were not affected by either age or the apoE genotype (not shown).
Table 1.
The effects of apoE4 and age on hippocampal cholinergic biochemical parameters
| Parameter | apoE3 (4 months) | apoE4 (4 months) | apoE3 (8 months) | apoE4 (8 months) |
|---|---|---|---|---|
|
Muscarinic receptors' density (fmol/mg protein) |
195 ± 21 (6) |
265 ± 16*
(5) |
468 ± 19 (12) |
438 ± 14 (12) |
|
Carbachol‐stimulated GTP‐γ35S binding EC50 (μM) E max (fold increase) |
3.11 ± 0.46 1.66 ± 0.06 (6) |
1.84 ± 0.25 1.60 ± 0.05 (5) |
1.07 ± 0.37 1.78 ± 0.07 (4) |
1.92 ± 0.32 1.75 ± 0.20 (4) |
| ChAT (pmol/μg prot.*15 min) |
0.96 ± 0.11 (11) |
1.00 ± 0.11 (11) |
0.47 ± 0.02 (6) |
0.48 ± 0.02 (6) |
| AChE (Δ abs./mg prot.*15 min) |
27.9 ± 2.6 (10) |
28.8 ± 1.7 (11) |
23.7 ± 1.3 (6) |
19.1 ± 0.7*
(6) |
| BuChE (Δ abs./mg prot.*15 min) |
1.39 ± 0.08 (11) |
1.46 ± 0.04 (11) |
1.01 ± 0.05 (6) |
0.74 ± 0.06*
(6) |
Results are mean ± SEM of the number of observations (mice) given in parentheses. *P < 0.05; significantly different from the apoE3 genotype of the same age by t‐test. abs., absorbance at 405 nm. Two‐way anova showed the age effect for ChAT (P < 0.0001) and AChE (P < 0.005), the age effect and interaction between age and genotype for BuChE (P < 0.0001 and P < 0.02, respectively), and the age effect and interaction between age and genotype for muscarinic receptor density (P < 0.0001 and P < 0.02, respectively).
Discussion
The main finding of the present study is that human apoE4 age dependently impairs the release of ACh in the hippocampus of male mice. This effect, which is not present in young 4‐month‐old mice but becomes apparent in adult 12‐month‐old mice, is specific for hippocampus and not observed in cortex. Its association with an apoE4‐driven decrease in VAChT but not ACh‐synthesizing enzyme ChAT suggests impairment of release‐related processes and not the synthesis of ACh. This is in line with demonstration of similar concentrations of ACh in forebrain tissue of adult apoE3 and apoE4 TR mice (Chan et al. 2008) and supported by the observation that [3H]choline uptake by hippocampal slices, which is coupled to ACh synthesis (Okuda et al. 2000), was not affected by apoE4. The decrease in ACh release is accompanied by apoE4‐driven reductions in the levels of the AChE and BuChE but with no change in the M2 receptor‐mediated autoregulation of evoked [3H]ACh release. These findings provide the first direct evidence that apoE4 impairs evoked hippocampal ACh release in apoE‐TR mice and suggests that the cholinergic transmission‐related decrease in hippocampal LTP in these mice (Yun et al. 2005) is due to impaired ACh release. In accordance with the susceptibility of cholinergic system to aging (for reviews see Pepeu and Giovannelli 1994; Schliebs and Arendt 2011), apoE genotype‐independent reductions of ChAT, AChE, and BuChE level with age were also observed.
Intactness of the M2 receptor‐mediated autoregulation of 3H‐ACh release is consistent with the notion that there is no major effect of apoE4 on pre‐synaptic M2 receptors. The decrease in the proportion of the M1 subtype in the hippocampus of 8‐month‐old apoE3 mice points to a post‐synaptic effect of the apoE genotype. This is specific to the hippocampus and was not observed in the cerebral cortex (data not shown) and most likely contributes to the impairment of LTP (Shinoe et al. 2005). Further pharmacological and electrophysiological studies are required to delineate the effects of the apoE genotype and age on the different classes of the post‐synaptic muscarinic receptors.
The mechanism underlying the effects of apoE4 on [3H]ACh release and the associated reduction in the levels of VAChT are not known but may be driven by a common ‘cholinergic gene locus’ (Erickson et al. 1994). The finding that, unlike the evoked ACh release, the uptake of choline and radiolabeling of ACh were not affected by apoE4 suggests that the age‐dependent effects of apoE4 are due to the effects of apoE4 on the release process and not on choline uptake and the number of synapses. The present finding of differential regulation of VAChT and ChAT by apoE4 and aging is in accordance with reported differential regulation of these proteins (Holler et al. 1996).
Since apoE is the main lipoprotein in the brain, it is possible that the apoE4‐driven cholinergic deficits are related to the diminished availability of phosphatidylcholine at the nerve terminal and subsequently to reductions in the levels of choline (Wurtman 2008). Indeed, prenatal supplementation with choline promotes hippocampal cholinergic transmission up to adulthood (Cermak et al. 1998). However, since apoE4 also affects non‐cholinergic synaptic parameters, e.g. reduction of the pre‐synaptic glutamatergic transporter VgluT in hippocampus (Liraz et al. 2013) or increase in VgluT transporter and disturbance of the glutamate–glutamine cycle in whole brain (Dumanis et al. 2013), additional brain structure‐specific mechanisms may play a role in mediating the synaptic pathological effects of apoE4.
The pathological effects of apoE4 depend on structural differences between apoE4 and apoE3 and on the susceptibility of mice to ‘on‐target’ effects of apoE. These mechanisms, particularly the susceptibility to on‐target effects of apoE4, are likely to be affected by the genetic background of the mice employed. Indeed, the cholinergic system is not affected by apoE4 in transgenic mice expressing apoE4 under the control of the Thy‐1 promoter on a FVB/N mouse background (Bronfman et al. 2000). Note that the present line of apoE4‐TR mice was crossed with C57Bl mice from Harlan, which lack the pre‐synaptic protein synuclein (Specht and Schoepfer 2001). Preliminary findings utilizing apoE4 mice crossed with C57Bl mice, which have normal synuclein levels, suggest that phenotypes not directly related to the nerve terminal (e.g., apoE4‐driven accumulation of Aβ and hyperphosphorylated tau; Liraz et al. 2013) are not affected by synuclein deficiency. The extent to which the apoE4‐driven cholinergic impairments are accentuated by the lack of synuclein remains to be determined.
In conclusion, the present study showed that the evoked release of ACh from hippocampal nerve terminals is impaired age dependently in apoE4‐TR mice. This animal model observation is in accordance with findings obtained in AD and during aging. These mice thus serve as an attractive model for studying mechanisms underlying the pathological effects of apoE4 on the cholinergic system.
Acknowledgments and conflict of interest disclosure
We thank Alex Smolar for technical assistance. This work was supported, in part, by a joint grant from the Czech‐Israel Binational Science Foundation (grant from the Ministry of Education, Youth, and Sports Kontakt II LH13269), and from the Legacy Heritage Bio‐Medical Program of the Israel Science Foundation (grant no. 1575/14).
All experiments were conducted in compliance with the ARRIVE guidelines. The authors have no conflict of interest to declare.
References
- Belinson H. and Michaelson D. M. (2009) Pathological synergism between amyloid‐beta and apolipoprotein E4–the most prevalent yet understudied genetic risk factor for Alzheimer's disease. J. Alzheimers Dis. 17, 469–481. [DOI] [PubMed] [Google Scholar]
- Braga I. L., Silva P. N., Furuya T. K., Santos L. C., Pires B. C., Mazzotti D. R., Bertolucci P. H., Cendoroglo M. S. and Smith M. C. (2015) Effect of APOE and CHRNA7 genotypes on the cognitive response to cholinesterase inhibitor treatment at different stages of Alzheimer's disease. Am. J. Alzheimers Dis. Other Demen. 30, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronfman F. C., Tesseur I., Hofker M. H., Havekens L. M. and Van Leuven F. (2000) No evidence for cholinergic problems in apolipoprotein E knockout and apolipoprotein E4 transgenic mice. Neuroscience 97, 411–418. [DOI] [PubMed] [Google Scholar]
- Cermak J. M., Holler T., Jackson D. A. and Blusztajn J. K. (1998) Prenatal availability of choline modifies development of the hippocampal cholinergic system. FASEB J. 12, 349–357. [DOI] [PubMed] [Google Scholar]
- Chan A., Tchantchou F., Graves V., Rozen R. and Shea T. B. (2008) Dietary and genetic compromise in folate availability reduces acetylcholine, cognitive performance and increases aggression: critical role of S‐adenosyl methionine. J. Nutr. Health Aging 12, 252–261. [DOI] [PubMed] [Google Scholar]
- Chapman S., Sabo T., Roses A. D. and Michaelson D. M. (2000) Reversal of presynaptic deficits of apolipoprotein E‐deficient mice in human apolipoprotein E transgenic mice. Neuroscience 97, 419–424. [DOI] [PubMed] [Google Scholar]
- Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L. and Pericak‐Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Craig L. A., Hong N. S. and McDonald R. J. (2011) Revisiting the cholinergic hypothesis in the development of Alzheimer's disease. Neurosci. Biobehav. Rev. 35, 1397–1409. [DOI] [PubMed] [Google Scholar]
- Dumanis S. B., DiBattista A. M., Miessau M., Moussa C. E. and Rebeck G. W. (2013) APOE genotype affects the pre‐synaptic compartment of glutamatergic nerve terminals. J. Neurochem. 124, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson J. D., Varoqui H., Schafer M. K., Modi W., Diebler M. F., Weihe E., Rand J., Eiden L. E., Bonner T. I. and Usdin T. B. (1994) Functional identification of a vesicular acetylcholine transporter and its expression from a “cholinergic” gene locus. J. Biol. Chem. 269, 21929–21932. [PubMed] [Google Scholar]
- Holler T., Berse B., Cermak J. M., Diebler M. F. and Blusztajn J. K. (1996) Differences in the developmental expression of the vesicular acetylcholine transporter and choline acetyltransferase in the rat brain. Neurosci. Lett. 212, 107–110. [DOI] [PubMed] [Google Scholar]
- Janickova H., Rudajev V., Zimcik P., Jakubik J., Tanila H., El‐Fakahany E. E. and Dolezal V. (2013) Uncoupling of M1 muscarinic receptor/G‐protein interaction by amyloid beta(1‐42). Neuropharmacology 67, 272–283. [DOI] [PubMed] [Google Scholar]
- Lazareno S., Dolezal V., Popham A. and Birdsall N. J. (2004) Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: receptor subtype selectivity via cooperativity rather than affinity. Mol. Pharmacol. 65, 257–266. [DOI] [PubMed] [Google Scholar]
- Liraz O., Boehm‐Cagan A. and Michaelson D. M. (2013) ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol. Neurodegener. 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machova E., Jakubik J., Michal P., Oksman M., Iivonen H., Tanila H. and Dolezal V. (2008) Impairment of muscarinic transmission in transgenic APPswe/PS1dE9 mice. Neurobiol. Aging 29, 368–378. [DOI] [PubMed] [Google Scholar]
- Machova E., Rudajev V., Smyckova H., Koivisto H., Tanila H. and Dolezal V. (2010) Functional cholinergic damage develops with amyloid accumulation in young adult APPswe/PS1dE9 transgenic mice. Neurobiol. Dis. 38, 27–35. [DOI] [PubMed] [Google Scholar]
- Okuda T., Haga T., Kanai Y., Endou H., Ishihara T. and Katsura I. (2000) Identification and characterization of the high‐affinity choline transporter. Nat. Neurosci. 3, 120–125. [DOI] [PubMed] [Google Scholar]
- Pepeu G. and Giovannelli L. (1994) The central cholinergic system during aging. Prog. Brain Res. 100, 67–71. [DOI] [PubMed] [Google Scholar]
- Pomara N., Willoughby L. M., Wesnes K. and Sidtis J. J. (2004) Increased anticholinergic challenge‐induced memory impairment associated with the APOE‐epsilon4 allele in the elderly: a controlled pilot study. Neuropsychopharmacology 29, 403–409. [DOI] [PubMed] [Google Scholar]
- Reinvang I., Espeseth T. and Westlye L. T. (2013) APOE‐related biomarker profiles in non‐pathological aging and early phases of Alzheimer's disease. Neurosci. Biobehav. Rev. 37, 1322–1335. [DOI] [PubMed] [Google Scholar]
- Saunders A. M., Strittmatter W. J., Schmechel D. et al (1993) Association of apolipoprotein E allele epsilon 4 with late‐onset familial and sporadic Alzheimer's disease. Neurology 43, 1467–1472. [DOI] [PubMed] [Google Scholar]
- Schliebs R. and Arendt T. (2011) The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. [DOI] [PubMed] [Google Scholar]
- Shinoe T., Matsui M., Taketo M. M. and Manabe T. (2005) Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J. Neurosci. 25, 11194–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht C. G. and Schoepfer R. (2001) Deletion of the alpha‐synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P. M., Mezdour H., Aratani Y., Knouff C., Najib J., Reddick R. L., Quarfordt S. H. and Maeda N. (1997) Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet‐induced hypercholesterolemia and atherosclerosis. J Biol Chem 272, 17972–17980. [DOI] [PubMed] [Google Scholar]
- Sullivan P. M., Han B., Liu F., Mace B. E., Ervin J. F., Wu S., Koger D., Paul S. and Bales K. R. (2011) Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol. Aging 32, 791–801. [DOI] [PubMed] [Google Scholar]
- Wurtman R. J. (2008) Synapse formation and cognitive brain development: effect of docosahexaenoic acid and other dietary constituents. Metabolism 57(Suppl 2), S6–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S. H., Park K. A., Sullivan P., Pasternak J. F., Ladu M. J. and Trommer B. L. (2005) Blockade of nicotinic acetylcholine receptors suppresses hippocampal long‐term potentiation in wild‐type but not ApoE4 targeted replacement mice. J. Neurosci. Res. 82, 771–777. [DOI] [PubMed] [Google Scholar]