

Abstract
Background
The equine infectious anemia virus (EIAV) vaccine is the only attenuated lentiviral vaccine applied on a large scale that has been shown to be effective in controlling the prevalence of EIA in China. This vaccine was developed by successive passaging of a field-isolated virulent strain in different hosts and cultivated cells. To explore the molecular basis for the phenotype alteration of this vaccine strain, we systematically analyzed its genomic evolution during vaccine development.
Results
Sequence analysis revealed that the genetic distance between the wild-type strain and six representative strains isolated from key development stages gradually increased with the number of passages. Env gene, but not gag and pol, showed a clear evolutionary flow similar to that of the whole genomes of different generations during the attenuation. Stable mutations were identified in multiple regions of multiple genes along with virus passaging. The adaption of the virus to the growth environment of cultured cells with accumulated genomic and genetic variations was positively correlated with the reduction in pathogenicity and rise of immunogenicity. Statistical analyses revealed significant differences in the frequency of the most stable mutations between in vivo and ex vivo-adapted strains and between virulent and attenuated strains.
Conclusions
These data indicate that EIAV evolution during vaccine development generated an accumulation of mutations under the selective drive force, which helps to better understand the molecular basis of lentivirus pathogenicity and immunogenicity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12977-016-0240-6) contains supplementary material, which is available to authorized users.
Keywords: EIAV, Genetic Evolution, Lentiviral vaccine
Background
Vaccination is the most effective means of controlling infectious diseases. However, the development of a safe and effective lentiviral vaccine, such as a human immunodeficiency virus-1 (HIV-1) vaccine, remains a huge scientific challenge. Studies on the development of lentiviral vaccines targeting HIV-1, simian immunodeficiency virus (SIV), chimeric simian-human immunodeficiency virus (SHIV), equine infectious anemia virus (EIAV) and feline immunodeficiency virus (FIV) have demonstrated that the live attenuated formulation is the most effective formulation [1, 2]. Because lentiviral genomes integrate into host chromosomes and feature a high frequency of genomic mutations, attenuated live vaccines are generally not considered as an option for practical lentiviral vaccines. However, the study of immune responses induced by attenuated vaccines can provide a useful reference to elucidate the protective immune responses to lentiviral infections [3].
EIAV is a member of the Lentivius genus in the Retroviridae family. The major features of EIAV, including its genomic structure, life cycle, in vivo antigen evolution, cell tropism and the interaction between the virus and host, are similar to HIV-1. Most horses infected with EIAV exhibit a repeated high-titer viremia with typical clinical features such as fever, thrombocytopenia and anemia. Some infected horses may eventually control viral replication and become asymptomatic carriers after several months or years of acute or chronic infection. EIAV infection is life-long.
EIAVLN40, which is an EIAV strain that is highly lethal to horses (experimental lethality of 80–100 %), was developed by passaging a field isolate in horses for 16 passages. The pathogenicity of this virus in donkeys was largely enhanced (raised from generally asymptomatic to near 100 % lethality) by the continuous passaging of EIAVLN40 in donkeys for 117 rounds (Fig. 1a). The resultant strain (EIAVDV117) was subsequently continuously acclimatized in cultivated primary donkey monocyte-derived macrophages (dMDM). The lethality of EIAVDV117 to horses and donkeys was gradually decreased by passaging in dMDM and was finally reduced to the point of not causing any clinical symptoms in either horses or donkeys after 90 passages (Fig. 1b). The ability to induce protective immunity in inoculated horses was detected in viruses that were attenuated in dMDM for approximately 110–125 passages. The resultant viral strain (termed EIAVDLV121) was able to elicit resistance to challenge with EIAVLN40 (the average genomic divergence to EIAVDLV121 was 2.8 %) in >75 % of the inoculated horses and >95 % of the inoculated donkeys. This attenuated viral strain was used as a vaccine to massively immunize 61 million horses and mules to prevent EIA infection in China from 1975 to the 1990s [4]. This nationwide vaccination program ended the incidence of equine infectious anemia (EIA) in this country. The successful application of the EIAV vaccine has provided an important and unique reference model for studies on lentivirus immunity and vaccines. EIAVDLV121 was further adapted to cultivated fetal donkey dermal (FDD) cells (termed EIAVFDDV13) to reduce the preparation costs of this attenuated EIAV strain. Part of the historic data on EIAVDLV121 protection of disease in laboratory infected horses indicated that the protection efficiency to challenge with the parental virulent strain (EIAVLN40, the average variation in Env amino acid sequences is 7.1 %) and an American strain (EIAVWyoming, the average variation in Env amino acid sequences is 37.8 %) was 81 % (25/31) and 80 % (8/10), respectively [4]. Our resent experiments on the immunogenicity of EIAVDLV121 and EIAVFDDV13 demonstrated a 50 % (2/4) and 83 % (5/6) similar protection of disease (Additional file 1: Table S1) [4, 5].
Fig. 1.
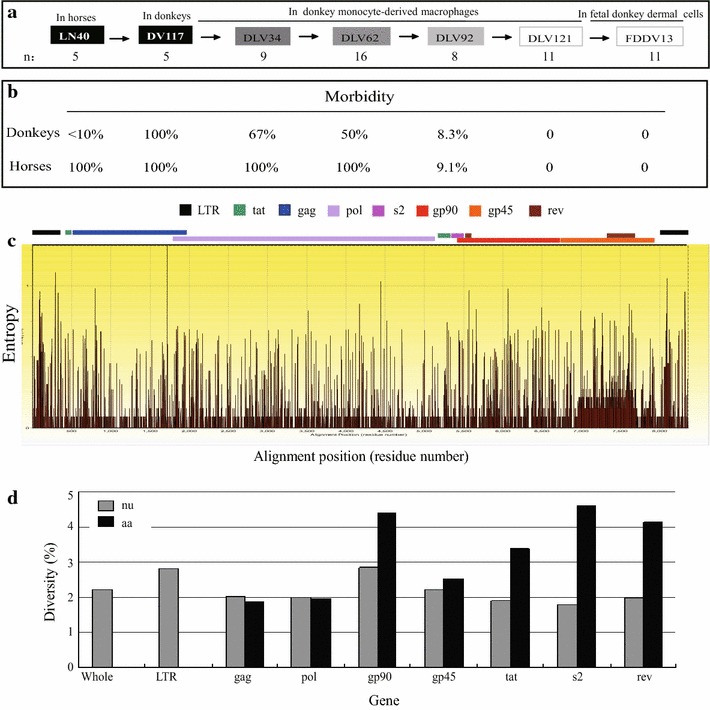
Genome variants of EIAV strains during vaccine development. a The flowchart of EIAV vaccine development. Four major stages were included the process of vaccine development: the in vivo passages in horses and donkeys and the passages in cultured dMDM and fetal donkey dermal (FDD) cells. The representative strains isolated from each of these stages and the numbers of clones used for the proviral genome analysis are indicated. These viral strains included the virulent strains EIAVLN40 and EIAVDV117, the vaccine strains EIAVDLV121 and EIAVFDDV13 and three strains of EIAVDV117 passaged in dMDM (the 34th passage strain EIAVDLV34, the 62nd passage strain EIAVDLV62 and the 92nd passage strain EIAVDLV92). b The pathogenicity attenuation of EIAV strains during vaccine development. The percentages indicate the morbidity of horses or donkeys that were experimentally infected with the indicated viral strains. c Shannon entropy (SE) plots of EIAV complete genomic sequences depict the mutation frequency at each nucleotide position. Full genomic sequences of 65 clones from seven EIAV strains were isolated from different key stages of vaccine development. Refer to Fig. 1a for detailed information of these viral strains. Larger entropy values indicate higher mutation frequencies. The relative positions of EIAV individual genes are labeled. d The variations in the EIAV genome and individual genes and their encoded proteins among the 65 clones from the seven representative strains. The diversities were calculated by computing the overall mean distances using the p-distance method in MEGA 5 software
During the development process of the attenuated EIAV vaccine, a series of virus strains with different pathogenicities or immunogenicities were obtained. These strains provided a useful resource for the study of essential factors that induce protective immunity to lentiviruses. In this article, we analyzed the proviral genomic characteristics and the evolutionary trend of representative strains from key stages of the process, including EIAVLN40, EIAVDV117, EIAVDLV34, EIAVDLV62, EIAVDLV92, EIAVDLV121 and EIAVFDDV13. Among these strains, EIAVDLV34, EIAVDLV62 and EIAVDLV92 were collected from the 34th, 62nd and 92nd passages of EIAVDV117 in dMDM, respectively (Fig. 1a).
Results and discussion
Analysis of viral genome variants over the course of the development of an attenuated EIAV vaccine
To examine the overall contributions of genes or gene fragments of the EIAV genome to the evolution of this virus during the development of the vaccine strains EIAVDLV121 and EIAVFDDV13, the frequency of each nucleotide of 65 full genomic sequences of EIAV strains sampled from key stages of the process was analyzed using Shannon Entropy (SE). As presented in Fig. 1c, nucleotides with high SE values were not randomly distributed but were clustered as different-sized islands. These nucleotides were largely located in the LTR and env regions.
The proviral genome size of different EIAV strains ranged from 7549 to 8277 bp. A detailed analysis of the average mutation rates of different genes and the LTR revealed that the overall diversity among the total 65 genomes of the seven EIAV strains was 2.0 %. The diversity was highest in the LTR (2.95 ± 0.26 %), followed by env (consisting of the gp90 and gp45 genes, which were 2.90 ± 0.19 and 2.11 ± 0.26 %, respectively) (Fig. 1d). The variation in the encoded proteins among these EIAV strains was considerably higher. The S2 accessary protein exhibited the highest diversity in the amino acid sequence, reaching 5.02 ± 1.58 %, followed by the surface unit (SU or gp90) of the envelope protein (Env) and Rev, which were 4.81 ± 0.48 and 3.99 ± 0.84 %, respectively (Fig. 1d). Moreover, the genetic distances between the different viral strains and the parental strains EIAVLN40 and EIAVDV117 gradually increased with the increasing passage numbers (Table 1).
Table 1.
Comparison of nucleotide and amino acid genetic distances of the various EIAVs compared to EIAVLN40 and EIAVDV117
| EIAVLN40 | EIAVDV117 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EIAVDV117 | EIAVDLV34 | EIAVDLV62 | EIAVDLV92 | EIAVDLV121 | EIAVFDDV13 | EIAVDLV34 | EIAVDLV62 | EIAVDLV92 | EIAVDLV121 | EIAVFDDV13 | ||
| LTR | nu | 2.90 (0.85)a | 3.30 (0.84) | 4.18 (1.11) | 4.18 (1.16) | 4.06 (1.01) | 5.86 (1.40) | 1.94 (0.58) | 2.97 (0.90) | 3.68 (1.01) | 2.59 (0.77) | 3.71 (1.09) |
| Gag | nu | 2.02 (0.35) | 1.72 (0.26) | 1.96 (0.29) | 2.16 (0.31) | 2.30 (0.32) | 2.47 (0.37) | 2.13 (0.29) | 2.31 (0.31) | 2.58 (0.34) | 2.62 (0.32) | 2.57 (0.36) |
| aa | 1.91 (0.51) | 1.69 (0.42) | 2.09 (0.53) | 2.08 (0.53) | 2.73 ± (0.58) | 2.31 (0.57) | 1.98 (0.45) | 2.12 (0.50) | 2.25 (0.53) | 2.73 (0.54) | 2.14 (0.50) | |
| Pol | nu | 1.99 (0.24) | 1.93 (0.16) | 2.00 (0.10) | 2.06 (0.19) | 2.40 (0.20) | 2.75 (0.23) | 1.65 (0.16) | 1.77 (0.16) | 1.74 (0.17) | 2.13 (0.19) | 2.38 (0.23) |
| aa | 2.03 (0.41) | 2.13 (0.34) | 2.27 (0.34) | 2.38 (0.36) | 2.49 (0.38) | 2.84 (0.43) | 1.35 (0.23) | 1.69 (0.26) | 1.61 (0.26) | 1.79 (0.27) | 2.00 (0.32) | |
| gp90 | nu | 3.13 (0.43) | 3.28 (0.39) | 3.40 (0.41) | 3.51 (0.41) | 3.94 (0.44) | 4.01 (0.48) | 3.43 (0.40) | 3.46 (0.42) | 3.60 (0.43) | 3.99 (0.43) | 3.78 (0.46) |
| aa | 4.20 (0.91) | 5.17 (0.82) | 5.17 (0.87) | 5.55 (0.91) | 6.07 (0.90) | 6.71 (1.11) | 6.38 (0.93) | 6.20 (0.94) | 6.38 (0.97) | 7.05 (1.04) | 7.59 (1.18) | |
| gp45 | nu | 2.45 (0.38) | 2.28 (0.29) | 2.36 (0.26) | 2.40 (0.29) | 2.60 (0.31) | 2.86 (0.41) | 1.64 (0.23) | 1.89 (0.23) | 1.86 (0.27) | 2.18 (0.29) | 2.32 (0.40) |
| aa | 3.31 (0.69) | 3.14 (0.55) | 3.56 (0.50) | 3.22 (0.53) | 3.42 (0.53) | –b | 1.78 (0.41) | 2.60 (0.44) | 2.14 (0.47) | 2.71 (0.53) | – | |
| Tat | nu | 1.62 (0.73) | 1.13 (0.36) | 1.40 (0.45) | 1.74 (0.57) | 2.04 (0.67) | 2.27 (0.79) | 2.28 (0.72) | 2.44 (0.75) | 2.59 (0.78) | 2.61 (0.80) | 2.85 (0.90) |
| aa | 2.05 (1.26) | 2.26 (0.97) | 2.93 (1.27) | 3.76 (1.51) | 4.38 (1.91) | 5.11 (2.11) | 2.98 (1.36) | 3.29 (1.45) | 3.81 (1.53) | 3.19 (1.61) | 3.93 (1.83) | |
| S2 | nu | 1.97 (0.91) | 1.72 (0.48) | 2.14 (0.77) | 2.45 (0.88) | 2.94 (1.01) | 2.45 (0.98) | 3.00 (0.96) | 3.31 (1.11) | 3.12 (1.07) | 4.10 (1.29) | 2.85 (1.10) |
| aa | 3.60 (2.20) | 6.85 (2.51) | 8.08 (3.23) | 7.57 (2.99) | 10.61 (3.81) | 6.38 (3.10) | 4.17 (1.36) | 5.62 (2.47) | 6.78 (2.75) | 7.97 (3.09) | 6.38 (3.13) | |
| Rev | nu | 1.62 (0.50) | 1.61 (0.37) | 1.81 (0.33) | 1.50 (0.34) | 1.81 (0.35) | 2.23 (0.65) | 1.57 (0.39) | 1.94 (0.39) | 1.59 (0.39) | 1.86 (0.38) | 1.58 (0.54) |
| aa | 2.52 (0.77) | 3.29 (0.72) | 2.48 (0.63) | 3.64 (0.84) | 5.70 (1.87) | 3.47 (0.12) | 4.25 (0.11) | 3.66 (0.11) | 4.45 (0.10) | 4.77 (0.63) | 2.52 (0.77) | |
| Complete | 2.29 (0.12) | 2.22 (0.13) | 2.37 (0.17) | 2.56 (0.11) | 2.78 (0.10) | 3.15 (0.11) | 2.10 (0.11) | 2.25 (0.15) | 2.38 (0.12) | 2.57 (0.13) | 2.81 (0.17) | |
aThe number in parenthesis indicate standard deviation
bThe gp45 amino acid genetic distances of EIAVFDDV13 with EIAVLN40 and EIAVDV117 no showed, because existence of truncated gp45
Phylogenetic analysis of strains collected during the development of an attenuated EIAV vaccine
Phylogenetic trees were constructed based on the 65 full-length proviral genomic sequences (Fig. 2). The clusters of each individual genome clearly demonstrated the evolutionary direction from EIAVLN40 to EIAVFDDV13. Changes in the viral growth environment inevitably created new branches in the phylogenetic tree [i.e., changing the host from horse to donkey (between EIAVLN40 and EIAVDV117) and shifting the cultivated cell type from dMDM to FDD (between EIAVDLV121 and EIAVFDDV13)]. Conversely, the evolution of EIAV was rather gradual and smooth under the same growth environment conditions, as shown by the overlap of the sequence distributions of species from EIAVDLV34, EIAVDLV62 and EIAVDLV92 in the map. This result indicated that viral evolution was a long-term process with selective mutations accumulating during continuous passages in cultured cells. Furthermore, dramatic alterations of the growth environment could speed up viral genomic mutations.
Fig. 2.
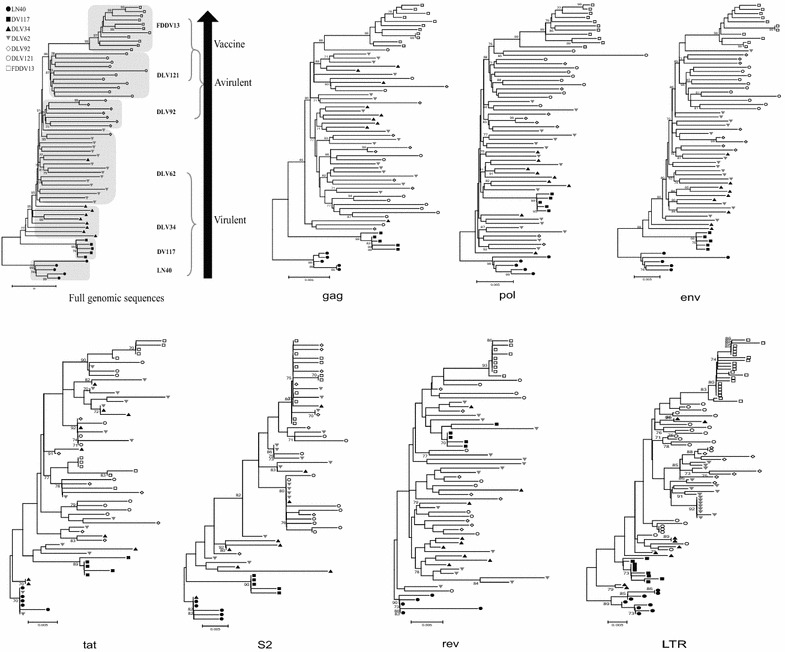
Phylogenetic tree analysis of EIAV proviral genomes and individual genes (gag, pol, env, tat, s2, rev and LTR). Sequence data were collected from 65 complete proviral genomes of seven viral strains. The phylogenetic trees were constructed using the neighbor-joining method and calculated with the Kimura 2 parameter in MEGA 5. Bootstrap values above 70 % are indicated. The virulent strains, including EIAVLN40, EIAVDV117, EIAVDLV34 and EIAVDLV62, are viruses that cause typical EIA symptoms (rectal temperature >39 °C, the platelet count <100,000/μL and the plasma viral load >106 copies/mL)
The evolutionary behavior of each individual EIAV gene and the LTR region were also phylogenetically analyzed. Three different evolutionary patterns were observed, as presented in Fig. 2. The first pattern was observed for the env gene and clearly demonstrated a direction toward the flow of the vaccine development process similar to that identified for the total viral genome, with a remarkable clustering of sequences from attenuated strains (EIAVDLV92, EIAVDLV121 and EIAVFDDV13) (Fig. 2). This result implied that there was a close association of the env evolution with the EIAV phenotypic change during the in vivo and ex vivo passages. The pol sequences also displayed a separated distribution between the EIAV virulent and attenuated strains (Fig. 2). However, the genes were not well-separated among strains with a similar pathogenicity, suggesting a close association with pathogenicity. The second pattern was observed for LTR and gag (Fig. 2), which showed separated branches of sequences that clustered according to the different EIAV growth environments. Specifically, sequences of EIAVLN40 (from horses), EIAVDV117 (from donkeys) and EIAVFDDV13 (from FDD cells) each formed a clearly separated branch. Although clones from strains grown in eMDM (EIAVDLV strains 34, 62, 92 and 121) were well-distinguished from the other strains, sequences within these EIAVDLV strains were mixed in the sub-branches of their clusters. This phylogenetic pattern suggested the association of LTR and gag with EIAV adaption to the environmental change and the reduced importance of pathogenicity. The third genetic evolutionary pattern was observed for the genes of three accessory proteins (tat, s2 and rev) (Fig. 2). For tat and rev in particular, the sequence distribution in the phylogenetic tree did not follow the trend of in vivo and ex vivo passaging of EIAV. Viral genes in different passages were largely mixed within different branches, demonstrating no obvious evolutionary direction. However, s2 exhibited a separation between sequences from strains replicated either in vivo or cultured cells (Fig. 2).
Combined with Fig. 1d, which depicts the higher amino acid mutation rate, our data on EIAV genetic evolution suggest that: (1) Gag, Pol and accessary proteins have higher percentages of silent nt substitutions or substitutions that are not consistent with the substitution patterns of the whole genome during the alteration of growth environment, implying that they are less driven by selective pressures; and (2) these mutations do not significantly contribute to the attenuation of pathogenicity. The above results suggest that viral genome evolution is a comprehensive result of different gene changes and mutations during the process of vaccine development and that the env may play crucial roles in the attenuation of pathogenicity [6]. The env is closely related to the virus’s targeting ability and immunogenicity. Multiple lines of evidence have demonstrated that the Env of highly virulent lentiviral strains contributes to stronger neutralization escape capabilities, enterotoxic activities and the ability to activate signaling pathways of inflammatory factors compared to the Env of attenuated strains [7].
Stable mutation rates during the development of an EIAV attenuated vaccine
To elucidate the meaning of the EIAV genome evolution and the contributions of the mutations in each gene to the altered viral functions during long-term in vivo and ex vivo passaging, the mutational characteristics of the LTR and viral genes were extensively analyzed using the predominant sequence of EIAVLN40 as the reference (Figs. 3, 4 and Additional file 2: Figure S2, Table 2). Some sequences contained premature stop codons in the coding region; these genes might play some roles in the quasispecies reservoir but were defective in the virus and were excluded from the analysis. For the convenience of description, we classified the seven viral strains according to their pathogenicity and origins into the following groups: virulent strains (EIAVLN40, EIAVDV117, EIAVDLV34 and EIAVDLV62); in vivo highly virulent strains (EIAVLN40 and EIAVDV117); attenuated strains (EIAVDLV92, EIAVDLV121 and EIAVFDDV13); cell culture adapted viral strains (EIAVDLV34, EIAVDLV62, EIAVDLV92, EIAVDLV121 and EIAVFDDV13); and vaccine strains (EIAVDLV121 and EIAVFDDV13). Some strains could be placed in more than one group because of different classification categories.
Fig. 3.
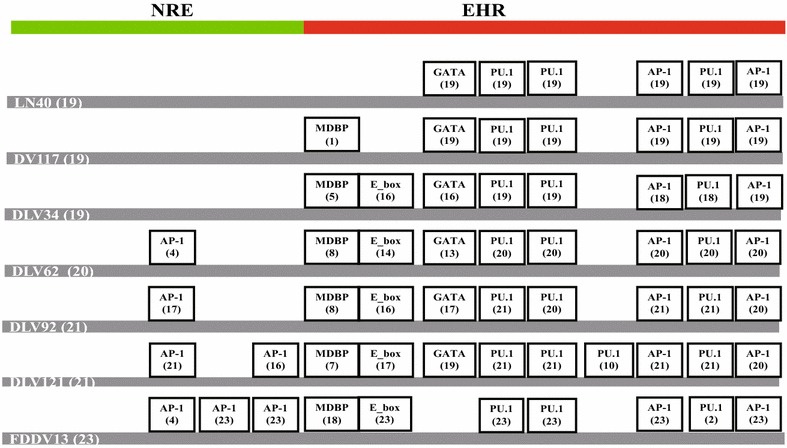
Alteration of the transcription factor binding motifs in the LTR U3 region. Sequences of 134 clones of the LTR from seven EIAV strains were randomly isolated from PCR-amplified fragments and analyzed. NRE indicates the negative regulation element and EHR indicates the enhancer region. The boxes define transcription factor binding sites. The numbers at the left end of the gray lines indicate the detected clones of each strain. The numbers in parentheses refer to the numbers of clones that contain the indicated transcription factor binding site
Fig. 4.
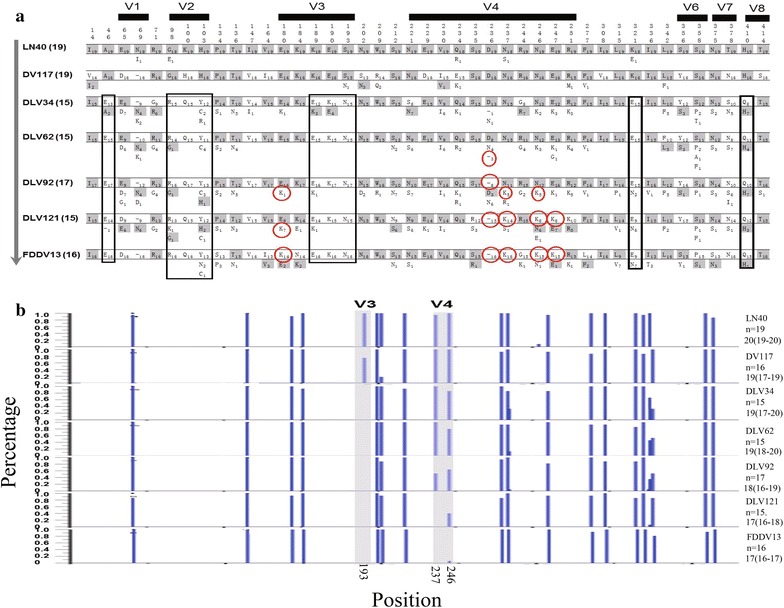
The stable mutations of EIAV gp90. a Stable mutations in gp90 proteins generated during different vaccine development stages. The amino acid sequences were deduced from the gene sequences originating from either the proviral genomes or from the directly cloned PCR products after removing sequences containing premature stop codons. The gp90 sequences were aligned to the reference sequence EIAVLN40. The shadowed residues and white background residues are identical to or different from the reference sequence, respectively. Stable mutation sites detected primarily in virus strains adapted to cultivated cells are boxed, whereas those limited in the attenuated strains are marked with red circles. V1–V8 designate the eight variable regions. The numbers on the top of the graphs show the positions of stable mutation sites, and those at the left side indicate the sequences applied for the analysis. The downward arrows indicate the direction of the vaccine development process. b The changes in gp90 glycosylation sites during vaccine development. The 116 gp90 sequences of seven different EIAV strains were analyzed using the N-GlycoSite program (http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html). The letter “n” in the labels on the right side indicates the total clone number. The numbers below show the average, minimum and maximum values of predicted N-glycosylation sites. The Y-axis shows the percentage of each glycosylation site in the detected clones
Table 2.
The proportion of stable amino acid substitutions of each gene in various EIAVs
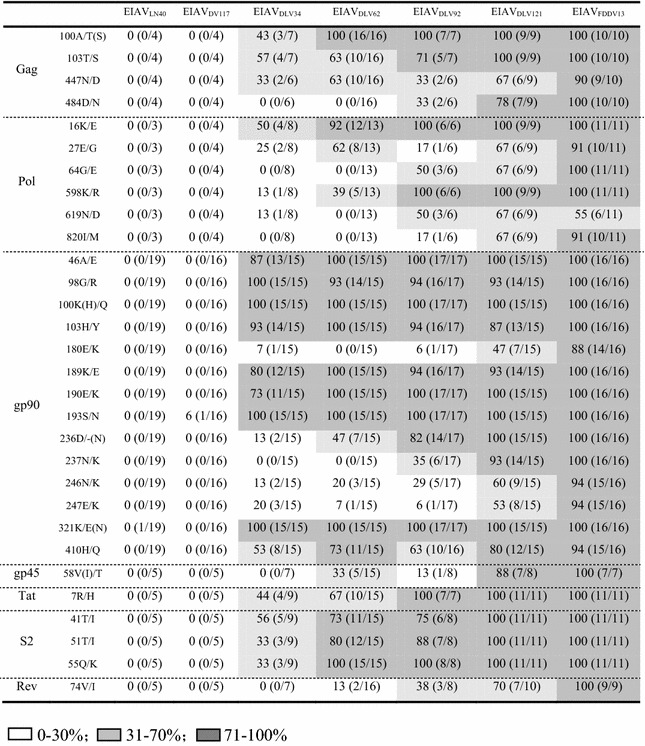
The LTR region contained the highest mutation rate in the genomic sequence because of insertions, deletions and point substitutions. The mutations were largely clustered in the U3 region, which consisted of the negative regulation element (NRE) and enhancer region (EHR). Most of the deletion and insertion mutations were identified in this region and were presumed to result in changes in the number and specificity of binding sites for cellular transcription factors (Fig. 3). The E_box is the bHLH transcription factor binding sequence. It was originally absent in the two in vivo virulent strains, but it appeared in the EHR in all of the cell culture adapted virus strains. The E_box sequence in the LTR of HIV-1 and human T cell leukemia virus-1 (HTLV-1) was found to significantly decrease or delay the transcription of the provirus [8]. Additionally, sequences with AP-1 binding sites in the NRE region began to appear and gradually increased concomitantly with the successive passages in cultured cells. The clones of the most attenuated strain (EIAVFDDV13) contained two to three AP-1 binding sites. The transcription factor AP-1 is known to regulate cell differentiation, proliferation and apoptosis in multiple cell types. Studies of FIV revealed that the AP-1 binding sites in the U3 region improved viral replication in feline kidney cell lines but decreased viral propagation in feline T lymphocytes and PBMCs [9]. In this study, the replication ability in cultured cells of the EIAV strains was positively correlated with the AP-1 copy number and the percentage of clones with high-copy AP-1 but was negatively correlated with viral pathogenicity. The fibroblast (FDD cells)-adapted EIAVFDDV13 lost the GATA binding site, which was present in the EHR region in the other strains. The transcription factor GATA regulates hematopoietic differentiation and is necessary for the development, differentiation and maturation of erythrocytes [10].
A predominant mutation in coding sequences is defined as that more than 10 % of clones in different viral strains exhibit mutations causing the change of same amino acid or more than 2/3 of clones in the same viral strain display the identical mutation sites. The EIAV Gag protein is a precursor that is subsequently hydrolyzed into the p15 matrix, p26 capsid, p11 nucleocapsid and p9 proteins. Four out of 14 predominant mutations were specifically generated during the period of ex vivo attenuation (Additional file 2: Figure S2A). Among these four sites, 100A/T(S), 103T/S and 447N/D were mostly observed in the cell culture adapted viruses; the first two were located in the CTL epitope region in the E2 domain of p15 and the third was located in p9. In contrast, the 484D/N mutation was only identified in p9 in the three fully attenuated virus strains. The p9 protein is important for provirus formation and viral budding [11].
The cleavage of the EIAV Pol precursor protein generates several viral enzymes that are essential for the viral life cycle, including the reverse transcriptase (RT)/RNaseH, a viral protease, a dUTPase and an integrase. During the EIAV vaccine development process, 24 predominant mutation sites were observed in Pol (Additional file 2: Figure S2B). Among these sites, three were primarily identified in the viral strains adapted in cultured cells (16 K/E, 27E/G and 598 K/R), with the first two located in the leader sequences. Additionally, another three mutations were primarily confined to the three attenuated strains (64G/E, 619 N/D and 820I/M), with two located in the RT/RNaseH region and one in the dUTPase region. The dUTPase plays important roles in influencing pathogenicity [12]. Indeed, Pol is closely associated with viral replication and the induction of immune responses. The outcomes of these viral strain-associated mutations in EIAV Pol are less understood than the outcomes of mutations in other proteins such as Env and S2 [7, 13–15], and therefore they require further investigation.
The env gene was the structural gene with the highest mutation rate during vaccine development. Most of these mutations were located in the envelope surface unit gp90, which contained 43 predominant mutation sites (Fig. 4a). Impressively, nine of these mutations were confined to the cell culture adapted viral strains. Five were primarily observed in the attenuated strains (180E/K in the V3 region and 236D/-, 237N/K, 246N/K and 247E/K in the V4 region), including four that resulted in a change of an acidic amino acid to a basic amino acid. The deletion of negatively charged 236D in the V4 region may lead to a polarity change of the V4 region. Liang et al. previously demonstrated that the reverse mutation of these nine substituted residues in gp90 in the vaccine strain EIAVFDDV13 did significantly alter the pathogenicity of EIAV [16]. Heavy glycosylation is a common feature of lentiviral envelope proteins, and the locations and numbers of glycosylation sites are associated with viral biological characteristics [17]. We found that the EIAV strains exhibited a decrease in gp90 glycosylation sites with the increasing passages in cultured cells. The average number of glycosylation sites in the virulent strains was 19–20 compared to an average of 18 in the initially attenuated strain EIAVDLV92 and 17 for the vaccine strains EIAVDLV121 and EIAVFDDV13 (Fig. 4b).
The 237N/K and 246N/K substitutions in the gp90 of the attenuated strains resulted in the loss of two potential glycosylation sites (237NNTW240 and 246NETW249) in the V4 region (Fig. 4b). Additionally, all cell culture adapted viral strains lost the glycosylation site 191NSSN194 in the V3 region because of the 193S/N substitution (Fig. 4b). Han et al. reported that these substitutions reduced viral replication and sensitivity to neutralizing antibodies in cultured cells [18]. Howe et al. demonstrated that the structure of the V4 region was important for EIAV evasion of immune surveillance, and the glycosylation sites in the V4 region blocked the principle neutralizing domain (PND) in the V3 region [19]. These structural features improved the resistance to host immune responses. The EIAV V3/V4 regions and the HIV-1 V1/V2 regions are topologically similar [20]. Recently, an analysis of the HIV-1 vaccine that was assessed in the Thailand RV-144 trial suggested that antibodies targeting the V1/V2 regions of gp120, which together form a five-strand beta barrel, were correlated with immune protection [21]. Therefore, the loss of glycosylation sites in the V4 region in attenuated EIAV strains may cause viruses to expose more epitopes for immune recognition (particularly the PND in the V3 region), leading to stronger stimulation of immune responses.
Our sequencing data displayed that the diversity of gp90 a.a was the highest among other EIAV structural proteins, ranging from 1.85 ± 0.25 % for EIAVLN40 to 4.14 ± 0.50 % for EIAVDV117, which implicated a wide variation in the surface antigens in different viral clones of EIAV quasispecies. Together with constant antigen shifting, the complexity in EIAV antigen composition results in the difficulty in vaccine development. We previously reported that a proviral derivate from the vaccine strain EIAVFDDV12 failed to elicit immune protection like its parental strain. The reduction of gp90 variation was considered the major difference between these two types of vaccine [4].
The EIAV trans membrane protein gp45 displayed a total of 10 predominant mutations, among which 58V(I)/T was primarily detected in the vaccine strains (Additional file 2: Figure S2C). We previously demonstrated that this mutation decreased the temperature sensitivity of gp45 [22], which might affect viral infection. Furthermore, all seven analyzed EIAVFDDV13 genomes contained a G/A mutation at the 795th nucleotide that created a premature stop codon (793TGA795) in the gp45 gene, resulting in a truncated gp45. The viruses expressing truncated gp45 grew significantly better in FDD cells than in horse macrophage. However, there was no significant difference in replication in horses between the two EIAV strains with different gp45 configurations [23]. The C-terminal truncated mutant may be positively selected because of its benefits for viral replication in FDD cells.
EIAV encodes three accessory proteins (Tat, S2 and Rev). Corresponding functional domains in Tat and the LTR bind to the TAR region of the EIAV genomic RNA to increase gene transcription efficiency. Our analysis revealed that Tat contained four predominant mutations, among which 7R/H was primarily found in the cell culture adapted viral strains (Additional file 2: Figure S2D). S2, which was the EIAV-encoded protein with the highest amino acid mutation rate in the vaccine strains compared to the initial EIAVLN40, contained six predominant mutations. Of these, 6K/R was only observed in the highly virulent EIAVDV117 strain, whereas 41T/I, 51T/I and 55Q/K were mostly located in the cell culture adapted adapted strains (Additional file 2: Figure S2D). We previously demonstrated that the reverse mutation of these four substituted residues in the vaccine strain EIAVFDDV13 compared to the residues from EIAVDV117 did not significantly change the replication features of this vaccine strain in cultured cells [24]. Rev assists viral RNA transportation out of the nucleus and is an important factor influencing lentivirus pathogenicity. Eleven predominant mutations were generated in Rev during the vaccine development process (Additional file 2: Figure S2D). Compared to the initial strain EIAVLN40, most clones of other strains displayed a deletion of residue N at position 22. Two stable mutations were principally observed in EIAVDV117, and a third mutation (74V/I) was primarily detected in the vaccine strains.
In most cases, 33 of the aforementioned predominant mutations were stably maintained during the attenuating process in cultured cells, but were maintained in only a portion of the clones detected from related viral strains. The percentages generally increased as the passaging in cultured cells continued and were correlated with the decrease in pathogenicity (Fig. 3 and Table 2). This evidence suggested the shifting of predominant clones in the pool of EIAV quasispecies. To evaluate the relationship between these stable mutations and the attenuation of pathogenicity and the alternation of the growth environment (from in vivo to ex vivo), a statistical analysis was performed to examine the differences between virulent and attenuated strains and between in vivo-originated and cell culture adapted strains (Table 3). With the exception of 447N/D, all other stable mutations in Gag presented in Table 3 were significantly different between the virulent and attenuated strains. Furthermore, all stable mutation sites summarized in Table 3 were significantly different between the in vivo-originated strains and the cell culture adapted strains, with the exception of a few mutations including 484D/N in Gag, 16K/E, 598K/R and 820I/M in Pol and 58V(I)/T in gp45. These data suggest that most of these stable mutations are responsible for EIAV adaptation to cultured cells and are correlated with changes in pathogenicity.
Table 3.
Differences between various amino acid sequences or LTR sequences derived from virulent and avirulent EIAV and in vivo and cell culture derived EIAV
| Virulenta | Avirulentb | p | Vivoc | Vitrod | p e | ||
|---|---|---|---|---|---|---|---|
| Gag | 100A/T | 19 (31) | 25 (36) | <0.01 | 0 (8) | 45 (49) | <0.01 |
| 103T/S | 14 (31) | 24 (26) | <0.01 | 0 (8) | 41 (49) | <0.01 | |
| 447N/D | 12 (31) | 16 (26) | >0.05 | 0 (8) | 28 (49) | <0.01 | |
| 484D/N | 0 (31) | 17 (26) | <0.01 | 0 (8) | 19 (49) | >0.05 | |
| Pol | 16K/E | 16 (28) | 26 (26) | <0.01 | 0 (8) | 19 (47) | >0.05 |
| 27E/G | 10 (28) | 17 (26) | 0.05 < p < 0.01 | 0 (7) | 42 (47) | <0.01 | |
| 64G/E | 0 (28) | 20 (26) | <0.01 | 0 (7) | 27 (47) | 0.05 < p < 0.01 | |
| 598K/R | 6 (28) | 26 (26) | <0.01 | 0 (7) | 20 (47) | >0.05 | |
| 619N/D | 1 (28) | 15 (26) | <0.01 | 0 (7) | 23 (47) | 0.05 < p < 0.01 | |
| 820I/M | 0 (28) | 17 (26) | <0.01 | 0 (7) | 16 (47) | >0.05 | |
| gp90 | 46A/E | 28 (65) | 48 (48) | <0.01 | 0 (35) | 76 (78) | <0.01 |
| 98G/R | 29 (65) | 45 (48) | <0.01 | 0 (35) | 75 (78) | <0.01 | |
| 100K/(H)Q | 46 (65) | 48 (48) | <0.01 | 16 (35) | 78 (78) | <0.01 | |
| 103H/Y | 30 (65) | 45 (48) | <0.01 | 0 (35) | 75 (78) | <0.01 | |
| 180E/K | 1 (65) | 22 (48) | <0.01 | 1 (35) | 22 (78) | <0.01 | |
| 189K/E | 27 (65) | 46 (48) | <0.01 | 0 (35) | 73 (78) | <0.01 | |
| 190E/K | 27 (65) | 48 (48) | <0.01 | 0 (35) | 74 (78) | <0.01 | |
| 193S/N | 31 (65) | 48 (48) | <0.01 | 1 (35) | 78 (78) | <0.01 | |
| 236D/-(N) | 9 (65) | 45 (48) | <0.01 | 0 (35) | 54 (78) | <0.01 | |
| 237N/K | 0 (65) | 36 (48) | <0.01 | 0 (35) | 67 (78) | <0.01 | |
| 246N/K | 5 (65) | 29 (48) | <0.01 | 0 (35) | 33 (78) | <0.01 | |
| 247E/K | 4 (65) | 24 (48) | <0.01 | 0 (35) | 28 (78) | <0.01 | |
| 321K/E(N) | 31 (65) | 48 (48) | <0.01 | 1 (35) | 78 (78) | <0.01 | |
| 410H/Q | 19 (65) | 37 (48) | <0.01 | 0 (35) | 56 (78) | <0.01 | |
| gp45 | 58V(I)/T | 5 (32) | 15 (23) | <0.01 | 0 (10) | 26 (45) | >0.05 |
| Tat | 7R/H | 14 (34) | 29 (29) | <0.01 | 0 (10) | 43 (53) | <0.01 |
| S2 | 41T/I | 16 (34) | 28 (30) | <0.01 | 0 (10) | 44 (54) | <0.01 |
| 51T/I | 15 (34) | 28 (30) | <0.01 | 0 (10) | 41 (54) | <0.01 | |
| 55Q/K | 18 (34) | 30 (30) | <0.01 | 0 (10) | 46 (54) | <0.01 | |
| Rev | 74V/I | 2 (35) | 19 (27) | <0.01 | 0 (10) | 21 (52) | <0.01 |
| LTR | AP-1 | 4 (77) | 57 (65) | <0.01 | 0 (38) | 61 (104) | <0.01 |
| MDBP | 14 (77) | 33 (65) | <0.01 | 1 (38) | 46 (104) | <0.01 | |
| E_box | 30 (77) | 56 (65) | <0.01 | 0 (38) | 86 (104) | <0.01 | |
aVirulent strains including EIAVLN40, EIAVDV117, EIAVDLV34, and EIAVDLV62
bAvirulent strains including EIAV DLV92, EIAVDLV121, and EIAVFDDV13
cIn vivo strains including EIAVLN40 and EIAVDV117
dEx vivo strains including EIAVDLV34, EIAVDLV62, EIAV DLV92, EIAVDLV121, and EIAVFDDV13
eDetermined by the Chi square test of the SAS 9.2
Conclusions
The vaccine development process of the attenuated EIAV vaccine is based on successive passages in different hosts and cultivated permissive cells to reduce pathogenicity while retaining immunogenicity. Viral strains collected at key stages of this process provided an important source of genomic information for studies on the molecular basis of the altered virological and immunological features. Our data demonstrated that different regions of the viral genome that are associated with decreased pathogenicity exhibited a series of stable mutations and that the percentage of mutated sites in the viral population gradually increased. These results indicated that these mutations were closely involved in pathogenicity attenuation and immunogenicity enhancement. The evolutionary analysis revealed that although the direction of evolution of the whole genome was consistent with the extent of attenuation, not all of the individual genes followed the same evolutionary pattern. This finding implicates the different contributions of each gene to the adaption of EIAV to changes in the growth environment and attenuation. Furthermore, our results suggest that there is a cumulative effect of the mutations in each individual gene on phenotypic alterations, including pathogenicity and immunogenicity. The stable mutation data presented in this article provide genetic information for future studies to determine the factors involved in viral infectivity.
Methods
Viruses and cells
EIAV strains were stocked in the Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences. EIAVLN40 and EIAVDV117 were originally obtained from peripheral blood mononuclear cells (PBMCs) collected during febrile episodes from horses or donkeys infected with EIAVLN40 or EIAVDV117, respectively. The strains were stored at −80 °C prior to analysis. EIAVDLV34, EIAVDLV62, EIAVDLV92, EIAVDLV121 and EIAVFDDV13 were obtained as lyophilized cell culture supernatants and reconstituted in sterile cattle serum. EIAVDLV34, EIAVDLV62, EIAVDLV92 and EIAVDLV121 were rejuvenated in dMDM, and the cells were harvested when they showed cytopathic effects. EIAVFDDV13 was rejuvenated in FDD, and the cells were harvested 10 days after inoculation. Dynamic replication curves were performed for several passages of EIAV strains, including EIAVDLV34, EIAVDLV62, EIAVDLV92, EIAVDLV121 and EIAVFDDV13. The same copy number of EIAVDLV34, EIAVDLV62, EIAVDLV92, EIAVDLV121 and EIAVFDDV13 was used to infect cultured dMDM, which was determined by quantitative real-time revers transcription (RT)-PCR of the gag gene fragment [4, 6]. The supernatants were collected from the infected cells at 2, 4, 6, 8 and 10 days post-infection to measure the viral quantity by assessing the reverse transcriptase (RT) activity (Additional file 2: Figure S1) [21].
Amplification and sequencing
The proviral DNA of EIAVLN40 and EIAVDV117 were prepared from peripheral blood mononuclear cells (PBMC) taken during febrile episodes of horses or donkeys infected by EIAVLN40 or EIAVDV117, respectively. Proviral DNA of EIAVDLV34, EIAVDLV62, EIAVDLV92 and EIAVDLV121 were obtained from cultured dMDM, EIAVFDDV13 proviral DNA was from FDD cells, infected by respective EIAV strains. Total cellular DNA was extracted using the OMEGA Blood DNA Kit (OMEGA, Shanghai, China) according to the manufacturer’s protocols. Proviral genomes of EIAVLN40, EIAVDV117, EIAVDLV121 and EIAVFDDV13 were cloned and sequenced from two amplified fragments using long and accurate PCR (LA-PCR) [25, 26]. The near entire proviral genome (7.9 kb of 8.2 kb) of EIAVDLV34, EIAVDLV62 and EIAVDLV92 proviral genomes were amplified by LA-PCR. To analyze the evolutionary characteristics of the long terminal repeat (LTR) region and the gp90 gene in detail, specific gene fragments were also directly amplified from proviral DNA of individual virus strains and cloned individually in addition to the whole genomes. After T-A cloning of PCR products, positive clones were sent for sequencing to obtain 5–16 whole-genome sequences, 19–23 LTR sequences and 16–21 gp90 sequences from different viral strains. All clones were obtained from indicated viral genomic fragments amplified by three to five independent PCR reactions.
Sequence and phylogenetic analysis
Nucleotide sequences were edited and assembled using the SeqManII tool of the Lasergene DNAStar program 7.1 (DNAStar) (Madison, WI, USA, 2006). The Megalign tool (DNAStar) was used for pairwise and multiple alignments of DNA sequences and for deducing amino acid sequences. Shannon entropy was calculated using BioEdit software [27]. Diversity and distance were calculated using the Molecular Evolutionary Genetics Analysis (MEGA) program 5 and a pairwise deletion option. Phylogenetic and molecular evolutionary analyses were conducted using MEGA5 (Center for Evolutionary Functional Genomics Biodesign Institute, Arizona State University, Tempe, AZ, USA, 2011) by the neighbor joining method [28]. Bootstrap values (based on 1000 replicates) for each node were provided if they were at least 70 %. N-glycosylation sites were predicted using N-GlycoSite (http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html). The stable mutation rates (marginal frequencies at each position of the amino acid sequence) were calculated using the MargFreq software (http://sray.med.som.jhmi.edu/SCRoftware/MargFreq/). The statistical analysis of the sequence variations was performed using SAS (Statistical Analysis System) (version 9.2, SAS Institute Inc, SAS Campus Drive, Cary, NC, USA, 2008).
Nucleotide sequence accession numbers
Sequences generated in this study have been submitted to GenBank (accession numbers: AF327878, AF327878, HM141919, GU385362–GU385365, GU385353–GU385362, HM141909–HM141912, HM1419113–HM141918, HM1419120–HM1419123, KT806081–KT806112).
Authors’ contributions
JHZ, XFW and YZL designed the experiments and wrote the paper, JHZ and XW conceived the project, XFW, YZL, QLi, QLiu, WWZ, CD and JC performed the experiments and analyzed the data. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (31302066 to X-F W, 31572537 to C D), the National Key Laboratory of Veterinary Biotechnology (SKLVBP201312 and SKLVBP201427) to X-F W, the Postdoctoral Foundation of Heilongjiang province, China (LBH-Z14033), and the Chinese Postdoctoral Science Foundation Project (2015M571386).
Competing interests
The authors declare that they have no competing interests.
Additional files
10.1186/s12977-016-0240-6 Protective efficacy of EIAVDLV121 and EIAVFDDV13.
10.1186/s12977-016-0240-6 Replication efficacies of EIAV strains. The growth rates of five EIAV strains (EIAVDLV34, EIAVDLV62, EIAVDLV92, EIAVDLV121 and EIAVFDDV13) in cultivated dMDM for 2, 4, 6, 8 and 10 days were compared by measuring reverse transcriptase (RT) activity. Data were collected from three independent experiments. Figure S2. Stable mutations in Gag (A), Pol (B), gp45 (C), Tat, S2 and Rev (D) generated during different vaccine development stages. The amino acid sequences were deduced from the gene sequences originating from either the proviral genomes or from the directly cloned PCR products after removing sequences containing premature stop codons. The sequences of each gene were aligned to the reference sequence EIAVLN40. The shadowed residues and white background residues are identical to or different from the reference sequence, respectively. Stable mutation sites detected primarily in virus strains adapted to cultivated cells are boxed, whereas those limited in the attenuated strains are marked with red circles. The numbers on the top of the graphs show the positions of stable mutation sites, and those at the left side indicate the sequences applied for the analysis. The downward arrows indicate the direction of the vaccine development process.
Footnotes
Xue-Feng Wang and Yue-Zhi Lin contributed equally to this work
Contributor Information
Xue-Feng Wang, Email: xuefengwang1982@126.com.
Yue-Zhi Lin, Email: sndhr@163.com.
Qiang Li, Email: liqiang007200@sina.com.
Qiang Liu, Email: liuqiang_yyy@163.com.
Wei-Wei Zhao, Email: vivilnic@163.com.
Cheng Du, Email: chengdu1981@163.com.
Jie Chen, Email: 18047113976@163.com.
Xiaojun Wang, Email: xjw@hvri.ac.cn.
Jian-Hua Zhou, Phone: +86-13796085512, Email: jianhua_uc@126.com.
References
- 1.Koff WC, Johnson PR, Watkins DI, Burton DR, Lifson JD, Hasenkrug KJ, McDermott AB, Schultz A, Zamb TJ, Boyle R, Desrosiers RC. HIV vaccine design: insights from live attenuated SIV vaccines. Nat Immunol. 2006;7:19–23. doi: 10.1038/ni1296. [DOI] [PubMed] [Google Scholar]
- 2.Craigo JK, Ezzelarab C, Cook SJ, Liu C, Horohov D, Issel CJ, Montelaro RC. Protective efficacy of centralized and polyvalent envelope immunogens in an attenuated equine lentivirus vaccine. PLoS Pathog. 2015;11:e1004610. doi: 10.1371/journal.ppat.1004610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craigo JK, Montelaro RC. Lessons in AIDS vaccine development learned from studies of equine infectious, anemia virus infection and immunity. Viruses. 2013;5:2963–2976. doi: 10.3390/v5122963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma JA, Shi N, Jiang CG, Lin YZ, Wang XF, Wang SA, Lv XL, Zhao LP, Shao YM, Kong XG, et al. A proviral derivative from a reference attenuated EIAV vaccine strain failed to elicit protective immunity. Virology. 2011;410:96–106. doi: 10.1016/j.virol.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 5.Lin YZ, Shen RX, Zhu ZY, Deng XL, Cao XZ, Wang XF, Ma J, Jiang CG, Zhao LP, Lv XL, et al. An attenuated EIAV vaccine strain induces significantly different immune responses from its pathogenic parental strain although with similar in vivo replication pattern. Antiviral Res. 2011;92:292–304. doi: 10.1016/j.antiviral.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Shen T, Liang H, Tong X, Fan X, He X, Ma Y, Xiang W, Shen R, Zhang X, Shao Y. Amino acid mutations of the infectious clone from Chinese EIAV attenuated vaccine resulted in reversion of virulence. Vaccine. 2006;24:738–749. doi: 10.1016/j.vaccine.2005.08.084. [DOI] [PubMed] [Google Scholar]
- 7.Ball JM, Swaggerty CL, Pei X, Lim WS, Xu X, Cox VC, Payne SL. SU proteins from virulent and avirulent EIAV demonstrate distinct biological properties. Virology. 2005;333:132–144. doi: 10.1016/j.virol.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 8.Terme JM, Calvignac S, Duc Dodon M, Gazzolo L, Jordan A. E box motifs as mediators of proviral latency of human retroviruses. Retrovirology. 2009;6:81. doi: 10.1186/1742-4690-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyazawa T, Tomonaga K, Kawaguchi Y, Kohmoto M, Inoshima Y, Maeda K, Mikami T, Maedadel K. Effects of insertion of multiple AP-1 binding sites into the U3 region of the long terminal repeat of feline immunodeficiency virus. Arch Virol. 1994;139:37–48. doi: 10.1007/BF01309453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pimkin M, Kossenkov AV, Mishra T, Morrissey CS, Wu W, Keller CA, Blobel GA, Lee D, Beer MA, Hardison RC, Weiss MJ. Divergent functions of hematopoietic transcription factors in lineage priming and differentiation during erythro-megakaryopoiesis. Genome Res. 2014;24:1932–1944. doi: 10.1101/gr.164178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin S, Chen C, Montelaro RC. Equine infectious anemia virus Gag p9 function in early steps of virus infection and provirus production. J Virol. 2005;79:8793–8801. doi: 10.1128/JVI.79.14.8793-8801.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Payne SL, Elder JH. The role of retroviral dUTPases in replication and virulence. Curr Protein Pept Sci. 2001;2:381–388. doi: 10.2174/1389203013381008. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Wu L, Li H, Hu X, Sun J, Ma L, Wan Y, Shao Y. Broader HIV-1 neutralizing antibody responses induced by envelope glycoprotein mutants based on the EIAV attenuated vaccine. Retrovirology. 2010;7:71. doi: 10.1186/1742-4690-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tagmyer TL, Craigo JK, Cook SJ, Even DL, Issel CJ, Montelaro RC. Envelope determinants of equine infectious anemia virus vaccine protection and the effects of sequence variation on immune recognition. J Virol. 2008;82:4052–4063. doi: 10.1128/JVI.02028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F, Craigo JK, Howe L, Steckbeck JD, Cook S, Issel C, Montelaro RC. A live attenuated equine infectious anemia virus proviral vaccine with a modified S2 gene provides protection from detectable infection by intravenous virulent virus challenge of experimentally inoculated horses. J Virol. 2003;77:7244–7253. doi: 10.1128/JVI.77.13.7244-7253.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang H, He X, Shen RX, Shen T, Tong X, Ma Y, Xiang WH, Zhang XY, Shao YM. Combined amino acid mutations occurring in the envelope closely correlate with pathogenicity of EIAV. Arch Virol. 2006;151:1387–1403. doi: 10.1007/s00705-005-0718-3. [DOI] [PubMed] [Google Scholar]
- 17.Vigerust DJ, Shepherd VL. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 2007;15:211–218. doi: 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han X, Zhang P, Wang X, Kong X, Xiang W, Zhou J. Construction of an Infectious Clone within the V3 Region of gp90 Surface Protein of EIAV by N- glycosylation Revers-mutations. Acta Veterinaria et Zootechnica Sinica. 2008;39:1731–1736. [Google Scholar]
- 19.Howe L, Leroux C, Issel CJ, Montelaro RC. Equine infectious anemia virus envelope evolution in vivo during persistent infection progressively increases resistance to in vitro serum antibody neutralization as a dominant phenotype. J Virol. 2002;76:10588–10597. doi: 10.1128/JVI.76.21.10588-10597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Zhang X, Fan X, Shen T, Tong X, Shen R, Shao Y. A conservative domain shared by HIV gp120 and EIAV gp90: implications for HIV vaccine design. AIDS Res Hum Retroviruses. 2005;21:1057–1059. doi: 10.1089/aid.2005.21.1057. [DOI] [PubMed] [Google Scholar]
- 21.Edlefsen PT, Rolland M, Hertz T, Tovanabutra S, Gartland AJ, deCamp AC, Magaret CA, Ahmed H, Gottardo R, Juraska M, et al. Comprehensive sieve analysis of breakthrough HIV-1 sequences in the RV144 vaccine efficacy trial. PLoS Comput Biol. 2015;11:e1003973. doi: 10.1371/journal.pcbi.1003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du J, Wang X, Ma J, Wang J, Qin Y, Zhu C, Liu F, Shao Y, Zhou J, Qiao W, Liu X. Structural and biochemical insights into the V/I505T mutation found in the EIAV gp45 vaccine strain. Retrovirology. 2014;11:26. doi: 10.1186/1742-4690-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang CG, Gao X, Ma J, Lin YZ, Wang XF, Zhao LP, Hua YP, Liu D, Zhou JH. C-terminal truncation of the transmembrane protein of an attenuated lentiviral vaccine alters its in vitro but not in vivo replication and weakens its potential pathogenicity. Virus Res. 2011;158:235–245. doi: 10.1016/j.virusres.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Jiang CG, Wang XF, Lin YZ, Ma J, Han XE, Zhao LP, Shen RX, Xiang WH, Zhou JH. Reverse mutation of the virulence-associated S2 gene does not cause an attenuated equine infectious anemia virus strain to revert to pathogenicity. Virology. 2013;443:321–328. doi: 10.1016/j.virol.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Wang S, Lin Y, Jiang C, Ma J, Zhao L, Lv X, Wang F, Shen R, Kong X, Zhou J. Genomic comparison between attenuated Chinese equine infectious anemia virus vaccine strains and their parental virulent strains. Arch Virol. 2011;156:353–357. doi: 10.1007/s00705-010-0877-8. [DOI] [PubMed] [Google Scholar]
- 26.Qi X, Wang X, Wang S, Lin Y, Jiang C, Ma J, Zhao L, Lv X, Shen R, Wang F, et al. Genomic analysis of an effective lentiviral vaccine-attenuated equine infectious anemia virus vaccine EIAV FDDV13. Virus Genes. 2010;41:86–98. doi: 10.1007/s11262-010-0491-6. [DOI] [PubMed] [Google Scholar]
- 27.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 28.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]