

Abstract
Treg cells hold enormous promise for therapeutic application in GVH disease, a lethal complication of allogeneic HSC transplantation. Mouse studies showed that donor‐derived recipient‐specific Treg (rsTreg) cells are far more efficient than polyclonal Treg cells in suppressing GVH disease. However, clinical grade preparations of rsTreg cells carries the risk of containing significant numbers of highly pathogenic recipient‐specific effector T cells. We hypothesized that an alternative approach using Treg cells specific for an exogenous (i.e. nondonor, nonrecipient) Ag (exoTreg cells) can overcome this risk by taking advantage of the bystander suppressive effect of Treg cells. For this, we used a murine model for aggressive GVH disease. We expanded ex vivo exoTreg cells that are primed against the HY Ag, which is only expressed in males. ExoTreg cells supressed GVH disease as efficiently as rsTreg cells in recipient male mice. We also applied this strategy in female mice that do not express this Ag. While exoTreg cells were not effective in female recipients when applied alone, providing the cognate HY Ag in vivo along side effectively activated exoTreg cells and completely abrogated GVH disease, establishing a targeted on/off system to provide a suppressive effect on alloreactive effector T cells.
Keywords: GVH disease, Immune regulation, Inducible activation of regulatory T (Treg) cells, Tolerance, Transplantation
Introduction
Allogeneic (allo) HSC transplantation is the treatment of choice for several types of malignant and nonmalignant hematological disorders 1. Donor T cells present within the graft are essential for: (i) engraftment 2, (ii) immune reconstitution 3, and (iii) graft‐versus‐leukemia/tumor effect 4. GVH disease is a life‐threatening complication of allo‐HSC transplantation 5. This immunological disorder is due to effector donor T (Teff) cells present within the graft that recognize allo‐Ags and attack target tissues of GVH disease 6. To date, in order to prevent GVH disease, graft recipients receive an immunosuppressive regimen, classically based on calcineurin inhibitors associated with an antimetabolite, methotrexate 7. These treatments are effective in many patients with a marked impact on outcomes, and represent today the “gold standard” of GVH disease prevention/treatment. Unfortunately, a significant proportion of patients still develop GVH disease, even after intensification of the immunosuppressive regimen 8. Furthermore, immunosuppressive treatment negatively impacts both antileukemic effect and immune reconstitution 6, 9, 10. Thus, developing innovative therapeutic strategies to limit the pathological effects of donor alloreactive T cells is crucial.
For the past decade, we and others have methodically studied strategies for controlling acute 5, 11, 12, 13, 14, 15, 16 and chronic 17 GVH disease in mice by transfer of CD4+CD25+Foxp3+ Treg cells. Recent reports of two clinical trials suggest that adding high numbers of polyclonal Treg (polyTreg) cells prior to 18 or after allo‐HSC transplantation 19 reduces GVH disease occurrence. Additionally, several groups have shown that Treg cells specific for Ags expressed by the target tissue suppress more effectively organ‐specific autoimmune diseases or graft rejection, compared with polyTreg cells 20, 21, 22. Similarly, in experimental models of allo‐HSC transplantation, we and others initially observed that donor‐derived recipient‐specific Treg (rsTreg) cells control GVH disease better than polyTreg cells, while preserving graft‐versus‐infection and graft‐versus‐tumor effects 11, 12, 13, 23. Unfortunately, due to the difficulty in sorting Treg cells to a high purity under clinical grade practice conditions, rsTreg cells would be contaminated with highly pathogenic recipient‐specific effector T cells, precluding their therapeutic utilization. Here, we explored an alternative strategy that uses Treg cells specific for an exogenous (i.e. nondonor, nonrecipient) Ag (exoTreg cells) which may be powerfully activated in vivo by providing the exogenous Ag. In this setting, contaminating Teff cells are expected to be non‐ or poorly pathogenic as the exogenous Ag would be presented only transiently by few host APCs and not expressed by target organs of GVH disease. For this strategy to function, two requirements must be met: (i) exoTreg cells specific for a single Ag must be able to suppress donor Teff cells specific for recipient‐type allo‐Ags (bystander effect); (ii) these same exoTreg cells can be efficiently reactivated in vivo by administration of the exogenous Ag. We have addressed these two questions in this work.
Results
Single Ag‐specific Treg cells suppress allogeneic Teff cells and prevent GVH disease
As a model for GVH disease, lethally irradiated (C57BL/6 (B6) X C3H) F1 (B6C3F1) recipient male mice were grafted with BM cells and T cells collected from B6 male LNs as illustrated in Fig. 1A. More than 80% of mice receiving BM cells and T cells rapidly developed lethal GVH disease (Fig. 1B) confirmed by a typical clinical presentation and by a typical histological appearance of the colon and liver (Fig. 1C and D). We next aimed to test the ability of two populations of Treg cells (single Ag‐specific Treg cells and rsTreg cells) to prevent GVH disease. For this purpose, we generated HY‐Treg cells (Treg cells primed against the single minor histocompatibility Ag HY, an Ag expressed only in males) from purified Treg cells of B6 female mice, following a procedure that we previously described 24. As a control, we produced C3H‐specific rsTreg cells following a selection and expansion procedure also previously described 12, 13, 16, 23. Both types of Treg cells expanded robustly during culture while maintaining both the expression of Foxp3 and CD25, as well as the ability to suppress T‐cell proliferation in vitro (Supporting Information Fig. 1A–C). The specificity of HY‐Treg cells was confirmed in vitro, as only stimulation with APCs expressing the HY Ag (derived from male mice) resulted in proliferation of HY‐Treg cells (Supporting Information Fig. 1D).
Figure 1.
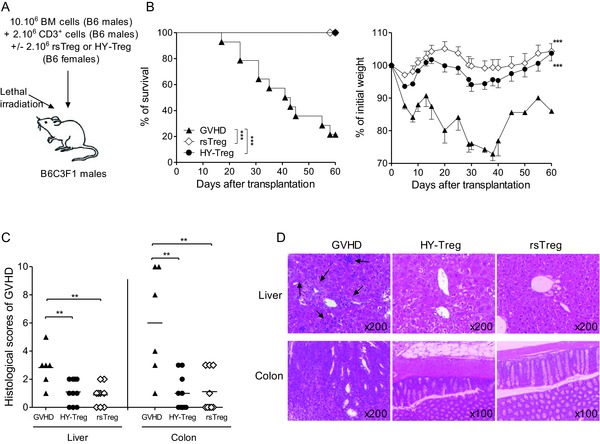
HY‐Treg cells exert the same immunosuppressive effect in vivo as rsTreg cells. (A) Experimental GVH disease model representing semiallogenic HSC transplantation testing different Treg‐cell populations. (B) Kaplan–Meier survival and weight (mean ± SEM) curves from mice injected with BM cells plus T cells alone (GVH disease group, n = 16) or with HY‐Treg cells (n = 13) or rsTreg cells (n = 11). (C) Histopathological scores of colon and liver after semiallogeneic HSC transplantation. Grading of GVH disease was performed 60 days after transplantation. Symbols correspond to histopathological score of individual mice, and horizontal bars show the mean histopathological score for each group. (B, C) Data shown are pooled from four experiments performed. **p < 0.01, ***p < 0.001, two‐tailed unpaired Student's t‐test and log‐rank (Mantel–Cox) test for survival between two mice groups. (D) H&E‐stained histological sections of colon and liver from one representative mouse per group 60 days after allo‐HSC transplantation. Arrows indicate cell infiltrates. Data shown are representative of four experiments performed.
After generating the two populations of Treg cells, their ability to prevent GVH disease was tested. Treg cells were co‐transferred with BM cells and T cells into male mice (harboring the HY Ag). In this experimental model, HY‐Treg cells are supposed to be activated only by HY whereas Teff cells that come from male mice are only activated by allogeneic Ags, but not by HY (Fig. 1A). Of note, in this particular context, HY‐Treg cells cannot be considered as exoTreg cells since the HY Ag is constitutively expressed in male recipients. Remarkably, HY‐Treg cells prevented clinical manifestations of GVH disease (Fig. 1B) and also resulted in a significant reduction of lesions in target organs (Fig. 1C and D), comparably to the prevention of GVH disease by rsTreg cells. Furthermore, like rsTreg cells 16, HY‐Treg cells promoted a strong inhibition of the expansion, activation, and differentiation of donor T cells. First, the donor T‐cell number (CD45.1+) was markedly reduced in the presence of HY‐Treg cells (Fig. 2A, left). Second, among dividing T cells, attested by CFSE dilution (Fig. 2A, right), the proportions (Supporting Information Fig. 2) and absolute numbers (Fig. 2B) of activated T cells with a CD25+, CD44high, CD62Llow phenotype, as well as cells producing IL‐2, IFN‐γ, and TNF‐α, were markedly reduced. It is worthwhile to note that lethally irradiated mice protected from GVH disease with HY‐Treg cells were not functionally immunodeficient, since they were able to reject third‐party skin grafts from BALB/c mice with an accelerated memory‐type immune response when a second skin graft was performed 90 days after the first one (Fig. 2C), in accordance with our previous observations using rsTreg cells 23. Finally, this effect was TGF‐β dependent, as treatment with anti‐TGF‐β mAb (days 0–3) resulted in loss of HY‐Treg cells’ protective function (Fig. 3), as previously shown in different models of disease protection by Treg cells 25. In conclusion, in the presence of their cognate Ag, HY‐Treg cells were as effective as rsTreg cells primed against the entire Ag repertoire of the MHC haplotype (C3H) in preventing GVH disease, and had similar cellular effects 16.
Figure 2.
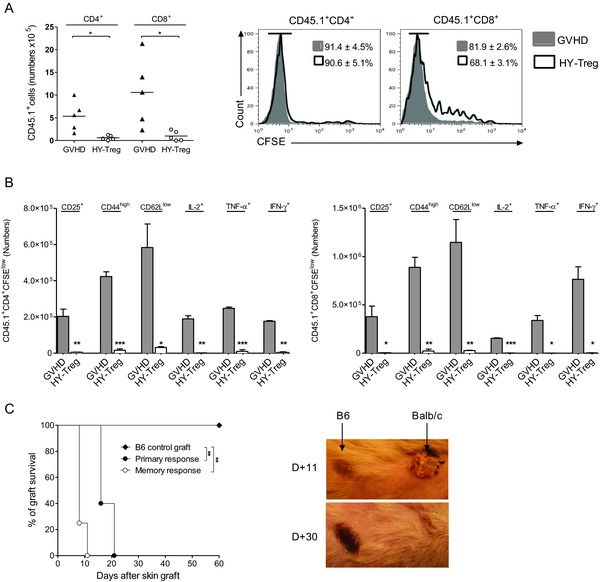
HY‐Treg cells prevent GVH disease by reducing activation and differentiation of donor Teff cells. Donor CD4+ and CD8+ Teff cells are analyzed at day 6 posttransplantation in the spleen of animals grafted as described in Fig. 1A and identified using the CD45.1+ congenic marker. (A) Mean absolute numbers and CFSE dilution of CD4+ and CD8+ donor CD45.1+ T cells when injected with or without HY‐Treg cells (n = 6 in each group). (B) Absolute numbers +SEM of CD4+ and CD8+ divided donor T cells (CD45.1+CFSElow) expressing membrane CD25, CD44, CD62L, or intracellular IL‐2, IFN‐γ, and TNF‐α with (white bars) or without (gray bars) HY‐Treg cells (n = 4 in each group). (C) Rejection of a third‐party allogeneic skin graft in mice protected from GVH disease with HY‐Treg cells. In order to evaluate the primary response of HY‐Treg cells protected mice, tail‐skin grafts from BALB/c mice were transplanted at day 60 onto the lateral thoracic wall (primary response, n = 6). On skin‐grafted mice, the memory response was tested by a second BALB/c skin grafted at day 150 (memory response, n = 5). Control group consists of mice treated with HY‐Treg cells and grafted with donor‐type B6 skin (B6 control, n = 5). Skin acceptance and rejection observed at days 11 and 30 in mice receiving a second skin graft is shown (right). Data shown are pooled from two experiments performed. *p < 0.05, **p < 0.01, ***p < 0.001, two‐tailed unpaired Student's t‐test.
Figure 3.
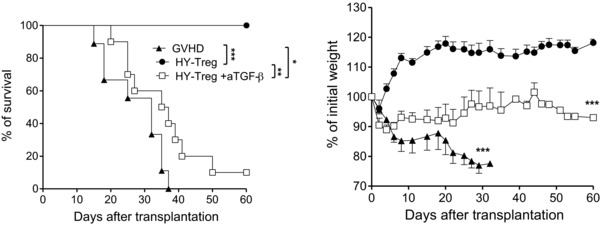
Prevention of GVH disease by HY‐Treg cells is TGF‐β dependent. Kaplan–Meier survival curves and weight (mean ± SEM) curves of GVH disease control group (n = 10) and HY‐Treg cells without (n = 10), or with (n = 10) four intraperitoneal injections of 25 mg/kg of anti‐TGF‐β mAb (2G7) from D0 to D3. Data shown are pooled from two experiments performed. *p < 0.05, **p < 0.01, ***p < 0.001, two‐tailed unpaired Student's t‐test (weight change), and log‐rank (Mantel–Cox) test (survival).
Lethal irradiation induces profound lymphopenia associated with a cytokine storm. These events may lead to a nonspecific activation of Treg cells, a phenomenon called lymphopenia‐induced proliferation (LIP) 26. To evaluate the impact of LIP on the suppressive effect of HY‐Treg cells, we repeated the experiment in nonirradiated B6C3F1 male recipients. Indeed, this is rendered possible due to the fact that in this parent into F1 strain combination, there is no donor cell rejection. This particular combination mimics the very aggressive clinical scenario of haplo‐mismatch HSC transplant, though grafted patients typically received irradiation and T‐cell‐depleted grafts. Thus, although the model is less relevant from the clinical perspective, it is helpful in assessing the potential contribution of LIP to Treg‐mediated GHV disease control. When mice were grafted with B6 donor T cells, more than 70% of mice developed lethal GVH disease. In this model, clinical signs of GVH disease resemble the presentation observed in irradiated mice, namely body weight lost (Fig. 4), diarrhea, hunched posture, and dull furs (not shown). The co‐transfer of HY‐Treg cells or rsTreg cells resulted in the absence of clinical signs of GVH disease during at least 2 months (the duration of these experiments, Fig. 4). This was seen even in a model that does not involve LIP, suggesting that the protective effect conferred by HY‐Treg cells is indeed due to their in vivo reactivation by their cognate Ag and not to LIP‐dependent activation.
Figure 4.
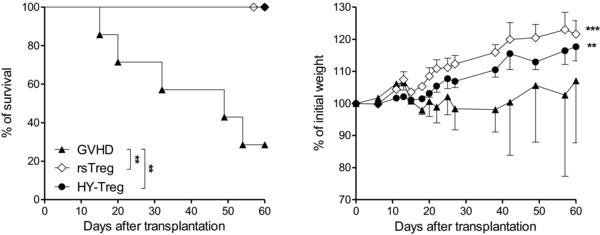
Prevention of GVH disease is not due to the lymphopenia‐induced proliferation of HY‐Treg cells. Kaplan–Meier survival curves and mean ± SEM weight curves after nonirradiated mice received B6 CD3+ cells (GVH disease group, n = 7) plus HY‐Treg cells ( n = 7) or rsTreg cells (n = 6). Data shown are pooled from three experiments performed. **p < 0.01, two‐tailed unpaired Student's t‐test (weight change), and log‐rank (Mantel–Cox) test (survival).
Treg cells specific for an exogenous Ag prevent GVH disease upon in vivo reactivation
We then set forth to test the second requirement: that these Treg cells can be efficiently reactivated in vivo by providing the exogenous Ag. In the previous experiments, the recipients were male mice that harbor the HY Ag, and thus, in this context, HY cannot be considered exogenous. We therefore attempted to reactivate HY‐Treg cells in female mice, which do not express the HY Ag. In this context, HY‐Treg cells could formally be considered as exoTreg cells. We used the same GVH disease model, modifying only the gender of recipient mice (previously male, now female, Fig. 5A). As expected, co‐transfer of exoTreg cells in female recipients had no effect on GVH disease. The mice displayed clinical and histological signs of GVH disease and died with a kinetic comparable with that of mice that received donor T cells alone (Fig. 5B and D). The failure of exoTreg cells to prevent GVH disease was also supported by lower expression of ICOS and glucocorticoid‐induced TNF receptor activation markers on exoTreg cells 6 days after transfer in female compared to male recipients (Supporting Information Fig. 3), indicating a reduced activation of exoTreg cells in the absence of their cognate Ag, as well as their survival at least at day 6 in the absence of any stimulation. In contrast, rsTreg cells maintained full efficacy in female recipients, resulting in complete abrogation of GVH disease. Subsequently, we tested whether we can reactivate in vivo exoTreg cells after transfer by providing the exogenous Ag to female recipients. We injected ex vivo HY‐pulsed donor DCs or the HY peptide alone, at time of GVH disease induction as well as at days 3 and 6. In these two groups, mice survived and had no signs of GVH disease. In contrast, control mice that received no Treg cells or were co‐injected with exoTreg cells followed by injection of DCs not pulsed with HY developed lethal GVH disease (Fig. 5C and D). Delaying the injection of the peptide to day 5 also allows controlling the GVH disease (Supporting Information Fig. 4). Thus, in these last experiments, we demonstrated that following in vivo re‐stimulations (with HY‐pulsed DCs or the sole HY peptide), exoTreg cells are as effective as rsTreg cells in preventing GVH disease.
Figure 5.
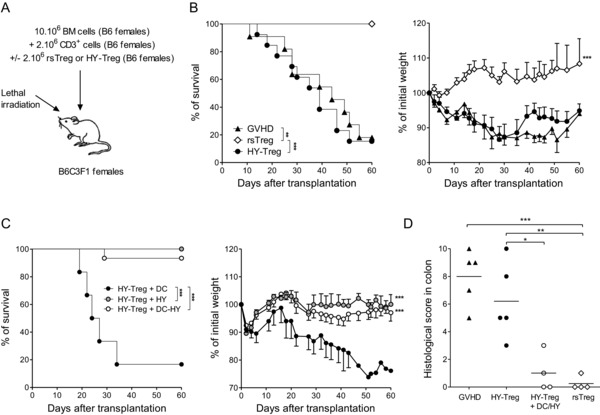
Prevention of GVH disease by HY‐Treg cells requires in vivo reactivation. (A) Experimental GVH disease model in female recipients representing semiallogenic HSC transplantation testing different Treg‐cell populations. (B) Kaplan–Meier survival curves and weight (mean ± SEM) curves demonstrate that in female mice (i.e. not expressing HY Ag) injected with BM cells plus T cells (GVH disease group, n = 10), HY‐Treg cells conferred no protection from GVH disease (n = 11) whereas rsTreg cells fully protected from GVH disease (n = 7). (C) In contrast, the Kaplan–Meier survival curves and weight (mean ± SEM) curves demonstrate that when female mice are injected at D0, D3, and D6 with B6 DC cells loaded with HY peptide (n = 10) or only by HY peptide (n = 10), HY‐Treg cells are able to prevent GVH disease as opposed to mice injected with HY‐Treg cells plus DC without peptide as control (n = 4). (D) Histopathological scores of colon after semiallogeneic HSC transplantation. Grading of GVH disease was performed 60 days after transplantation. Each symbol represents an individual animal and bars represent means. Data shown are pooled from two experiments performed. *p < 0.05, **p < 0.01, ***p < 0.001, two‐tailed unpaired Student's t‐test, and log‐rank (Mantel–Cox) test (survival).
Discussion
The therapeutic potential of Treg cells is substantial and still in its infancy. Two clinical trials have recently suggested that Treg‐cell‐based therapy could reduce the occurrence of acute GVH disease in allo‐HSC transplantation 18, 19. In the same line, a recent report demonstrated that Treg cells could also control ongoing chronic GVH disease in mice 17. Additionally, it was shown that the adoptive transfer of a particular donor‐derived CD4−CD8− Treg‐cell population prior to allo‐BM transplantation, induced a stable‐mixed chimerism and specific acceptance of donor skin allografts, thus suggesting that the therapeutic use of Treg cells could be extended to organ/BM combined transplantations 27.
Whatever the setting and disease, the hallmark of Treg cells is that they have to be activated to exert their immunosuppressive effect in vitro 28. In this report, we used a strategy of ex vivo selected and expanded Ag‐specific Treg cells to test whether exoTreg cells, once reactivated in vivo, act solely on T cells having the same Ag specificity or, alternatively, mediate a suppressive effect on a larger repertoire of donor T cells. Here, we clearly demonstrate that exoTreg cells could exert a suppressive effect on the whole allogeneic donor T‐cell population. Our data also demonstrate that LIP had virtually no contribution in eliciting the suppressive function of exoTreg cells, without stimulation by their cognate Ag. This is supported by lower expression of ICOS and glucocorticoid‐induced TNF receptor activation markers on exoTreg cells after transfer in female recipients, attesting for a reduced activation of exoTreg cells in the absence of their cognate Ag, even in lymphopenic recipients. This is in accordance with our previous publication showing that LIP‐induced T‐cell activation was much reduced compared to allogeneic‐induced T‐cell activation 29. Thus, it seems that LIP‐dependent Treg activation was not sufficient to trigger enough suppressive activity to suppress GVH disease.
Initially, the bystander suppressive effect of Ag‐specific Treg cells was demonstrated in autoimmune‐prone NOD mice and defined as the capacity for Treg cells responsive to a single auto‐Ag to inhibit diabetes mediated by Teff cells reactive to multiple Ags. In these two reports 21, 22, Treg cells were obtained from a CD4+ T‐cell receptor (TCR) transgenic line that is known to be diabetogenic, the BDC2.5 T cells 30. Bystander suppressive effect only developed when a co‐localized co‐activation of both Treg cells and Teff cells occurred. Similar observations were confirmed in models of EAE using Treg cells from transgenic mice expressing the myeline‐basic protein‐specific TCR 31 or in experimental BSA‐induced arthritis using Treg cells retrovirally transduced to express the OVA‐specific class II restricted TCR 32. Of note, all these reports rely on genetically engineered Treg cells expressing a specific TCR. In the field of transplantation tolerance, Treg cells collected from mice treated with human gamma globulin plus anti‐CD4 Ab and adoptively transferred in naïve mice could prevent allogeneic skin rejection providing that Treg cells were adequately reactivated before their transfer 33. However, in these experiments, the nature (induced or natural) of the Treg cells was not documented and again their suppressive effect was local.
A systemic bystander suppressive effect of Treg cells was suggested in a model of HSC transplantation 34. Continuous administration of an HY peptide in female mice allows for induced HY‐specific Treg generation from naïve conventional T cells. When these female mice were sublethally irradiated and grafted with HSC transplantation from syngeneic male donors, HSC transplantation was not rejected.
Here, using natural Treg cells derived from WT mice, and not a TCR‐transgenic line, we demonstrate for the first time a successful application of this principle of bystander suppressive effect of Ag‐specific natural Treg cells to prevent one of the most catastrophic immune‐mediated processes, GVH disease. For this “bystander suppressive effect” to be clinically relevant and consequently applicable in Treg‐cell therapy of GVHD, it would need to satisfy three requirements: (i) the effect is achieved with WT Treg cells (i.e. not from TCR‐transgenic mice), (ii) the Treg cells are specific for third‐party exogenous Ag (i.e. nondonor, nonrecipient Ag), and (iii) it functions systemically to address the systemic nature of the disease. Thus, in our hands, a “systemic bystander effect” was seen when DCs presenting an exogenous Ag (nondonor, nonrecipient Ag) properly reactivated in vivo exoTreg cells. This population of highly activated specific HY‐Treg cells subsequently fully suppressed the diverse repertoire of alloreactive Teff cells through a TGF‐β‐dependent mechanism.
From the clinical perspective, it is worthwhile to consider the importance of the “systemic bystander effect” in the context of the ongoing efforts to harness the power of Treg cells for therapeutic applications. As seen in this and other reports, Treg cells offer an unparalleled promise for auto‐ and alloimmune disorders. They appear not only extremely potent, but capable of inducing tolerance while reducing the risk of immunodeficiency. However, in order to optimize their efficacy, efforts have been made to enhance the purity as well as the specificity of Treg cells, with only limited success 35, 36, 37, 38, 39, 40, 41, 42, 43, 44. We present an alternative approach by using Treg cells specific for a “third‐party” exogenous Ag. We showed that these exoTreg cells generated ex vivo from a polyTreg cells population, prevented experimental GVH disease via potent suppression of pathogenic Teff cells providing that HY‐Treg cells were reactivated in vivo upon immunization of recipient mice with their cognate Ag. The potential for exoTreg cells based therapy is substantial. These experiments suggest that by using a “third‐party” exogenous Ag, we can maintain both the efficacy and specificity of Treg cells, while eliminating the risk of pathogenicity of putative contaminating Ag‐specific Teff cells. With this universal Ag‐unrelated immunosuppression through Treg cells approach, we can consider treating any T‐cell‐dependent disease, without the need to know the Ag involved. In HSC transplantation, a Treg‐cell‐based therapy would be conducted with controlled amounts of T cells and Treg cell. To replicate ratios reported in this work in humans, standard doses of 106–107 donor T cells/kg will require co‐administration of 106–107 Treg cell/kg. A recent publication shows that such Treg‐cell expansion can be performed in good manufacturing practice conditions 43. In addition, because of the short half‐life of mature DCs, one may envision that the suppressive action of injected Treg cells is transient, conferring an on/off property to the system. Such property may be critical in regards to the possibility to suppress GVH disease but not graft‐versus‐leukemia/tumor response by re‐stimulation of donor Treg cells at first signs of GVH disease. Indeed, delaying DC or peptide administration several days after transplantation should permit to donor T cells to eliminate residual tumor cells through alloreactivity and then controlling an ongoing GVH disease. Such versatility is not possible with rsTreg cells due to their constant activation by allogeneic Ags and their immediate and constant suppressive effect on alloreactive T cells. As for the delayed activation of HY‐Treg cells, we have performed an experiment that showed that delaying HY administration on day 5 did not result in differences in term of survival or clinical signs of GVHD disease compared with mice treated at day 0. Nevertheless, additional experiments need to be performed in order to test whether or not Treg could be activated in a delayed manner and in which settings they could eliminate tumor cells. In conclusion, the “systemic bystander effect”, demonstrated here, holds a tremendous promise for the therapeutic application of exoTreg cells in preventing GVH disease, a fatal complication of over 30% of allo‐HSC transplantation 6. Furthermore, the same strategy can be applied to other autoimmune or solid organ transplant settings, thus removing one of the major obstacles on the way of this important therapeutic strategy from the bench to the bedside.
Materials and methods
Mice
Six‐ to ten‐week‐old (B6 × C3H) F1 (H2kb) and B6 (H‐2b) were obtained from Harlan Laboratories (France) and C3H (H‐2k) from Charles River Laboratories (France). B6 Ly5.1 were bred in our animal facility under specific pathogen‐free conditions. Experiments were performed according to the European Union guidelines and approved by our institutional review board (N° p3/2007/018, CREEA Ile de France n°3).
Ex vivo expansion of Ag‐specific Treg cells
Cell suspensions were obtained from spleen and peripheral LNs cells of B6 mice. Cells were first labeled with biotin‐coupled anti‐CD25 mAb (7D4, BD Bioscience, Le pont de Claix, France), followed with antibiotin microbeads (Miltenyi Biotec, Paris, France) and enriched in CD25+ cells using magnetic cell large selection columns (Miltenyi Biotec). Cells were then stained with FITC‐labeled anti‐CD4 (GK1.5), PE‐labeled anti‐CD62L (MEL‐14) and streptavidin‐Cy‐Chrome, which bound to free biotin‐labeled CD25 molecules (all obtained from BD Bioscience). The CD4+CD25highCD62Lhigh T cells were sorted by flow cyto‐metry using a FACSAria (BD Bioscience), yielding a purity of 98%. Purified Treg cells were cultured 4 weeks in the presence of recombinant murine IL‐2 (10 ng/mL, R&D Systems, Lille, France) as previously described13 and 20‐Gy‐irradiated recipient‐type C3H splenocytes to expand rsTreg cells. HY‐Treg cells were cultivated in presence of CD8+ DCs loaded with HY peptide (10 μg/mL, N‐15‐S, NY, PolyPeptide, Strasbourg, France). DC was obtained from spleen cells of C57BL/6 mice. After digestion with liberase (0.4 mg/mL, Roche, Meylan, France) and DNase (0.1 mg/mL, Roche), cells were labeled with anti‐CD11c‐coated microbeads (Miltenyi Biotec), followed by two consecutives magnetic cell separation using LS columns (Miltenyi Biotec). Cells were stained with FITC‐labeled anti‐CD11b (M1/70, BD Biosciences), PE‐labeled anti‐CD11c (HL3, BD Biosciences), and Cy‐Chrome‐labeled CD8a (53–6.7, BD Biosciences) and the CD11chighCD8+CD11b− cells (CD8+ DC) were sorted by flow cytometry using a FACSAria yielding a purity of 99%, as previously described 24.
In vitro suppression assay
After removal of dead cells by a gradient of Lymphocyte Separation Medium (Eurobio, Les Ulis, France), and five washes to remove residual IL‐2, up to 1 × 105 Treg cells that had been expanded for 4 weeks were added to the culture of 1 × 105 fresh CD25‐depleted T cells (purified from C57BL/6 spleen) stimulated by 2 × 105 irradiated B6 splenocytes and by 5 μg/mL soluble anti‐CD3 mAb (BD Biosciences). Cells cultured in round‐bottom, 96‐well plates for 96 h were pulsed with 3H‐methyl‐thymidine for the last 18 h.
Experimental GVH disease
Donor T cells were collected from LNs of donor animals and the percentage of CD3+ cells was determined by flow cytometry at time of infusion. Irradiated (10 Gy) or nonirradiated 7‐ to 12‐week‐old (B6 × C3H) F1 recipient mice were injected i.v. in the retro‐orbital sinus with 10 × 106 B6 BM cells (control group), 2 × 106 B6 CD3+ T cells alone to induce GVH disease, or with 2 × 106 cultured rsTreg cells or HY‐Treg cells. In nonirradiated recipients, 10 × 106 B6 CD3+ T cells alone are required to induce GVH disease. The same number of HY‐Treg cells was then added to test their clinical effect. Clinical signs of GVH disease (body weight loss, diarrhea, skin lesions, hunched posture) were monitored regularly. Body weight loss of more than 30% of the initial weight led to euthanasia of sick mice.
Histology
After mouse death or sacrifice, liver and colon samples were fixed in 4% formaldehyde solution for several days and embedded in paraffin. For both organs, 5 μm sections were stained with H&E for histological examination. One pathologist analyzed slides in a blinded fashion to assess the intensity of GVH disease. GVH disease lesions in each bowel sample were graded according to a semiquantitative scoring system described by Hill and Ferrara with minor modifications 45. Six parameters were scored for the colon (surface of the crypts, inflammation of the chorion, crypt regeneration, crypt epithelial cell apoptosis, crypt loss, mucosal ulceration), and seven for the liver (bile ducts, periportal necrosis, endothelitis, acidophilic body, confluent necrosis, sinusoidal lymphocytosis, inflammatory cell infiltrate). Each parameter was scored as follows: 0 as normal; 1 as focal and rare; 2 as focal and mild; 3 as diffuse and mild; 4 as diffuse and moderate; and 5 as diffuse and severe.
Flow cytometry
The following Abs were used for FACS analysis: anti‐CD3, anti‐CD4, anti‐CD8, anti‐CD90.2, anti‐H2Kk, anti‐CD25, anti‐CD62L, anti‐CD44, anti‐CD45.1, anti‐IL‐2, anti‐TNF‐α, and anti‐IFN‐γ, labeled with FITC, PE, allophycocyanin, PerCP, or Alexa Fluor 700. All mAbs were purchased from BD Biosciences. The PE‐ or Efluor 450‐labeled anti‐Foxp3 staining was performed using the eBioscience kit and protocol. For intracellular cytokine staining, cells were re‐stimulated with 1 μg/mL PMA (Sigma Aldrich, Saint Quentin Fallavier, France) and 0.5 μg/mL Ionomicyn (Sigma Aldrich) for 4 h, in the presence of GolgiPlug (1 μL/mL; BD Biosciences). After cell surface staining, intracellular staining was performed using the CytoFix/CytoPerm kit (BD Biosciences). For latency associated protein staining, mice were injected intravenously with 3 mg/kg Brefeldin A (Sigma Aldrich) 6 h prior to sacrifice. Events were acquired on an LSRII or FACSCanto II flow cytometer (BD Biosciences) and analyzed using FlowJo (Tree Star, Ashland, OR, USA) software.
In vivo activation of Treg cells
DCs were isolated from B6 female mice after magnetic sorting, and cultured at 37°C during 12 h in the presence of GM‐CSF (20 ng/mL) and HY peptide (10 μg/mL). B6C3F1 female recipient were immunized i.v. in the retro‐orbital sinus with 1 × 106 B6‐pulsed DCs or 100 μg of HY peptide at D0, D3, and D6 post graft.
Skin grafting
At 2 and 5 months after HSC transplant, tail‐skin grafts from female B6 and BALB/c mice were transplanted onto lateral thoracic wall of the recipients under ketamine (75 mg/kg) and xylazine (15 mg/kg) anesthesia. Skin grafts were monitored regularly by visual and tactile inspection. Rejection was defined as loss of viable donor epithelium.
Statistical analyses
Statistical significances were calculated using the two‐tailed unpaired Student's t‐test for cell analysis. Log‐rank (Mantel–Cox) test was used to compare survival between two groups of mice. When statistically significant, p values were indicated.
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Abbreviations
- exoTreg cell
Treg cell specific for an exogenous (i.e. nondonor, nonrecipient) Ag
- polyTreg cell
polyclonal Treg cell
- rsTreg
donor‐derived recipient‐specific Treg cell
- Teff cell
effector T cell
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Figure S1. Expansion of antigen‐specific Treg cells. (A) Isolation of highly purified Treg cells. Representative dot plot obtained before (left) and after Treg cells sorting (right). Highly purified Treg cells were cultured with C3H APC to select and expand rsTreg cells or with autologous B6 CD8+ DCs loaded with HY peptide to expand HY specific Treg cells (HY‐Treg). (B‐C) Ex vivo expansion of rsTreg cells (B) and HY‐Treg cells (C) during 30 days (left), phenotype of Treg cells by flow cytometry at the end of the culture and expression of CD25+Foxp3+ cells among CD4+ cells is shown (middle). Suppressive activity of Treg cells was measured by in vitro proliferation of T cells cultured with DC and anti‐CD3 and then determined by the incorporation of 3H methyl‐thymidine. Histograms represent mean ± SEM (right). (D) In vitro specific proliferation assay of HY‐Treg cells in presence of APC obtained from B6 male or female was determined by 3H‐methyl‐thymidine incorporation. Data shown are pooled from 2 experiments performed. **p < 0.01, two‐tailed unpaired Student's t test.
Figure S2. HY‐Treg cells prevent GVH disease by reducing activation and differentiation of donor Teff cells. Donor CD4+ and CD8+ Teff cells are analyzed at day 6 post‐transplantation in the spleen of animals grafted as described in Fig. 1A and identified using the CD45.1+ congenic marker. Mean percentage ± SEM of CD4+ (A) and CD8+ (B) divided donor T cells (CD45.1+CFSElow) expressing membrane CD25, CD44, CD62L or intracellular IL‐2, IFN‐g and TNF‐a with or without HY‐Treg cells (n = 4 in each group). Data shown are pooled from 2 experiments performed. *p < 0.05, **p < 0.01, two‐tailed unpaired Student's t test.
Figure S3. Prevention of GVH disease is associated with high expression of activation markers on HY‐Treg cells in the presence of their cognate antigen. HY‐Treg cells are collected at day 6 post‐transplantation in the spleen of female or male grafted animals, identified by their CD90.2+CD4+H2Kk‐ phenotype and analyzed for ICOS and GITR expression. (A) Representative histograms of ICOS (left) and GITR (right) expression by HY‐Treg cells in male (grey) or in female (white) B6C3F1 recipients. (B) Mean MFI ± SEM (left) and percentage (right) of ICOS and GITR expressing HY‐Treg cells in male (n = 4, grey) or in female (n = 4, white) B6C3F1 recipients. *p < 0.05, **p < 0.01, two‐tailed unpaired Student's t test.
Figure S4. Treatment of GVH disease by delayed HY‐Treg cells activation. Female B6C3F1 were lethally irradiated and grafted with BM cells, Teff cells and HY‐Treg cells as described in Fig. 5A. Kaplan‐Meier survival curves from control mice injected with Teff cells and HY‐Treg cells (black circle), mice intravenously injected with 100mg HY peptide at D0, D2 and D5 (grey circle) or at D5, D7 and D9 (white circle) post‐transplantation.
Acknowledgments
GHM was supported by the French government then by the “Ligue Nationale contre le cancer”. CP was partially supported by the region Ile de France, “DIM STEM‐Pôle,” and IFRNT. This work was supported by “Agence de la Biomédecine”, l'Institut National de la santé et de la recherche médicale, le centre national de la recherche scientifique.
Authors are indebted to Pr. Daniel Azoulay and Dr. Eliane Piaggio for critical reading of the manuscript, and to Pierric Parent for animal care and Louis Perol for technical help.
The copyright line has been changed since first published on 9 JUL 2013
References
- 1. Thomas, E. , Storb, R. , Clift, R. A. , Fefer, A. , Johnson, F. L. , Neiman, P. E. , Lerner, K. G. et al., Bone‐marrow transplantation (first of two parts). N. Engl. J. Med. 1975. 292: 832–843. [DOI] [PubMed] [Google Scholar]
- 2. Martin, P. J. , Hansen, J. A. , Buckner, C. D. , Sanders, J. E. , Deeg, H. J. , Stewart, P. , Appelbaum, F. R. et al., Effects of in vitro depletion of T cells in HLA‐identical allogeneic marrow grafts. Blood 1985. 66: 664–672. [PubMed] [Google Scholar]
- 3. Mackall, C. L. and Gress, R. E. , Pathways of T‐cell regeneration in mice and humans: implications for bone marrow transplantation and immunotherapy. Immunol. Rev. 1997. 157: 61–72. [DOI] [PubMed] [Google Scholar]
- 4. Horowitz, M. M. , Gale, R. P. , Sondel, P. M. , Goldman, J. M. , Kersey, J. , Kolb, H. J. , Rimm, A. A. et al., Graft‐versus‐leukemia reactions after bone marrow transplantation. Blood 1990. 75: 555–562. [PubMed] [Google Scholar]
- 5. Amarnath, S. , Costanzo, C. M. , Mariotti, J. , Ullman, J. L. , Telford, W. G. , Kapoor, V. , Riley, J. L. et al., Regulatory T cells and human myeloid dendritic cells promote tolerance via programmed death ligand‐1. PLoS Biol. 2010. 8: e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferrara, J. L. , Levine, J. E. , Reddy, P. and Holler, E. , Graft‐versus‐host disease. Lancet 2009. 373: 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ratanatharathorn, V. , Nash, R. A. , Przepiorka, D. , Devine, S. M. , Klein, J. L. , Weisdorf, D. , Fay, J. W. et al., Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft‐versus‐host disease prophylaxis after HLA‐identical sibling bone marrow transplantation. Blood 1998. 92: 2303–2314. [PubMed] [Google Scholar]
- 8. Storb, R. , Deeg, H. J. , Pepe, M. , Appelbaum, F. , Anasetti, C. , Beatty, P. , Bensinger, W. , et al., Methotrexate and cyclosporine versus cyclosporine alone for prophylaxis of graft‐versus‐host disease in patients given HLA‐identical marrow grafts for leukemia: long‐term follow‐up of a controlled trial. Blood 1989. 73: 1729–1734. [PubMed] [Google Scholar]
- 9. Bhatia, S. , Francisco, L. , Carter, A. , Sun, C. L. , Baker, K. S. , Gurney, J. G. , McGlave, P. B. et al., Late mortality after allogeneic hematopoietic cell transplantation and functional status of long‐term survivors: report from the Bone Marrow Transplant Survivor Study. Blood 2007. 110: 3784–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Socie, G. , Stone, J. V. , Wingard, J. R. , Weisdorf, D. , Henslee‐Downey, P. J. , Bredeson, C. , Cahn, J. Y. et al., Long‐term survival and late deaths after allogeneic bone marrow transplantation. Late Effects Working Committee of the International Bone Marrow Transplant Registry. N. Engl. J. Med. 1999. 341: 14–21. [DOI] [PubMed] [Google Scholar]
- 11. Taylor, P. A. , Lees, C. J. and Blazar, B. R. , The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft‐versus‐host disease lethality. Blood 2002. 99: 3493–3499. [DOI] [PubMed] [Google Scholar]
- 12. Cohen, J. L. , Trenado, A. , Vasey, D. , Klatzmann, D. and Salomon, B. L. , CD4(+)CD25(+) immunoregulatory T cells: new therapeutics for graft‐versus‐host disease. J. Exp. Med. 2002. 196: 401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trenado, A. , Charlotte, F. , Fisson, S. , Yagello, M. , Klatzmann, D. , Salomon, B. L. and Cohen, J. L. , Recipient‐type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft‐versus‐host disease while maintaining graft‐versus‐leukemia. J. Clin. Invest. 2003. 112: 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor, P. A. , Panoskaltsis‐Mortari, A. , Swedin, J. M. , Lucas, P. J. , Gress, R. E. , Levine, B. L. , June, C. H. et al., L‐Selectin(hi) but not the L‐selectin(lo) CD4+25 +T‐regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood 2004. 104: 3804–3812. [DOI] [PubMed] [Google Scholar]
- 15. Ermann, J. , Hoffmann, P. , Edinger, M. , Dutt, S. , Blankenberg, F. G. , Higgins, J. P. , Negrin, R. S. et al., Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood 2005. 105: 2220–2226. [DOI] [PubMed] [Google Scholar]
- 16. Trenado, A. , Sudres, M. , Tang, Q. , Maury, S. , Charlotte, F. , Gregoire, S. , Bonyhadi, M. et al., Ex vivo‐expanded CD4+CD25+ immunoregulatory T cells prevent graft‐versus‐host‐disease by inhibiting activation/differentiation of pathogenic T cells. J. Immunol. 2006. 176: 1266–1273. [DOI] [PubMed] [Google Scholar]
- 17. Sagoo, P. , Ratnasothy, K. , Tsang, Y. , Barber, L. D. , Noble, A. , Lechler, R. I. and Lombardi, G. , Alloantigen‐specific regulatory T cells prevent experimental chronic graft‐versus‐host disease by simultaneous control of allo‐ and autoreactivity. Eur. J. Immunol. 2012. 42: 3322–3333. [DOI] [PubMed] [Google Scholar]
- 18. Di Ianni, M. , Falzetti, F. , Carotti, A. , Terenzi, A. , Castellino, F. , Bonifacio, E. , Del Papa, B. et al., prevent GVHD and promote immune reconstitution in HLA‐haploidentical transplantation. Blood 2011. 117: 3921–3928. [DOI] [PubMed] [Google Scholar]
- 19. Brunstein, C. G. , Miller, J. S. , Cao, Q. , McKenna, D. H. , Hippen, K. L. , Curtsinger, J. , Defort, T. et al., Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 2011. 117: 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sagoo, P. , Ali, N. , Garg, G. , Nestle, F. O. , Lechler, R. I. and Lombardi, G. , Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci. Transl. Med. 2011. 3: 83ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang, Q. , Henriksen, K. J. , Bi, M. , Finger, E. B. , Szot, G. , Ye, J. , Masteller, E. L. et al., In vitro‐expanded antigen‐specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 2004. 199: 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tarbell, K. V. , Yamazaki, S. , Olson, K. , Toy, P. and Steinman, R. M. , CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 2004. 199: 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaidot, A. , Landau, D. A. , Martin, G. H. , Bonduelle, O. , Grinberg‐Bleyer, Y. , Matheoud, D. , Gregoire, S. et al., Immune reconstitution is preserved in hematopoietic stem cell transplant co‐administered with regulatory T cells for GVHD prevention. Blood 2011. 117: 2975–2983. [DOI] [PubMed] [Google Scholar]
- 24. Fisson, S. , Djelti, F. , Trenado, A. , Billiard, F. , Liblau, R. , Klatzmann, D. , Cohen, J. L. et al., Therapeutic potential of self‐antigen‐specific CD4+ CD25+ regulatory T cells selected in vitro from a polyclonal repertoire. Eur. J. Immunol. 2006. 36: 817–827. [DOI] [PubMed] [Google Scholar]
- 25. Tang, Q. and Bluestone, J. A. , The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol. 2008. 9: 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boyman, O. , Letourneau, S. , Krieg, C. and Sprent, J. , Homeostatic proliferation and survival of naive and memory T cells. Eur. J. Immunol. 2009. 39: 2088–2094. [DOI] [PubMed] [Google Scholar]
- 27. Su, Y. , Huang, X. , Wang, S. , Min, W. P. , Yin, Z. , Jevnikar, A. M. and Zhang, Z. X. , Double negative Treg cells promote nonmyeloablative bone marrow chimerism by inducing T‐cell clonal deletion and suppressing NK cell function. Eur. J. Immunol. 2012. 42: 1216–1225. [DOI] [PubMed] [Google Scholar]
- 28. Thornton, A. M. and Shevach, E. M. , Suppressor effector function of CD4+CD25+immunoregulatory T cells is antigen nonspecific. J. Immunol. 2000. 164: 183–190. [DOI] [PubMed] [Google Scholar]
- 29. Maury, S. , Salomon, B. , Klatzmann, D. and Cohen, J. L. , Division rate and phenotypic differences discriminate alloreactive and nonalloreactive T cells transferred in lethally irradiated mice. Blood 2001. 98: 3156–3158. [DOI] [PubMed] [Google Scholar]
- 30. Katz, J. D. , Wang, B. , Haskins, K. , Benoist, C. and Mathis, D. , Following a diabetogenic T cell from genesis through pathogenesis. Cell 1993. 74: 1089–1100. [DOI] [PubMed] [Google Scholar]
- 31. Stephens, L. A. , Malpass, K. H. and Anderton, S. M. , Curing CNS autoimmune disease with myelin‐reactive Foxp3+ Treg. Eur. J. Immunol. 2009. 39: 1108–1117. [DOI] [PubMed] [Google Scholar]
- 32. Wright, G. P. , Notley, C. A. , Xue, S. A. , Bendle, G. M. , Holler, A. , Schumacher, T. N. , Ehrenstein, M. R. et al., Adoptive therapy with redirected primary regulatory T cells results in antigen‐specific suppression of arthritis. Proc. Natl. Acad. Sci. USA 2009. 106: 19078–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karim, M. , Feng, G. , Wood, K. J. and Bushell, A. R. , CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen‐specific reactivation in vivo is critical for bystander regulation. Blood 2005. 105: 4871–4877. [DOI] [PubMed] [Google Scholar]
- 34. Verginis, P. , McLaughlin, K. A. , Wucherpfennig, K. W. , von Boehmer, H. and Apostolou, I. , Induction of antigen‐specific regulatory T cells in wild‐type mice: visualization and targets of suppression. Proc. Natl. Acad. Sci. USA 2008. 105: 3479–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoffmann, P. , Eder, R. , Kunz‐Schughart, L. A. , Andreesen, R. and Edinger, M. , Large‐scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood 2004. 104: 895–903. [DOI] [PubMed] [Google Scholar]
- 36. Hoffmann, P. , Boeld, T. J. , Eder, R. , Albrecht, J. , Doser, K. , Piseshka, B. , Dada, A. et al., Isolation of CD4+CD25+ regulatory T cells for clinical trials. Biol. Blood Marrow Transplant 2006. 12: 267–274. [DOI] [PubMed] [Google Scholar]
- 37. Hoffmann, P. , Eder, R. , Boeld, T. J. , Doser, K. , Piseshka, B. , Andreesen, R. and Edinger, M. , Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T‐cell lines upon in vitro expansion. Blood 2006. 108: 4260–4267. [DOI] [PubMed] [Google Scholar]
- 38. Roncarolo, M. G. and Battaglia, M. , Regulatory T‐cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat. Rev. Immunol. 2007. 7: 585–598. [DOI] [PubMed] [Google Scholar]
- 39. Hoffmann, P. , Boeld, T. J. , Eder, R. , Huehn, J. , Floess, S. , Wieczorek, G. , Olek, S. et al., Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur. J. Immunol. 2009. 39: 1088–1097. [DOI] [PubMed] [Google Scholar]
- 40. Nadig, S. N. , Wieckiewicz, J. , Wu, D. C. , Warnecke, G. , Zhang, W. , Luo, S. , Schiopu, A. et al., In vivo prevention of transplant arteriosclerosis by ex vivo‐expanded human regulatory T cells. Nat. Med. 2010. 16: 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roncarolo, M. G. , Gregori, S. , Lucarelli, B. , Ciceri, F. and Bacchetta, R. , Clinical tolerance in allogeneic hematopoietic stem cell transplantation. Immunol. Rev. 2011. 241: 145–163. [DOI] [PubMed] [Google Scholar]
- 42. Tresoldi, E. , Dell'albani, I. , Stabilini, A. , Jofra, T. , Valle, A. , Gagliani, N. , Bondanza, A. et al., Stability of human rapamycin‐expanded CD4+CD25+ T regulatory cells. Haematologica 2011. 96: 1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hippen, K. L. , Merkel, S. C. , Schirm, D. K. , Sieben, C. M. , Sumstad, D. , Kadidlo, D. M. , McKenna, D. H. et al., Massive ex vivo expansion of human natural regulatory T cells (Tregs) with minimal loss of in vivo functional activity. Sci. Transl. Med. 2011. 3: 83ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Edinger, M. and Hoffmann, P. , Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr. Opin. Immunol. 2011. 23: 679–684. [DOI] [PubMed] [Google Scholar]
- 45. Hill, G. R. and Ferrara, J. L. , The primacy of the gastrointestinal tract as a target organ of acute graft‐versus‐host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood 2000. 95: 2754–2759. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Figure S1. Expansion of antigen‐specific Treg cells. (A) Isolation of highly purified Treg cells. Representative dot plot obtained before (left) and after Treg cells sorting (right). Highly purified Treg cells were cultured with C3H APC to select and expand rsTreg cells or with autologous B6 CD8+ DCs loaded with HY peptide to expand HY specific Treg cells (HY‐Treg). (B‐C) Ex vivo expansion of rsTreg cells (B) and HY‐Treg cells (C) during 30 days (left), phenotype of Treg cells by flow cytometry at the end of the culture and expression of CD25+Foxp3+ cells among CD4+ cells is shown (middle). Suppressive activity of Treg cells was measured by in vitro proliferation of T cells cultured with DC and anti‐CD3 and then determined by the incorporation of 3H methyl‐thymidine. Histograms represent mean ± SEM (right). (D) In vitro specific proliferation assay of HY‐Treg cells in presence of APC obtained from B6 male or female was determined by 3H‐methyl‐thymidine incorporation. Data shown are pooled from 2 experiments performed. **p < 0.01, two‐tailed unpaired Student's t test.
Figure S2. HY‐Treg cells prevent GVH disease by reducing activation and differentiation of donor Teff cells. Donor CD4+ and CD8+ Teff cells are analyzed at day 6 post‐transplantation in the spleen of animals grafted as described in Fig. 1A and identified using the CD45.1+ congenic marker. Mean percentage ± SEM of CD4+ (A) and CD8+ (B) divided donor T cells (CD45.1+CFSElow) expressing membrane CD25, CD44, CD62L or intracellular IL‐2, IFN‐g and TNF‐a with or without HY‐Treg cells (n = 4 in each group). Data shown are pooled from 2 experiments performed. *p < 0.05, **p < 0.01, two‐tailed unpaired Student's t test.
Figure S3. Prevention of GVH disease is associated with high expression of activation markers on HY‐Treg cells in the presence of their cognate antigen. HY‐Treg cells are collected at day 6 post‐transplantation in the spleen of female or male grafted animals, identified by their CD90.2+CD4+H2Kk‐ phenotype and analyzed for ICOS and GITR expression. (A) Representative histograms of ICOS (left) and GITR (right) expression by HY‐Treg cells in male (grey) or in female (white) B6C3F1 recipients. (B) Mean MFI ± SEM (left) and percentage (right) of ICOS and GITR expressing HY‐Treg cells in male (n = 4, grey) or in female (n = 4, white) B6C3F1 recipients. *p < 0.05, **p < 0.01, two‐tailed unpaired Student's t test.
Figure S4. Treatment of GVH disease by delayed HY‐Treg cells activation. Female B6C3F1 were lethally irradiated and grafted with BM cells, Teff cells and HY‐Treg cells as described in Fig. 5A. Kaplan‐Meier survival curves from control mice injected with Teff cells and HY‐Treg cells (black circle), mice intravenously injected with 100mg HY peptide at D0, D2 and D5 (grey circle) or at D5, D7 and D9 (white circle) post‐transplantation.