

Abstract
Purpose of review
Early and accurate diagnosis of transthyretin familial amyloid polyneuropathy (TTR-FAP) represents one of the major challenges faced by physicians when caring for patients with idiopathic progressive neuropathy. There is little consensus in diagnostic and management approaches across Europe.
Recent findings
The low prevalence of TTR-FAP across Europe and the high variation in both genotype and phenotypic expression of the disease means that recognizing symptoms can be difficult outside of a specialized diagnostic environment. The resulting delay in diagnosis and the possibility of misdiagnosis can misguide clinical decision-making and negatively impact subsequent treatment approaches and outcomes.
Summary
This review summarizes the findings from two meetings of the European Network for TTR-FAP (ATTReuNET). This is an emerging group comprising representatives from 10 European countries with expertise in the diagnosis and management of TTR-FAP, including nine National Reference Centres. The current review presents management strategies and a consensus on the gold standard for diagnosis of TTR-FAP as well as a structured approach to ongoing multidisciplinary care for the patient. Greater communication, not just between members of an individual patient's treatment team, but also between regional and national centres of expertise, is the key to the effective management of TTR-FAP.
Keywords: algorithm, diagnosis, Europe, management, TTR, FAP
INTRODUCTION
Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a highly debilitating and irreversible neurological disorder presenting symptoms of progressive sensorimotor and autonomic neuropathy [1▪,2▪,3]. TTR-FAP is caused by misfolding of the transthyretin (TTR) protein leading to protein aggregation and the formation of amyloid fibrils and, ultimately, to amyloidosis (commonly in the peripheral and autonomic nervous system and the heart) [4,5]. TTR-FAP usually proves fatal within 7–12 years from the onset of symptoms, most often due to cardiac dysfunction, infection, or cachexia [6,7▪▪].
The prevalence and disease presentation of TTR-FAP vary widely within Europe. In endemic regions (northern Portugal, Sweden, Cyprus, and Majorca), patients tend to present with a distinct genotype in large concentrations, predominantly a Val30Met substitution in the TTR gene [8–10]. In other areas of Europe, the genetic footprint of TTR-FAP is more varied, with less typical phenotypic expression [6,11]. For these sporadic or scattered cases, a lack of awareness among physicians of variable clinical features and limited access to diagnostic tools (i.e., pathological studies and genetic screening) can contribute to high rates of misdiagnosis and poorer patient outcomes [1▪,11]. In general, early and late-onset variants of TTR-FAP, found within endemic and nonendemic regions, present several additional diagnostic challenges [11,12,13▪,14].
Delay in the time to diagnosis is a major obstacle to the optimal management of TTR-FAP. With the exception of those with a clearly diagnosed familial history of FAP, patients still invariably wait several years between the emergence of first clinical signs and accurate diagnosis [6,11,14]. The timely initiation of appropriate treatment is particularly pertinent, given the rapidity and irreversibility with which TTR-FAP can progress if left unchecked, as well as the limited effectiveness of available treatments during the later stages of the disease [14]. This review aims to consolidate the existing literature and present an update of the best practices in the management of TTR-FAP in Europe. A summary of the methods used to achieve a TTR-FAP diagnosis is presented, as well as a review of available treatments and recommendations for treatment according to disease status.
Box 1.

no caption available
METHODOLOGY
This article is based on outcomes from two roundtable meetings of the European Network for TTR-FAP (ATTReuNET) (November 2012 and March 2014) and a comprehensive review of the published literature. The group comprised 14 TTR-FAP experts from 10 European countries (Bulgaria, Cyprus, France, Germany, Italy, the Netherlands, Portugal, Spain, Sweden, and Turkey; Tables S1 and S2), and includes nine National Reference Centres (NRCs). The experts completed a semistructured questionnaire on the local practice of TTR-FAP disease management in preparation for both meetings (Table S3). Group members are clinicians from a variety of specialties, including neurology, internal medicine, cardiology, and nephrology.
Electronic database searches (NCBI PubMed) formed the basis of the literature search within the time frame (1952 to December 2014). Key search terms included ‘transthyretin familial amyloid polyneuropathy’, ‘familial amyloid polyneuropathy’, ‘transthyretin amyloidosis’, ‘TTR-FAP’, ‘TTR-FAP and Europe’, and ‘TTR-FAP and Bulgaria/Cyprus/France/Germany/Italy/the Netherlands/Portugal/Spain/Sweden/Turkey’.
Misdiagnosis of transthyretin familial amyloid polyneuropathy
The diagnosis of TTR-FAP presents a significant challenge to the physician. Given its rarity within the general population, there is a reduced likelihood of the evaluating physician immediately recognizing the symptoms of TTR-FAP without first mistaking them for a more widely seen disorder. Common misdiagnoses made prior to correctly diagnosing TTR-FAP include idiopathic axonal polyneuropathy, chronic inflammatory demyelinating polyneuropathy, and lumbar spinal stenosis [1▪,15▪]. Diabetes or chronic alcoholism may induce polyneuropathies similar to TTR-FAP [10]. Further potential misdiagnoses include Charcot–Marie–Tooth neuropathy or motor neuron disease [1▪].
The commonly held misconception that TTR-FAP is a disease of young people with an established family history and a classic presentation means that patients exhibiting classic neurological symptoms without, or with an unknown, familial history may be overlooked or misdiagnosed [1▪]. It is often the case that patients presenting with inconsistent autonomic symptoms are later identified as new clinical phenotypes [11,12,13▪,14].
Diagnostic drivers
The diagnostic process is driven by two components. The first is clinical suspicion, which permits a tentative diagnosis of TTR-FAP through patient history and physical examination (symptoms and signs); the second is diagnostic confirmation using accurate diagnostic tools, including histopathology and genetic analysis (Table 1) [8,10,12,13▪,15▪–17▪]. Following a clinical suspicion, positive results from both biopsy and genetic analysis are essential to distinguish TTR-FAP from the large number of peripheral neuropathies [1▪,13▪,18], formally diagnose TTR-FAP, and specify the genetic variant [19].
Table 1.
Summary of diagnostic methods and respective aims for TTR amyloidosisa
| Investigation | Sensitivity | Specificity | Aim | References |
| Biopsy sites | ||||
| Sural nerve biopsy | 79–80% TTR | High | Detecting amyloid deposits | [12,13▀,15▀] |
| Labial salivary gland biopsy | 91% Val30Met early onset | High | Detecting amyloid deposits | [8] |
| Abdominal fat pad biopsy | 14–83% | High | Detecting amyloid deposits | [16▀] |
| Pathology test methods | [10] | |||
| Congo red staining | Medium–high | High | Detecting amyloid deposits | |
| Polarized microscopy examination | High | High | Green birefringence | |
| Immunohistochemistry with anti-TTR antibodies | High | Medium–high | Detecting TTR deposits | |
| Genetic tests | [10] | |||
| PCR-RFLP | High | High | Detecting predicted mutations in the TTR gene | |
| Real-time PCR (melting curve analysis) | High | High | Detecting predicted mutations in the TTR gene | |
| Sequencingb | High | High | Screening for unknown mutations in the TTR gene | |
| PCR-SSCP | Medium | Medium | Detecting predicted mutations in the TTR gene | |
| Mass spectrometry tests | [17▀] | |||
| LMD/MS | Approx. 100% | High | Determining specific type of amyloid deposits | |
LMD/MS, laser microdissection mass spectrometric-based proteomic analysis; PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism; SSCP, single-strand conformation polymorphism; TTR, transthyretin.
bSequencing is essential for diagnosis of TTR amyloidosis (sporadic cases).
Clinical diagnosis
Patients with TTR-FAP can present with a range of symptoms [11], and care should be taken to acquire a thorough clinical history of the patient as well as a family history of genetic disease. Delay in diagnosis is most pronounced in areas where TTR-FAP is not endemic or when there is no positive family history [1▪]. TTR-FAP and TTR-familial amyloid cardiomyopathy (TTR-FAC) are the two prototypic clinical disease manifestations of a broader disease spectrum caused by an underlying hereditary ATTR amyloidosis [19]. In TTR-FAP, the disease manifestation of neuropathy is most prominent and definitive for diagnosis, whereas cardiomyopathy often suggests TTR-FAC. However, this distinction is often superficial because cardiomyopathy, autonomic neuropathy, vitreous opacities, kidney disease, and meningeal involvement all may be present with varying severity for each patient with TTR-FAP.
Among early onset TTR-FAP with usually positive family history, symptoms of polyneuropathy present early in the disease process and usually predominate throughout the progression of the disease, making neurological testing an important diagnostic aid [14]. Careful clinical examination (e.g., electromyography with nerve conduction studies and sympathetic skin response, quantitative sensation test, quantitative autonomic test) can be used to detect, characterize, and scale the severity of neuropathic abnormalities involving small and large nerve fibres [10]. Although a patient cannot be diagnosed definitively with TTR-FAP on the basis of clinical presentation alone, symptoms suggesting the early signs of peripheral neuropathy, autonomic dysfunction, and cardiac conduction disorders or infiltrative cardiomyopathy are all indicators that further TTR-FAP diagnostic investigation is warranted. Late-onset TTR-FAP often presents as sporadic cases with distinct clinical features (e.g., milder autonomic dysfunction) and can be more difficult to diagnose than early-onset TTR-FAP (Table 2) [1▪,11,12,13▪,14,20].
Table 2.
ATTReuNET-recommended diagnosis of nonendemic (usually late-onset) TTR-FAP: Key points of note
| Typical clinical features of later disease (average 4 years post onset; the usual delay for diagnosis) |
| Progressive idiopathic polyneuropathy |
| Early walking difficulties, using aid support |
| Initial complaint: [20] |
| Sensory-motor neuropathic symptoms (80%) |
| Autonomic symptoms (10%) |
| Examination: All modality sensory deficit |
| Presence of family history (less than 50%) |
| Autonomic neuropathy without diabetes (uncommon at the onset) |
| Neurogenic orthostatic hypotension |
| Digestive symptoms (e.g., diarrhoea, constipation) |
| Urogenital symptoms (e.g., erectile dysfunction) |
| Unintentional major weight loss |
| Associated cardiac symptomatology (syncope, dyspnoea) |
| Diagnosis |
| DNA testing for TTR mutation (sequencing) first line in the future |
| Tissue biopsy confirms amyloid deposition |
ATTReuNET, European Network for TTR-FAP; TTR, transthyretin; TTR-FAP, transthyretin familial amyloid polyneuropathy.
Histopathology
The aim of histopathological analysis is to obtain direct evidence for amyloid deposits through biopsy on several possible tissue sites, including the labial salivary gland, abdominal subcutaneous adipose tissue, gastrointestinal tract, nerve tissue, and other organs with evidence of involvement (e.g., heart, kidney) (Table 1) [1▪,13▪,21▪▪,22–25]. Biopsy tissue is Congo red-stained in order to visualize the extracellular amyloid deposits by their characteristic apple-green birefringence after crosspolarized light examination [26,27]. Following biopsy, immunohistochemistry can be used to confirm that amyloid is formed by TTR.
Abdominal fat tissue biopsy and rectal biopsy are the most frequently performed according to the members of ATTReuNET, followed by sural nerve and labial salivary gland biopsy (Table 3). The selection of biopsy site varies widely between treatment centres and is largely dependent on the expertise of a particular team, whereas access to these facilities depends on the geographical area in which the patient resides. For example, biopsy sensitivity depends on the protocol for Congo red staining, the experience of the pathologist in reading the slides, and the variable sensitivity, as well as the specificity of the antibodies used for standard immunohistochemistry (Table 1) [1▪,10]. Therefore, the characterization of amyloid deposits should preferably be performed by experienced pathologists in order to minimize the risk of misdiagnosis [16▪,25,28,29]. Conducting repeat biopsy at different sites to confirm diagnosis is not uncommon (Table 3) (Adams, personal communication, 2014).
Table 3.
ATTReuNET-compiled type and frequency of TTR-FAP biopsies performed across Europe
| Country | ||||||||||
| Portugal | Cyprus | Sweden | Bulgaria | Germany | The Netherlands | Turkey | France | Spain | Italy | |
| Estimated number of TTR-FAP cases declared, n | 2000 | 50 | 250 | 87 | 120 | 80 | 20–30 | 500 | 500 | 200 |
| Site of biopsy, % performed in each centrea | ||||||||||
| Abdominal fat aspiration | 3 | – | 100 | – | 40–60 | 100 | 10–15 | – | 50 | 85 |
| Sural nerve | 2 | – | – | – | 15–20 | – | 75 | 40 | 15 | – |
| Rectal | – | 100 | – | 80 | 25–40 | 25 | – | – | 30 | – |
| Labial salivary gland | 95 | – | – | 6 | – | – | 10–15 | 100 | 5 | 5 |
| Gastric | – | – | 20–80b | – | – | – | – | 5 | – | – |
| Heart | – | – | 5 | 2 | – | 10 | – | < 5 | – | 10 |
| Others | – | – | 0–5c | 12 | – | – | – | – | – | – |
| Types of biopsies performed, n | 3 | 1 | 3 | 5 | 3 | 3 | 3 | 4 | 4 | 3 |
ATTReuNET, European Network for TTR-FAP; TTR-FAP, transthyretin familial amyloid polyneuropathy.
aWhere total numbers exceed 100%, more than one biopsy was performed.
bAs part of routine evaluation of gastric function.
cSkin, n = 6; renal, n = 6.
Genetic analysis
Genetic testing is carried out to allow detection of specific amyloidogenic TTR mutations (Table 1), using varied techniques depending on the expertise and facilities available in each country (Table S2). A targeted approach to detect a specific mutation can be used for cases belonging to families with previous diagnosis. In index cases of either endemic and nonendemic regions that do not have a family history of disease, are difficult to confirm, and have atypical symptoms, TTR gene sequencing is required for the detection of both predicted and new amyloidogenic mutations [26,27].
Postdiagnostic investigations
Following diagnosis, the neuropathy stage and systemic extension of the disease should be determined in order to guide the next course of treatment (Table 4) [3,30,31]. The three stages of TTR-FAP severity are graded according to a patient's walking disability and degree of assistance required [30]. Systemic assessment, especially of the heart, eyes, and kidney, is also essential to ensure all aspects of potential impact of the disease can be detected [10].
Table 4.
TTR-FAP stages of disease according to symptom severity
| Stage of disease | Symptoms | PND | Treatment suggestions | Comments |
| Stage 0 | Asymptomatic | Follow-up according to patient's age and mutation type | ||
| Stage I | Mild, ambulatory, symptoms at lower limbs limited | I. Sensory disturbances in extremities but preserved walking capacity | Confirm diagnosis | Best candidates for liver transplant are early onset Met30 (young with mild symptoms) |
| II. Difficulties in walking but without the need for a walking stick | First-line pharmacotherapy: tafamidis (EU approved) or diflunisal if not available | |||
| Liver transplant | ||||
| Follow up every 6 months for disease progression, especially cardiac | ||||
| Stage II | Moderate, further neuropathic deterioration, ambulatory but requires assistance | IIIa. One stick or one crutch required for walking | Diflunisal may slow progression of the disease | |
| IIIb. Two sticks or two crutches required for walking | ||||
| Stage III | Severe, bedridden/wheelchair-bound with generalized weakness | IV. Patient confined to a wheelchair or bed | No evidence for pharmacotherapy | |
| Treatment through clinical trial | ||||
| Recommended assessments | ||||
| Neuropathic: Modified Norris score, modified polyneuropathy disability (PND) score, neuropathy impairment score-weakness score (onset of orthostatic hypotension ++), scintigraphy with metaiodobenzylguanidine (mIBG) | ||||
| Electrophysiological: Quantitative sensation tests, quantitative autonomic tests, electromyographic test, sympathetic skin response | ||||
| Cardiac: ECG, Holter ECG, BNP/NT-proBNP, troponin, and echocardiography. When necessary: MRI, ‘bone’ DPD scintigraphy, intracardiac electrophysiological study | ||||
| Renal: Urinalysis | ||||
| General: Physical and clinical examination include weight (body mass index), blood test including s-albumin, quality of life | ||||
BNP, brain natriuretic peptide; DPD, 3,3-diphosphono-1,2-propanodicarboxylic acid; ECG, electrocardiography; NT-proBNP, N-terminal of the prohormone BNP; TTR-FAP, transthyretin familial amyloid polyneuropathy. Adapted from [3,30,31].
The goals of cardiac investigations are to detect serious conduction disorders with the risk of sudden death and infiltrative cardiomyopathy. Electrocardiograms (ECG), Holter-ECG, and intracardiac electrophysiology study are helpful to detect conduction disorders. Echocardiograms, cardiac magnetic resonance imaging, scintigraphy with bone tracers, and biomarkers (e.g., brain natriuretic peptide, troponin) can all help to diagnose infiltrative cardiomyopathy [10]. An early detection of cardiac abnormalities has obvious benefits to the patient, given that the prophylactic implantation of pacemakers was found to prevent 25% of major cardiac events in TTR-FAP patients followed up over an average of 4 years [32▪▪]. Assessment of cardiac denervation with 123-iodine meta-iodobenzylguanidine is a powerful prognostic marker in patients diagnosed with FAP [33].
Eyes
Ophthalmological assessment is warranted to identify any ocular manifestations of TTR-FAP presenting as keratoconjunctivitis sicca, secondary glaucoma, vitreous opacities, or pupillary abnormalities [34].
Kidney
It is also recommended that the monitoring of proteinuria and renal function form part of the TTR-FAP diagnostic approach. Microalbuminuria was detected in 75% of Portuguese patients with the Val30Met TTR mutation at different timepoints of the disease, with 21% eventually progressing to renal failure [35]. Following the onset of neuropathy, dialysis is generally required within 10 years [36].
Recommendations for the diagnosis of transthyretin familial amyloid polyneuropathy
The current members of ATTReuNET acknowledge that the existing diagnostic approach taken for TTR-FAP is suboptimal. Questionnaire data show that most patients tend to see between three and four physicians before they receive an accurate diagnosis, and that a gap of 2–3 years between the emergence of first symptoms and an accurate diagnosis is not uncommon [11].
The group calls for a pooling of expertise across Europe, specifically in relation to diagnosis, by standardizing techniques and methods in pathological laboratories and facilitating access to TTR gene testing by providing referral channels to laboratories with the capacity to confirm diagnosis of amyloid disease. A gold standard process for TTR-FAP diagnosis requires clinical, pathological and genetic evidence in networks under the banner of NRCs. These are the first steps towards providing greater urgency to, and improving the quality of, TTR-FAP diagnosis.
Accurate diagnosis of sporadic cases of TTR-FAP presents a significant challenge, and special attention should be given to patients presenting with progressive, length-dependent axonal polyneuropathy predominantly affecting temperature and pain sensation (Table 2). Particular attention should also be given to patients with autonomic dysfunction, involuntary major weight loss, carpal tunnel syndrome, and associated cardiac involvement [10,26].
Disease management strategies for transthyretin familial amyloid polyneuropathy
The management of TTR-FAP has expanded significantly in recent years; with the availability of pharmacotherapeutic alternatives, liver transplantation is no longer the only treatment option [26]. A comprehensive care package and a multidisciplinary approach are required to manage this multisystem disease. Targeted therapy is essential in the first instance to prevent further production of amyloid deposits. Thereafter, symptomatic therapy of sensorimotor and autonomic polyneuropathy and cardiac, renal, and ocular injury is required [6,10]. Finally, genetic counselling to patients and relatives is recommended [37].
Liver transplant
Prior to the pharmacotherapy era and as early as 1990, orthotopic liver transplant was the standard of care for patients with TTR-FAP [10,26,38]. A 20-year analysis of survival data from the Familial Amyloidotic Polyneuropathy World Transplant Registry of 2044 liver transplant patients reported a 20-year survival rate of 55.3% after treatment. Multivariate analysis revealed modified body mass index, onset of disease (<50 years of age), disease duration before liver transplant, and TTR mutation type (Val30Met vs non-Val30Met) as independent and significant factors for better survival outcomes (i.e., patients with an early onset, Val30Met mutation and shorter duration of the disease have improved prognosis) [39▪▪]. In addition, a large cohort study of 215 consecutive patients who were followed up for 18 years at the French NRC identified five pejorative factors for survival after liver transplant: polyneuropathy disability score score not less than III, orthostatic hypotension, New York Heart Association (NYHA) functional class more than I, QRS complex duration at least 120 ms and thickened interventricular septum [40]. Risks can be computed using the online calculator by the French Referral Center for FAP and Other Rare Peripheral Neuropathies (NNERF) [41]. However, liver transplant is not readily accessible to many patients. ATTReuNET members reported an average wait of up to 1 year in France, Spain, Italy, and Sweden, increasing to 2–3 years for patients in Germany, the Netherlands, Portugal, and Cyprus (liver transplant not performed in Turkey).
Whereas liver transplant removes the main source of mutated TTR[42–44], it does not prevent progression of cardiac disease because the wild-type TTR may continue to further expand existing amyloid deposits in the heart [45▪▪,46]. Therefore, continued scrutiny of the cardiac system is warranted, as some patients will develop atrioventricular blocks or infiltrative cardiomyopathy several years or decades later; a combined heart and liver transplant may be recommended in selected patients with non-Val30Met mutations and cardiomyopathy [47,48▪,49–51]. However, ocular and central nervous system involvements often progress and/or appear after liver transplant due to the local synthesis of mutated TTR in retinal epithelium and coroid plexus [52–54].
Tafamidis
Tafamidis is a first-in-class therapy that slows the progression of TTR amyloidogenesis by stabilizing the mutant TTR tetramer, thereby preventing its dissociation into monomers and amyloidogenic and toxic intermediates [55,56]. Tafamidis is currently indicated in Europe for the treatment of TTR amyloidosis in adult patients with stage I symptomatic polyneuropathy to delay peripheral neurological impairment [57].
In an 18-month, double-blind, placebo-controlled study of patients with early-onset Val30Met TTR-FAP, tafamidis was associated with a 52% lower reduction in neurological deterioration (P = 0.027), a preservation of nerve function, and TTR stabilization versus placebo [58▪▪]. However, only numerical differences were found for the coprimary endpoints of neuropathy impairment [neuropathy impairment score in the lower limb (NIS-LL) responder rates of 45.3% tafamidis vs 29.5% placebo; P = 0.068] and quality of life scores [58▪▪]. A 12-month, open-label extension study showed that the reduced rates of neurological deterioration associated with tafamidis were sustained over 30 months, with earlier initiation of tafamidis linking to better patient outcomes (P = 0.0435) [59▪]. The disease-slowing effects of tafamidis may be dependent on the early initiation of treatment. In an open-label study with Val30Met TTR-FAP patients with late-onset and advanced disease (NIS-LL score >10, mean age 56.4 years), NIS-LL and disability scores showed disease progression despite 12 months of treatment with tafamidis, marked by a worsening of neuropathy stage in 20% and the onset of orthostatic hypotension in 22% of patients at follow-up [60▪].
Tafamidis is not only effective in patients exhibiting the Val30Met mutation; it also has proven efficacy, in terms of TTR stabilization, in non-Val30Met patients over 12 months [61]. Although tafamidis has demonstrated safe use in patients with TTR-FAP, care should be exercised when prescribing to those with existing digestive problems (e.g., diarrhoea, faecal incontinence) [60▪].
Diflunisal
Diflunisal is a nonsteroidal anti-inflammatory drug (NSAID) that, similar to tafamidis, slows the rate of amyloidogenesis by preventing the dissociation, misfolding, and misassembly of the mutated TTR tetramer [62,63]. Off-label use has been reported for patients with stage I and II disease, although diflunisal is not currently licensed for the treatment of TTR-FAP.
Evidence for the clinical effectiveness of diflunisal in TTR-FAP derives from a placebo-controlled, double-blind, 24-month study in 130 patients with clinically detectable peripheral or autonomic neuropathy [64▪]. The deterioration in NIS scores was significantly more pronounced in patients receiving placebo compared with those taking diflunisal (P = 0.001), and physical quality of life measures showed significant improvement among diflunisal-treated patients (P = 0.001). Notable during this study was the high rate of attrition in the placebo group, with 50% more placebo-treated patients dropping out of this 2-year study as a result of disease progression, advanced stage of the disease, and varied mutations.
One retrospective analysis of off-label use of diflunisal in patients with TTR-FAP reported treatment discontinuation in 57% of patients because of adverse events that were largely gastrointestinal [65]. Conclusions on the safety of diflunisal in TTR-FAP will depend on further investigations on the impact of known cardiovascular and renal side-effects associated with the NSAID drug class [66,67].
Symptomatic management
The management of symptoms associated with sensory-motor neuropathy and autonomic dysfunction should be initiated immediately following diagnosis and should be tailored to the individual patient [10]. Symptomatic treatment may include painkillers, antidiarrhoeal drugs, treatment of symptomatic orthostatic hypotension, diuretics for patients with cardiac failure, prophylactic pacemaker implantation for severe cardiac conduction disorders [32▪▪], or vitrectomy/trabeculectomy for the treatment for ocular amyloidosis or glaucoma, respectively [10].
Emerging therapies
A number of new treatments for TTR-FAP are currently in phase II or III development. Posttranscriptional gene silencing is an approach that aims to inhibit the hepatic production of mutant and nonmutant TTR using small interfering RNAs [68▪▪] or antisense oligonucleotides [6]. Two phase III trials are ongoing, involving ALN-TTR02, an RNA interference (NCT01960348) [69], and ISIS 420915, an antisense oligonucleotide (NCT01737398) [70]. The removal of amyloid deposits has also been demonstrated in mouse models using a synergistic combination of doxycycline and tauroursodeoxycholic acid (TUDCA) [71], with ongoing clinical trials seeking to replicate these findings in patients (NCT01855360, NCT01171859) [72,73]. Preliminary data from the latter, a small phase II study, are promising, showing stabilization of neuropathy scores over 12 months of treatment with no clinical progression of cardiac involvement [74]. Immunotherapy produces a regulated immune response against the specific amyloid protein by enhancing the clearance of these deposits with monoclonal antibodies. Currently, a number of antibodies [e.g., monoclonal antibody NEOD001; the combination of a serum amyloid P (SAP) depleter (GSK2315698) and an anti-SAP antibody (GSK2398852)] are undergoing testing in patients with various forms of amyloidosis (NCT01707264, NCT01777243) [75,76].
A comprehensive care strategy
ATTReuNETrecommends a multidisciplinary approach to the management of TTR-FAP including not only the diagnosing physician, but also a neurologist and a cardiologist, and possibly an ophthalmologist, in the initial assessment and subsequent reviews. Treatment strategies should also extend beyond antiamyloid therapy (surgical or pharmacotherapeutic) to include symptomatic treatment, the management of complications (e.g., cardiac failure, end-stage renal disease), and genetic counselling (to be detailed in the next part of this supplement [37]). Figure 1 presents a comprehensive treatment algorithm for TTR-FAP, developed by the group. Physicians should note that existing treatments are most effective in patients with stage I TTR-FAP, with limited data available on the efficacy in more advanced stages, different genotypes, and late-onset variants.
FIGURE 1.

Strategy for specific therapy in TTR-FAP. CI, contraindications; LT, liver transplantation; TTR-FAP, transthyretin familial amyloid polyneuropathy. aCI for LT include: active and uncontrolled cancer; aged > 50 years for males and > 70 years for females [39▪▪,77], except for Italy (aged >65 years); modified body mass index below 800 kg/m2·g/L; some non-Val30Met TTR mutations; cardiac insufficiency. bStage I: walking unaided outside. cStage II: walking with aid. dProtocol clinical trial for antisense oligonucleotides, small interfering RNA, combination doxycycline–tauroursodeoxycholic acid; or diflunisal off-label. Adapted from [1▪].
Ongoing monitoring is crucial and permits systematic tracking of TTR-FAP disease progression. Figure 2 describes an algorithm for patient follow-up during active treatment. Clinical and biochemical assessment should take place no later than 3 months from treatment initiation, with a full, multidisciplinary consultation at 6 and 12 months. Patients should be assessed biannually or as required at their TTR-FAP clinic, with monitoring maintained throughout their lives. Ideally, the review should involve a neurologist, a cardiologist, and an internal medicine specialist (depending on local practice) at each visit. Regular follow-up with the multidisciplinary team can help to diminish a patient's anxiety and aid the acceptance of the diagnosis, as well as enable the detection of new manifestations of amyloid deposits and other disease symptoms. Patients with more advanced disease (stage II or III) should be seen on a quarterly basis, whereas those who respond well to treatment can be followed up less regularly.
FIGURE 2.
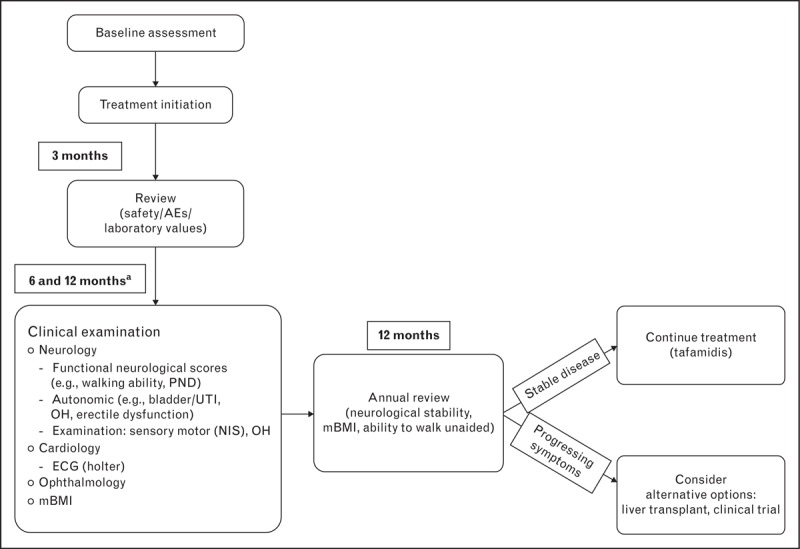
Algorithm for patient follow-up during treatment for TTR-FAP (compiled from clinical experience of ATTReuNET in March 2014). AE, adverse event; ECG, electrocardiogram; mBMI, modified body mass index; NIS, Neurological Impairment Scale; OH, orthostatic hypotension; PND, modified polyneuropathy disability score; TTR-FAP, transthyretin familial amyloid polyneuropathy; UTI, urinary tract infection. aQuarterly basis for those with more advanced (stage II, III) disease unless responding well to treatment.
CONCLUSION
TTR-FAP is a rare but life-threatening disease. Given the limited window for treatment effectiveness, an early and accurate diagnosis followed by appropriate therapeutic intervention is critical. The establishment of direct referral systems through NRCs across Europe allows the pooling of clinical resources and expertise to help reduce misdiagnosis and delay to treatment. Increased awareness of existing nonclassical TTR-FAP patient groups (e.g., sporadic cases) will help to direct and streamline the diagnostic process, enabling early implementation of specific strategies and better patient outcomes. The recent emergence of alternatives to liver transplantation has greatly enhanced the treatment options available to patients with TTR-FAP.
Acknowledgements
The authors would like to acknowledge Stayko Sarafov (Department of Neurology, Medical University – Sofia, Sofia, Bulgaria) for contributing towards the Bulgarian national data, Dr Antoine Rousseau and Dr Natalia Novais Ferreira on their contributions on ophthalmological assessments, and Hui Chin Teoh of PAREXEL for provision of writing assistance during the development of this article, with funding provided by Pfizer. Members of ATTReuNET who provided country-specific information for this article were David Adams [CHU Bicêtre (APHP), Service de Neurologie, Université Paris-Sud, Paris, France]; Juan Buades (Servicio de Medicina Interna, Hospital Son Llatzer, Palma de Mallorca, Spain); Josep M. Campistol (Instituto Clínico de Nefrología y Urología ICNU, Barcelona, Spain); Teresa Coelho (Hospital Santo António, Centro Hospitalar do Porto, Porto, Portugal); Lucía Galán (Servicio de Neurología, Hospital Clínico San Carlos, Madrid, Spain); Ivailo Tournev (Department of Neurology, Medical University – Sofia, and Department of Cognitive Science and Psychology, New Bulgarian University, Sofia, Bulgaria); Velina Guergueltcheva (University Hospital Sofiamed, Sofia, Bulgaria); Bouke P. Hazenberg (University Medical Center Groningen, University of Groningen, Groningen, the Netherlands); Ernst Hund (Universität Heidelberg, Heidelberg, Germany), Jan B. Kuks (Department of Neurology, University Medical Center Groningen, University of Groningen, Groningen, the Netherlands); Theodore Kyriakides (Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus); Laura Obici (Amyloidosis Research and Treatment Center, Fondazione IRCCS Policlinico S. Matteo, Pavia, Italy); Yesim Parman (Istanbul University, Istanbul, Turkey); Michel S. Slama (Hôpital Antoine Beclere, Université Paris-Sud, Clamart, France); and Ole B. Suhr (Department of Public Health and Clinical Medicine, Umeå University, Umeå, Sweden).
Financial support and sponsorship
This supplement was funded by Pfizer.
Conflicts of interest
Medical writing support was provided by PAREXEL and funded by Pfizer. The interpretation, discussion, and publication by the authors are independent of the funding organization, which sought no control over the content of the subsequent publications. Authors did not receive payment for any article within this supplement. D.A. received honoraria from Pfizer for organizing and participating in symposia and master classes, received funding from Alnylam for participating in scientific congresses, and is the principal investigator in clinical trials; sponsored by Alnylam and Isis Pharmaceuticals. O.B.S.'s department received honoraria from Pfizer for his contributions to educational activities and his services as clinical investigator in clinical trials, he is a member of the THAOS registry sponsored by Pfizer, and he participates as clinical investigator in clinical trials sponsored by Alnylam. E.H. received honoraria from Pfizer for organizing and participating in symposia. L.O. received financial support from Alnylam to attend scientific meetings, received honoraria from Pfizer for lectures, and is the principal investigator in a clinical trial sponsored by Alnylam. I.T. received honoraria from Pfizer for organizing and participating in symposia and for participation in scientific congresses, and is principal investigator in clinical trials sponsored by Alnylam and Isis Pharmaceuticals. M.S.S. received honoraria from Pfizer, Alnylam, and is the principal investigator in clinical trials sponsored by Alnylam and Pfizer. B.P.H. received honoraria from Pfizer, Alnylam, Isis Pharmaceuticals, GlaxoSmithKline, Millennium, Binding Site, Celgene, Proteotech, and Applied Spectral Imaging for organizing and participating in symposia. T.C. received financial support to attend scientific meetings from Pfizer, Alnylam, and Isis Pharmaceuticals, and received honoraria from Pfizer for work on a speaker's bureau. J.M.C. declares no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as follows:
▪ of special interest
▪▪ of outstanding interest
Supplementary Material
REFERENCES
- 1▪.Adams D, Lozeron P, Lacroix C. Amyloid neuropathies. Curr Opin Neurol 2012; 25:564–572. [DOI] [PubMed] [Google Scholar]; A recent and comprehensive review on the diagnosis and management of amyloid neuropathy.
- 2▪.Dohrn MF, Rocken C, De Bleecker JL, et al. Diagnostic hallmarks and pitfalls in late-onset progressive transthyretin-related amyloid-neuropathy. J Neurol 2013; 260:3093–3108. [DOI] [PubMed] [Google Scholar]; A recent article illustrating the late diagnosis of TTR FAP in Germany.
- 3.Coutinho P, da Silva AM, Lima JL. Glenner GG, e Costa PP, de Freitas AF, et al. Forty years of experience with type 1 amyloid neuropathy: review of 483 cases. Amyloid and Amyloidosis. Amsterdam: Excerpta Medica; 1980. 88–98. [Google Scholar]
- 4.Hou X, Aguilar MI, Small DH. Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J 2007; 274:1637–1650. [DOI] [PubMed] [Google Scholar]
- 5.Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007; 36:411–423. [DOI] [PubMed] [Google Scholar]
- 6.Adams D, Cauquil C, Theaudin M, et al. Current and future treatment of amyloid neuropathies. Expert Rev Neurother 2014; 14:1437–1451. [DOI] [PubMed] [Google Scholar]
- 7▪▪.Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from nonendemic areas. J Neurol Neurosurg Psychiatry 2012; 83:152–158. [DOI] [PubMed] [Google Scholar]; This paper described the major clinical landmarks and abnormalities of nerve conduction and cardiac-related indices that can be accurately assessed in patients with late-onset TTR-FAP in nonendemic areas.
- 8.Dardiotis E, Koutsou P, Papanicolaou EZ, et al. Epidemiological, clinical and genetic study of familial amyloidotic polyneuropathy in Cyprus. Amyloid 2009; 16:32–37. [DOI] [PubMed] [Google Scholar]
- 9.Ueda M, Ando Y. Recent advances in transthyretin amyloidosis therapy. Transl Neurodegener 2014; 3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013; 8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol 2015; (In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cappellari M, Cavallaro T, Ferrarini M, et al. Variable presentations of TTR-related familial amyloid polyneuropathy in seventeen patients. J Peripher Nerv Syst 2011; 16:119–129. [DOI] [PubMed] [Google Scholar]
- 13▪.Koike H, Hashimoto R, Tomita M, et al. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid 2011; 18:53–62. [DOI] [PubMed] [Google Scholar]; This paper focuses on the difficulties of diagnosing sporadic cases of TTR-FAP and the importance of using nerve biopsy to identify the amyloid deposit.
- 14.Plante-Bordeneuve V, Ferreira A, Lalu T, et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology 2007; 69:693–698. [DOI] [PubMed] [Google Scholar]
- 15▪.Adams D, Lozeron P, Theaudin M, et al. Regional difference and similarity of familial amyloidosis with polyneuropathy in France. Amyloid 2012; 19:61–64. [DOI] [PubMed] [Google Scholar]; This study examined the genotype and geographic distribution of various types of FAP patients in France.
- 16▪.van Gameren II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum 2006; 54:2015–2021. [DOI] [PubMed] [Google Scholar]; This paper recommends subcutaneous abdominal fat aspiration as the preferred method for detecting systemic amyloidosis, given its >80% accuracy.
- 17▪.Klein CJ, Vrana JA, Theis JD, et al. Mass spectrometric-based proteomic analysis of amyloid neuropathy type in nerve tissue. Arch Neurol 2011; 68:195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper focuses on mass spectrometric-based proteomic analysis of amyloid neuropathy of unknown origin, which is especially useful to detect and understand the properties of new biochemicals.
- 18.England JD, Asbury AK. Peripheral neuropathy. Lancet 2004; 363:2151–2161. [DOI] [PubMed] [Google Scholar]
- 19.Sipe JD, Benson MD, Buxbaum JN, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21:221–224. [DOI] [PubMed] [Google Scholar]
- 20.Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol 2002; 59:1771–1776. [DOI] [PubMed] [Google Scholar]
- 21▪▪.Do AB, Coelho T, Sousa A, Guimaraes A. Usefulness of labial salivary gland biopsy in familial amyloid polyneuropathy Portuguese type. Amyloid 2009; 16:232–238. [DOI] [PubMed] [Google Scholar]; This paper shows where a simple labial salivary gland biopsy was carried out for an early diagnosis of the disease in Portugal, and has a very high sensitivity to detect amyloid deposits in recently symptomatic TTR gene carriers.
- 22.Westermark P. Subcutaneous adipose tissue biopsy for amyloid protein studies. Methods Mol Biol 2012; 849:363–371. [DOI] [PubMed] [Google Scholar]
- 23.Kyle RA, Spencer RJ, Dahlin DC. Value of rectal biopsy in the diagnosis of primary systemic amyloidosis. Am J Med Sci 1966; 251:501–506. [DOI] [PubMed] [Google Scholar]
- 24.Rocken C, Sletten K. Amyloid in surgical pathology. Virchows Arch 2003; 443:3–16. [DOI] [PubMed] [Google Scholar]
- 25.Picken MM, Westermark P. Amyloid detection and typing: summary of current practice and recommendations of the consensus group. Amyloid 2011; 18:48–50. [DOI] [PubMed] [Google Scholar]
- 26.Adams D, Theaudin M, Cauquil C, et al. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep 2014; 14:435. [DOI] [PubMed] [Google Scholar]
- 27.Sekijima Y, Yoshida K, Tokuda T, Ikeda S. Familial transthyretin amyloidosis. Gene Reviews. http://www.ncbi.nlm.nih.gov/books/NBK1194/ [Accessed 2 June 2015] [Google Scholar]
- 28.Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002; 346:1786–1791. [DOI] [PubMed] [Google Scholar]
- 29.Schonland SO, Hegenbart U, Bochtler T, et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 2012; 119:488–493. [DOI] [PubMed] [Google Scholar]
- 30.Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord 2013; 6:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto S, Wilczek HE, Nowak G, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. Am J Transplant 2007; 7:2597–2604. [DOI] [PubMed] [Google Scholar]
- 32▪▪.Algalarrondo V, Dinanian S, Juin C, et al. Prophylactic pacemaker implantation in familial amyloid polyneuropathy. Heart Rhythm 2012; 9:1069–1075. [DOI] [PubMed] [Google Scholar]; The study investigated TTR-FAP patients with His-ventricular interval equivalent to 70 ms, or His-ventricular interval below 55 ms associated with a fascicular block, or a first-degree atrioventricular block, or a Wenckebach anterograde point equivalent to 100 bpm detected after an intracardiac electrophysiological investigation. Following prophylactic pacemaker implantation in 95 patients for a duration of over 45 ± 35 months, a high-degree atrioventricular block was documented in 25% of patients.
- 33.Coutinho MC, Cortez-Dias N, Cantinho G, et al. Reduced myocardial 123-iodine metaiodobenzylguanidine uptake: a prognostic marker in familial amyloid polyneuropathy. Circ Cardiovasc Imaging 2013; 6:627–636. [DOI] [PubMed] [Google Scholar]
- 34.Rousseau A, Kaswin G, Adams D, et al. Ocular involvement in familial amyloid polyneuropathy. J Fr Ophtalmol 2013; 36:779–788. [DOI] [PubMed] [Google Scholar]
- 35.Lobato L, Beirao I, Silva M, et al. Familial ATTR amyloidosis: microalbuminuria as a predictor of symptomatic disease and clinical nephropathy. Nephrol Dial Transplant 2003; 18:532–538. [DOI] [PubMed] [Google Scholar]
- 36.Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol 2012; 7:1337–1346. [DOI] [PubMed] [Google Scholar]
- 37.Obici L, Kuks JB, Buades J, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol 2015; (In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 1993; 341:1113–1116. [DOI] [PubMed] [Google Scholar]
- 39▪▪.Ericzon B-G, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation 2015; 99:1847–1854. [DOI] [PubMed] [Google Scholar]; This major study assessed the prognostic factors for long-term survival after liver transplantation (LT) in a cohort of 1940 patients, evaluating the 20-year rate of survival, and emphasized early-onset Val30Met with short duration of the disease as best candidates for LT.
- 40.Algalarrondo V, Antonini T, Théaudin M, et al. Prediction of long-term survival after liver transplantation for familial transthyretin amyloidosis. J Am Coll Cardiol 2015; (In press). [DOI] [PubMed] [Google Scholar]
- 41.French Referral Center for FAP and Other Rare Peripheral Neuropathies (NNERF). NNERF website [in French]. www.nnerf.fr/attrtransplantscore/ [Accessed 1 September 2015]
- 42.Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 1991; 40:242–246. [DOI] [PubMed] [Google Scholar]
- 43.Tsuchiya A, Yazaki M, Kametani F, et al. Marked regression of abdominal fat amyloid in patients with familial amyloid polyneuropathy during long-term follow-up after liver transplantation. Liver Transpl 2008; 14:563–570. [DOI] [PubMed] [Google Scholar]
- 44.Suhr OB. Impact of liver transplantation on familial amyloidotic polyneuropathy (FAP) patients’ symptoms and complications. Amyloid 2003; 10:77–83. [PubMed] [Google Scholar]
- 45▪▪.Yazaki M, Mitsuhashi S, Tokuda T, et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant 2007; 7:235–242. [DOI] [PubMed] [Google Scholar]; Postmortem study of two Val30Leu TTR-FAP patients who died after progression of the disease. Post–liver transplantation showed paradoxical wild-type TTR deposition preferentially in myocardium, which led to fatal cardiac dysfunction.
- 46.Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid 2007; 14:277–282. [DOI] [PubMed] [Google Scholar]
- 47.Gustafsson S, Ihse E, Henein MY, et al. Amyloid fibril composition as a predictor of development of cardiomyopathy after liver transplantation for hereditary transthyretin amyloidosis. Transplantation 2012; 93:1017–1023. [DOI] [PubMed] [Google Scholar]
- 48▪.Okamoto S, Zhao Y, Lindqvist P, et al. Development of cardiomyopathy after liver transplantation in Swedish hereditary transthyretin amyloidosis (ATTR) patients. Amyloid 2011; 18:200–205. [DOI] [PubMed] [Google Scholar]; This study highlighted that liver transplant does not slow the development of cardiomyopathy, and recommended combined heart and liver transplant in non-Val30Met patients.
- 49.Herlenius G, Wilczek HE, Larsson M, et al. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation 2004; 77:64–71. [DOI] [PubMed] [Google Scholar]
- 50.Arpesella G, Chiappini B, Marinelli G, et al. Combined heart and liver transplantation for familial amyloidotic polyneuropathy. J Thorac Cardiovasc Surg 2003; 125:1165–1166. [DOI] [PubMed] [Google Scholar]
- 51.Rapezzi C, Perugini E, Salvi F, et al. Phenotypic and genotypic heterogeneity in transthyretin-related cardiac amyloidosis: towards tailoring of therapeutic strategies? Amyloid 2006; 13:143–153. [DOI] [PubMed] [Google Scholar]
- 52.Hara R, Kawaji T, Ando E, et al. Impact of liver transplantation on transthyretin-related ocular amyloidosis in Japanese patients. Arch Ophthalmol 2010; 128:206–210. [DOI] [PubMed] [Google Scholar]
- 53.Sandgren O, Kjellgren D, Suhr OB. Ocular manifestations in liver transplant recipients with familial amyloid polyneuropathy. Acta Ophthalmol 2008; 86:520–524. [DOI] [PubMed] [Google Scholar]
- 54.Maia LF, Magalhaes R, Freitas J, et al. CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry 2015; 86:159–167. [DOI] [PubMed] [Google Scholar]
- 55.Johnson SM, Connelly S, Fearns C, et al. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol 2012; 421:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A 2012; 109:9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.EMA Committee for Medicinal Products for Human Use. Vyndaqel. European Medicines Agency. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/002294/WC500109204.pdf [Accessed 1 January 2015] [Google Scholar]
- 58▪▪.Coelho T, Maia LF, Martins da SA, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]; This phase III clinical trial assessed the effect of tafamidis in Val30Met TTR-FAP on NIS progression showing statistically significant efficacy, demonstrating a delay in peripheral neurologic impairment with tafamidis.
- 59▪.Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol 2013; 260:2802–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper confirmed the positive effect of tafamidis in slowing progression of the disease in the 12-month extension phase of the previous study.
- 60▪.Lozeron P, Theaudin M, Mincheva Z, et al. Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol 2013; 20:1539–1545. [DOI] [PubMed] [Google Scholar]; In an open-label prospective study, 29 patients with Val30Met TTR-FAP with NIS greater than 10 were evaluated by NIS and disability scores at 6 months and 13 patients at 12 months after receiving tafamidis. The disease stage worsened in 20%, and two of nine developed orthostatic hypotension.
- 61.Merlini G, Plante-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res 2013; 6:1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006; 13:236–249. [DOI] [PubMed] [Google Scholar]
- 63.Tojo K, Sekijima Y, Kelly JW, et al. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res 2006; 56:441–449. [DOI] [PubMed] [Google Scholar]
- 64▪.Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013; 310:2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]; A randomized, double-blind, placebo-controlled study conducted among 130 patients with TTR-FAP randomly assigned to receive diflunisal 250 mg or placebo twice daily for 2 years showed reduction of the rate of progression of neurological impairment score NIS + 7 and preserved quality of life.
- 65.Whelan CJ, Sattianayagam P, Dungu J, et al. Tolerability of diflunisal therapy in patients with transthyretin amyloidosis. XIIIth International Symposium on Amyloidosis; 6–10 May 2012. Abstract OP 56. [Google Scholar]
- 66.Castano A, Helmke S, Alvarez J, et al. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012; 18:315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harirforoosh S, Asghar W, Jamali F. Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J Pharm Pharm Sci 2013; 16:821–847. [DOI] [PubMed] [Google Scholar]
- 68▪▪.Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013; 369:819–829. [DOI] [PubMed] [Google Scholar]; Antitransthyretin small interfering RNA, encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles ALN-TTR01 and ALN-TTR02, suppressed the production of both mutant and nonmutant forms of transthyretin in TTR-FAP, establishing proof-of-concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
- 69.Alnylam Pharmaceuticals. APOLLO: The study of an investigational drug, patisiran (ALN-TTR02), for the treatment of transthyretin (TTR)-mediated. ClinicalTrials gov. https://www.clinicaltrials.gov/ct2/show/NCT01960348 [Accessed 1 January 2015] [Google Scholar]
- 70.Isis Pharmaceuticals. Efficacy and safety of ISIS-TTR Rx in famililal amyloid polyneuropathy. ClinicalTrials gov. www.clinicaltrials.gov/ct2/show/NCT01737398 [Accessed 1 January 2015] [Google Scholar]
- 71.Cardoso I, Martins D, Ribeiro T, et al. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 2010; 8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brigham and Women's Hospital. Tolerability and efficacy of a combination of doxycycline and TUDCA in patients with transthyretin in amyloid cardiomyopathy. ClinicalTrials gov [Accessed 1 January 2015] [Google Scholar]
- 73.IRCCS Policlinico S.Matteo. Safety, efficacy and pharmacokinetics of doxycycline plus tauroursodeoxycholic acid in transthyretin amyloidosis. ClinicalTrials gov. https://clinicaltrials.gov/ct2/show/NCT01171859 [Accessed 1 February 2015] [Google Scholar]
- 74.Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012; 19:34–36. [DOI] [PubMed] [Google Scholar]
- 75.Prothena Therapeutics Ltd. Phase 1/2, open label, dose escalation study of NEOD001 in subjects with light chain (AL) amyloidosis. ClinicalTrials gov. www.clinicaltrials.gov/ct2/show/NCT01707264 [Accessed 1 January 2015] [Google Scholar]
- 76.GlaxoSmithKline. A study to evaluate the safety of GSK2398852 when co-administered with GSK2315698 in patients with systemic amyloidosis. ClinicalTrials gov. www.clinicaltrials.gov/ct2/show/NCT01777243 [Accessed 1 January 2015] [Google Scholar]
- 77.Okamoto S, Wixner J, Obayashi K, et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transpl 2009; 15:1229–1235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.