

Abstract
Background
MicroRNAs (miRNAs) are important regulatory elements of gene expression. Similarly to coding genes, miRNA genes follow a birth and death pattern of evolution likely reflecting functional relevance and divergence. For instance, miRNA529 is evolutionarily related to miRNA156 (a highly conserved miRNA in land plants), but it is lost in Arabidopsis thaliana. Interestingly, both miRNAs target sequences overlap in some members of the SQUAMOSA promoter-binding protein like (SPL) family, raising important questions regarding the diversification of the miR156/miR529-associated regulatory network in land plants.
Results
In this study, through phylogenic reconstruction of miR156/529 target sequences from several taxonomic groups, we have found that specific eudicot SPLs, despite miRNA529 loss, retained the corresponding target site. Detailed molecular evolutionary analyses of miR156/miR529-target sequence showed that loss of miR529 in core eudicots, such as Arabidopsis, is correlated with a more relaxed selection of the miRNA529 specific target element, while miRNA156-specific target sequence is under stronger selection, indicating that these two target sites might be under distinct evolutionary constraints. Importantly, over-expression in Arabidopsis of MIR529 precursor from a monocot, but not from a basal eudicot, demonstrates specific miR529 regulation of AtSPL9 and AtSPL15 genes, which contain conserved responsive elements for both miR156 and miR529.
Conclusions
Our results suggest loss of functionality of MIR529 genes in the evolutionary history of eudicots and show that the miR529-responsive element present in some eudicot SPLs is still functional. Our data support the notion that particular miRNA156 family members might have compensated for the loss of miR529 regulation in eudicot species, which concomitantly may have favored diversification of eudicot SPLs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-016-0716-5) contains supplementary material, which is available to authorized users.
Keywords: miR156, miR529, Evolution, Arabidopsis, Eudicots
Background
MicroRNAs (miRNAs) are small RNAs important to transcriptional and post-transcriptional regulation in animals, plants, and viruses. MiRNAs bind complementarily to their target mRNA sequences, leading to translational repression, RNA degradation, or RNA cleavage [1]. Most plant miRNAs are encoded by gene families, and mature miRNAs often have multiple target genes with similar complementary motifs in their mRNAs. The almost perfect complementarity between miRNAs and targets facilitates computational prediction and could be due to their evolutionary origins. One well-accepted model for MIR gene evolution is the inverted duplication of target gene sequences in plant genomes. These duplicated regions become MIR genes over time through sequence variations, which permit the generation of hairpin-like transcripts to become substrates for DICER-LIKE enzymes [2]. Another interesting model suggests that MIR genes can randomly originate from various inverted repeats throughout the genome, independently of target gene duplications [3–6]. For instance, recent evidences indicate that MIR genes may be generated as a result of transposon activity [7, 8].
It has been recently shown that evolutionary patterns of miRNA genes (including lineage-specific gain or loss) can potentially influence the evolution of their target genes [9]. Moreover, synonymous codons in target genes have been found near conserved miRNA target sites in at least four plant genomes, indicating selection constraint on synonymous codons for efficient miRNA binding and proper miRNA function [10]. Several miRNAs are conserved throughout large evolutionary distances from embryophyta to core rosids. However, some miRNAs appear to be species or lineage specific [11–13]. For instance, miR156 is conserved in all Angiosperms studied thus far. Interestingly, miR156 is correlated at the nucleotide level with miR529, sharing 14–16 nt [12]. Curiously, although both miRNAs have a common ancestor in embryophytes, miR529 seems to have been lost in some taxonomic groups, including core eudicots such as Arabidopsis thaliana [12–14]. Both miRNAs share target genes, which are members of the SQUAMOSA promoter-binding protein like (SPL) family [15]. SPL genes are plant-specific transcription factors defined by a highly conserved region of 76 amino acids called SBP domain [16], and by their crucial and widespread functions in development [17–23]. SPL genes can be roughly separated into two major groups–long and short–the latter containing responsive elements for miR156 and, in some species, for miR156 and miR529. Interestingly, sites for miR156 reside in coding as well as untranslated regions of target sequences, whereas miR529 binding sites are chiefly located in coding regions and overlap with miR156 sites [24, 25].
In plant lineages containing both miRNA genes, differential expression of miR156 and miR529 in vegetative and reproductive organs/tissues might have favored lineage-specific retention of miR156/529 sequence variants in their genomes [26]. Another possibility is that the combinatory action of miR156 and miR529 leads to the regulation of distinct targets in specific lineages, such as in Physcomitrella patens [27]. Recently, the evolutionary differences between these two miRNA families in monocot species have been investigated [25], but the consequences of miR529 loss in core eudicots such as Arabidopsis are not yet clear. A broader evolutionary analysis of MIR156 and MIR529 genes and their targets including eudicot species should offer valuable insight into this issue.
In this study, we extended our knowledge regarding evolutionary and functional divergences between miR529 and miR156 regulation on their conserved targets. Through phylogenic reconstruction of target sequences, we confirmed that specific eudicot SPL family members retained miR529-target sites, independently of the presence of MIR529 genes in their genomes. Loss of miR529 function in eudicots is correlated with a more relaxed selection of the miRNA529-specific target element, while miRNA156-specific target sequence is under stronger selection. Additionally, we showed that A. thaliana plants overexpressing MIR529 precursor from a monocot, but not from a basal eudicot, display similar phenotypes as the spl9;spl15 mutant due to the specific down-regulation of these miR156/529-targeted SPLs. Based upon functional and evolutionary analyses, we proposed that the loss of MIR529 genes might have favored diversification of SPLs in eudicot species. It is also possible that new miR156 family member(s) have replaced miR529 functions in eudicots.
Results and discussion
Sequence and phylogenetic analyses reveal that a miR529-responsive element in SPL genes is broadly conserved across land plants
MIR529 genes are present in genomes of a number of species across land plants. Accordingly, in such species, transcripts of a subset of SPL family have responsive elements for both miR156 and miR529 [24]. To get a better view of the extent to which a miR529-responsive element is conserved in land plants, we searched for miR529-responsive elements in SPL genes from species with publicly available genome sequences, including those species in which miRNA529 is absent in their genomes or in transcribed sequences identified thus far. High-confidence prediction of miRNA targets was performed by psRNATarget based on sequence complementarity and evolutionary conservation [28]. We collected conserved mature miR529 sequences from three species (Physcomitrella patens, Oryza sativa, and Aquilegia coerulea), which are available in miRbase v. 21 (http://www.mirbase.org/) and shown in Fig. 1. We then employed more stringent cut-off threshold for the Maximum expectation (E) parameter (range 0–2.0; [28]) to minimize false positive target prediction (data not shown). Surprisingly, we found SPL genes from several species that retain a highly conserved and overlapping responsive element (25 nt in length) for both miRNAs (namely miR156/529-responsive element; Fig. 1a and Additional file 1), independently of the presence of MIR529 genes in their genomes. This suggests that, whereas MIR529 and MIR156 genes have undergone distinct evolutionary fates [25], their mutual targets (which contain the miR156/529-responsive element) have been more conservative even in species which apparently have lost MIR529 genes. For instance, the miR156/529-responsive element in eudicot SPLs resides only in coding regions, similarly to what is observed in monocots and bryophytes (Additional file 1; [24]).
Fig. 1.
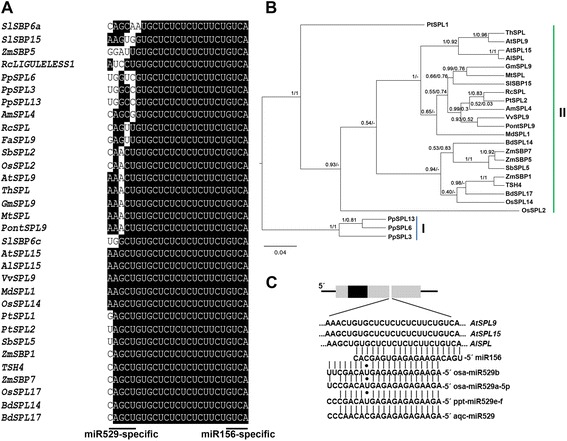
Alignment and phylogeny of SPLs containing the miR156/miR529-response element. a Alignments of the 25-nt miR156/529-response element were done using ClustalW (http://www.phytozome.net/). Black color denotes conserved nucleotides. b Rooted ML phylogenetic tree depicts the relationship between SPLs containing the miR156/529-response element from representative species. The numbers indicate branch support. Left numbers, posterior probability branch support. Right numbers, aLRT branch support (see Methods). “-”, indicates that a particular branch is not observed in Bayesian or maximum-likelihood trees. Blue line comprises moss SPLs and green line encompasses monocot and eudicot SPLs. c Target sites for miR156 and miR529 in A. thaliana SPL9 and SPL15 and A. lyrata SPL transcripts. At, Arabidopsis thaliana; Al, Arabidopsis lyrata; Th, Thellungiella halophila; Vv, Vitis vinifera; Sl, Solanum lycopersium; Md, Malus domestica; Fa, Fragaria ananassa; Pt, Populus trichocarpa; Gm, Glycine max; Mt, Medicago truncatula; Pont, Poncirus trifoliata; Am, Antirrhinum majus; Rc, Ricinus communis; Zm, Zea mays; Os, Oryza sativa; Bd, Brachypodium distachyon; Aqc, Aquilegia coerulea; Pp, Physcomitrella patens. Accession numbers are given in Additional file 1
To further explore the evolutionary relationship of miR156/529 common targets, phylogenetic inference of SBP-box genes containing the miR156/529-responsive element was estimated using maximum-likelihood (ML) and Bayesian inference methods. The percentage of pairwise identity of well-aligned sequence blocks (see Methods) was 73.3 %, and the substitution saturation test reported that the alignment was not saturated (data not shown). We observed two groups of SPLs in our consensus tree (Fig. 1b). Group I included known miR156/529 SPL targets in bryophyte, whereas group II contained various monocot and core eudicot SPLs harboring conserved binding sites for miR156/529. This analysis indicated that SPLs containing miR156/529 target sites have a common origin in land plants (Fig. 1b). A. thaliana SPL9 and SPL15 are closely related and most likely form a pair of paralogous genes [29, 30]. Accordingly, both SPL9 and SPL15 as well as their orthologs retained the miR156/529-responsive element (Fig. 1b and c).
It has been proposed for monocot species and P. patens that SPLs containing miR156/529 sites evolved conservatively with a slow rate when compared with SPLs harboring only the miR156-responsive element [24]. To further elucidate the evolutionary fates of eudicot SPLs containing the miR156/529-responsive element, we analyzed two blocks in SPL sequences: “SBP domain” block, which contains nucleotides of the SBP domain [16] plus few nucleotides upstream/downstream, and “target site” block, which contains nucleotides that comprise both miR156/529-responsive elements (see Methods). For the “SBP domain” block, we estimated nonsynonymous (Ka) and synonymous substitution (Ks) ratios (Ka/Ks) of representative SPLs. We chose Arabidopsis as a representative eudicot due to the fact that, even after extensive sequencing efforts, precursors or canonical mature sequences of miR529 have not been found in either A. thaliana or its closest relative A. lyrata [31]. Pairwise alignments of best-aligned blocks (249 to 2061 nt) among SPL genes from A. thaliana, monocots, and P. patens indicated that the “SBP domain” block is under purifying selection for all comparisons, reflecting functional constraints, which is expected since SBP is likely a DNA binding domain (Additional file 2; [16]).
Given that “target site” blocks are short (25 nt) and could give rise to unrealistic Ka/Ks ratios, we decided to compare their alignment more directly (Fig. 2). Considering the five first bases restricted to the miR529 target site, nonsynonymous substitutions including amino acids with different physicochemical features were found. In contrast, such drastic changes did not occur for the nucleotides specifying miR156 target site (Fig. 2). These observations indicated that different regions of the “target site” block might be under distinct evolutionary constraints. In fact, the bases that diverged more in the “target site” block specifically correspond to the miR529 target site (Fig. 2). Therefore, it is possible that loss of miR529 regulation in eudicots allowed a more relaxed or low purifying selection of the miRNA-responsive element region, which reflects its loss of functionality as a miR529 target. In line with this observation, the percentage of nucleotide identity within the “target site” block (including both miR156 and miR529 response elements) is lower among eudicot SPL genes (58.7 %) than among monocot and bryophyte ones (81.3 and 86.7 %, respectively) (Fig. 2). In monocots and P. patens, the “target site” block is more conserved, likely because it is functionally relevant as miR529 is still present in these species [24].
Fig. 2.
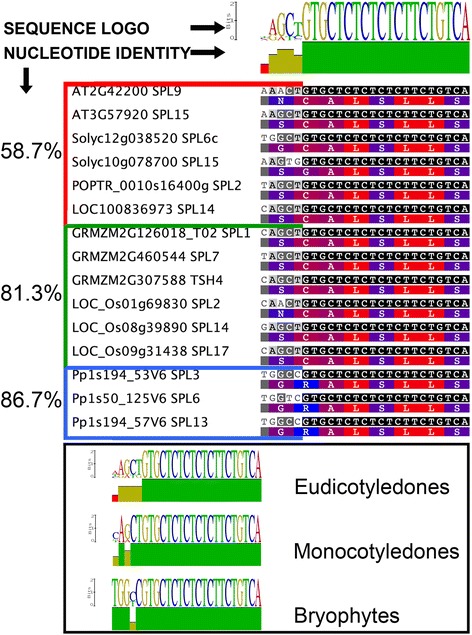
DNA Alignments of miR156 and miRNA156/529 Target Sites. Amino acids coded by miR156/529 target site are shown. Amino acids are colored according to their hydrophobicity; Red box, eudicot sequences; Green box, monocot sequences; Blue box, bryophyte sequences. Percentage of nucleotide identity for the 5′ four nucleotides is shown at the side of each colored box. Black lines underneath the topmost logo indicate miRNA binding sites. Box at the bottom shows logos constructed based on alignments of each plant group. Accession numbers are given in Additional file 1
Overexpression of a basal eudicot microRNA529 precursor in A. thaliana phenocopies miR156 overexpressor
Although miR156 is highly conserved, being present in all plant species assessed thus far, miR529 seems to be restricted to particular taxonomic groups [12]. To get a better view of the MIR156/529 gene evolution, the phylogenetic relationship of these miRNAs was accessed using the maximum-likelihood (ML) approach. For phylogenetic analyses, we included MIR156 and MIR529 precursors from Physcomitrella patens, monocot species (Oryza sativa, Zea mays, Brachypodium distachyon, and Sorghum bicolor), a basal eudicot (Aquilegia coerulea), and precursors of MIR156 genes from Arabidopsis thaliana. A consensus ML tree was generated in which two general, broad groups were readily observed (Additional file 3A). Group I comprised MIR156 precursors from different species, while group II contained MIR529 precursors from monocots and moss. Monocot MIR529 precursors were grouped into a distinct subset of group II (Additional file 3A), suggesting that evolutionary divergence occurred in a common ancestor of land plants, which led to the split between MIR529 genes of moss and flowering plants.
It had been proposed earlier that a key feature of miRNA evolution is that, once evolved, families and family members are rarely lost [2]. However, not all miRNAs are equally conserved and it has been recently shown that several miRNA losses occurred in families that evolved prior to the origin of spermatophytes [32]. Our data suggest that miR156 and miR529 families experienced dynamic duplications and losses across embryophytes, through which clade- or species-specific miRNA gene subgroups have arisen or were eliminated. For instance, A. thaliana has at least 10 MIR156 loci and 10 miR156-targeted SPLs, whereas rice has at least 12 MIR156 loci, two MIR529 loci, and eight miR156-targeted and four miR156/miR529-targeted SPLs [28].
Interestingly, the predicted MIR529 precursor from the basal eudicot A. coerulea [33] was grouped into group I, with A. thaliana MIR156h and Aquilegia MIR156a and b precursors, indicating a common origin of these miRNAs (Additional file 3A). Moreover, Aquilegia MIR529 seems to be highly conserved with Arabidopsis MIR156h at both nucleotide and secondary structure levels (Additional file 3B). These observations raised the question of whether this MIR precursor of Aquilegia defined as MIR529 is indeed a MIR156 homolog. To test this hypothesis, we investigated whether loci flanking Aquilegia pre-miR529 are localized into syntenic blocks when comparing with monocot species. We firstly searched for such syntenic groups among distinct monocot species, including Z. mays, O. sativa, B. distachyon, and S. bicolor (Additional file 4A). The colinearity of genes around pre-miR529 locus is relatively conserved, therefore defining conserved syntenic block in monocots. Conversely, we did not detect any conservation of syntenic block including pre-miR529 from Aquilegia, although some orthologous genes were found to be present in this species (Additional file 4B). Moreover, there is no detectable synteny between MIR156h-associated regions from eudicots (A. thaliana and V. vinifera) and the MIR529-associated region from basal eudicot A. coerulea (Additional file 4B, C). This complete loss of synteny, along with our phylogenetic and sequence analyses, suggests that the basal eudicot Aquilegia lost a bona-fide MIR529 gene, perhaps after the regulatory role of miR529 was perturbed due to mutations in the miR529 sequence as recently proposed [25].
To determine whether AqcMIR529 could be properly processed and could give rise to functional miRNAs, we constitutively expressed its foldback in A. thaliana plants under control of the viral 35S promoter, which confers strong and near-ubiquitous expression ([34]; such plants were hereafter referred to as 35S::AqcMIR529 lines; see Methods). We compared vegetative phenotypes among Col-0 or wild type, 35S:: AqcMIR529 lines, transgenic plants overexpressing the AtMIR156a precursor (35S::AtMIR156a; [35]), and the double mutant spl9;spl15 (Fig. 3a). This mutant contains T-DNA insertions in both AtSPL9 and AtSPL15 loci, which resulted in an accentuated reduction in their expression and function, although other AtSPLs are still functional. Transgenic A. thaliana plants transformed with the Aquilegia-overexpressing construct had phenotypes similar to 35S::MIR156a plants, such as a high number of smaller, more rounded rosette leaves (Fig. 3a). Moreover, overexpression of the MIR529 precursor from Aguilegia in A. thaliana led to the down-regulation not only of AtSPL9 and AtSPL15 genes (which contain both miR156 and miR529 response elements; Fig. 1), but also other SPL genes (Fig. 3b). Thus, 35S::AqcMIR529 lines showed a stronger tendency toward the phenotype of 35S::MIR156a plants (Fig. 3a).
Fig. 3.

Phenotypic, expression, and RACE analyses of Arabidopsis 35S::AqcMIR529 plants. a Morphology of 25-day-old plants (Col-0 or wild type, 35S::MIR156a, spl9;spl15, and 35S::AqcMIR529). Scale bar represents 1 cm. b Stem–loop pulsed RT-PCR to detect AqcMIR529 precursor and some AtSPL transcripts in Arabidopsis leaf tissues. Reactions without RT (−RT) and without cDNA (−) were used as negative controls. Numbers between brackets indicate PCR cycles. Actin-2 was used as an internal control. c Modified 5′-RACE analyzes of cleaved SPL15 transcripts in 35S::AqcMIR529 leaf tissues. The 5′-ends of the SPL15 cleavage products preferentially map to miR156
In line with the observed phenotypes and expression analyses, RACE analysis of SPL15 cleavage sites demonstrated that transcripts are chiefly targeted by miR156 in 35S::AqcMIR529 plants (Fig. 3c). Based on these observations and given that AqcMIR529 precursor is more conserved with MIR156-like precursors (Additional file 3), we propose at least two non-exclusive possibilities: (1) the predicted AqcMIR529 precursor is more likely paralogous to AqcMIR156a and b genes and would produce a miRNA156-like small RNA; (2) AqcMIR529 precursor might have accumulated mutations in the miR529 sequence as recently proposed [25], leading to a loss of miR529 biogenesis and/or function. As other basal eudicots seem to accumulate miR529-like small RNAs [36], we cannot rule out the possibility that miR529-like small RNAs still accumulate in specific A. coerulea tissues, since there is no available information regarding large-scale identification of small RNA populations in this basal eudicot.
MiR156 and miR529 show overlapping expression patterns during rice vegetative development
Distinct from the basal eudicot Aquilegia, it is well documented that monocot species retained both MIR156 and MIR529 precursors in their genomes [12, 26, 37], raising the question of whether miR529 regulation has been retained in monocot species because it is essential or it is just a classical case of redundancy reflecting subfunctionalization in which miR529 has a limited effect as compared with miR156. Rice has two copies of MIR529 precursors (a and b) in its genome. It has been shown that OsmiR529a* (or OsmiR529a-5p) is preferentially expressed in panicle, whereas OsmiR529b is ubiquitously expressed in roots, shoots, and panicle [26]. The authors, however, did not evaluate the expression of OsmiR156 or OsmiR529 in vegetative apices and young leaves, organs in which SPLs define an endogenous flowering pathway and control leaf maturation and initiation, respectively [19, 38]. To get more insights into the possible roles of OsmiR529 [26], we analyzed transcript accumulation patterns of OsmiR529b, OsmiR156a-j, and one of their common targets (OsSPL14) in vegetative apices, juvenile leaves (L3-L5), and young panicles. OsmiR529b and OsmiR156a-j were expressed in all tissues/organs evaluated, though at variable levels (Fig. 4a). OsmiR156a-j transcripts accumulated at high levels in juvenile leaf tissues, whereas OsSPL14 was down-regulated in leaf and young panicle tissues (Fig. 4a). OsSPL14 is targeted by both miRNAs in seedlings, whereas it seems to be predominantly targeted by miR529a-5p in panicle [26]. Likewise, OsmiR529b might also have a more prominent effect on the post-transcriptional regulation of OsSPL14 expression in young panicle.
Fig. 4.
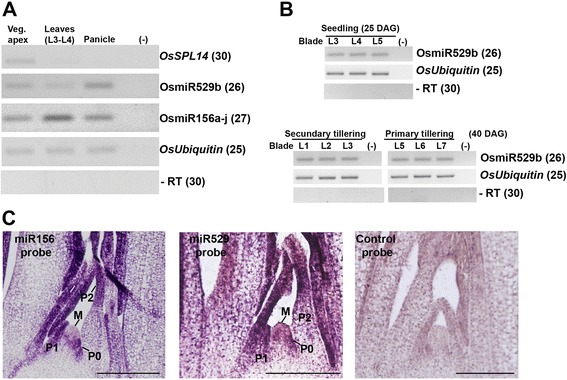
Expression patterns of miR529 and miR156 in Oryza sativa. a Stem–loop pulsed RT-PCR to detect OsmiR529b, OsmiR156a-j, and OsSPL14 transcripts in vegetative apices (Veg. apex), leaf (L), and young panicle tissues. Reactions without RT (−RT) and without cDNA (−) were used as negative controls. Numbers between brackets indicate PCR cycles. DAG, days after germination. Rice Ubiquitin (LOC_Os03g0234200) was used as an internal control. b Stem–loop pulsed RT-PCR to detect OsmiR529b transcripts in leaf (L) tissues (Blade) from rice seedlings and tillering developmental stages. Reactions without RT (−RT) and without cDNA (−) were used as negative controls. Numbers between brackets indicate PCR cycles. Rice Ubiquitin (LOC_Os03g0234200) was used as an internal control. c Spatiotemporal expression patterns of miR529 and miR156 in rice shoot apical meristem (SAM). Probes of 3′-labelled LNA-modified oligonucleotides detecting miR156 and miR529 as described [38] were hybridized with longitudinal sections of the SAM from 25-DAG rice seedlings. A scramble-miRNA 3′-labelled LNA probe was used as a negative control. Purple staining shows probe localization. M, meristem; P, leaf primordia. Bars: 10 μm
OsmiR156 is dynamically expressed during rice leaf development, and a gradual increase of OsmiR156 expression might be essential for regulating the temporal expression of target genes, including OsSPL14 [38]. We evaluated the expression pattern of OsmiR529b in similar developed leaves in seedling (L3-L5) and tillering stages. In contrast with the observed OsmiR156 expression patterns [38], OsmiR529b was ubiquitously expressed in all leaf developmental stages (Fig. 4b), suggesting that this miRNA has a minor or negligible contribution for temporal control of the expression of SPL genes during rice leaf maturation. Nevertheless, it is also possible that miR529 function as a dampening miRNA to establish the correct balance of SPL targets during temporal leaf development in monocots.
Given that OsmiR529b and OsmiR156a-j transcripts accumulated in the vegetative apex (Fig. 4a), we decided to evaluate their spatial expression patterns in the shoot apical meristem (SAM) via in situ hybridization using specific probes as described ([38]; see Methods). Both miRNAs are expressed in incipient (P0) and developing leaf primordia, but not in the meristem proper (i.e., the peripheral and central zones) (Fig. 4c). Such expression pattern strengthened the data from Arabidopsis, which showed that miRNA regulation of SPLs occurs mainly in leaf primordia and that SPL activity may nonautonomously inhibit initiation of new leaves at the SAM, perhaps via auxin signaling pathways [39]. Our in situ data showed that OsmiR529a-b and OsmiR156a-j have overlapping spatial expression patterns in leaf primordia, which suggest that these miRNAs can redundantly regulate or collaborate to fine-tune regulation of target expression in these organs. However, based on reported higher levels of OsmiR156 expression compared with OsmiR529 expression [26], the contribution for SPL expression modulation is unlikely to be comparable for both miRNAs, mainly during early stages of rice vegetative development.
Modified 5′-RACE procedure can be used to access cleavage products of miRNA targets as well as the processing of miRNA precursors [40, 41]. Parallel analysis of RNA end (PARE) signatures that are derived from rice degradome and that only mapped to the pre-miRNAs can give additional evidences of roles of miR156 and miR529 in rice development. We therefore collected PARE signatures of OsMIR156a-l and OsMIR529a-b precursors from publicly available resources (see Methods). Based on available data, it seems that rice MIR156 and MIR529 precursors are differentially processed, which may lead to differential miRNA accumulation across rice tissues/organs (Fig. 4 and Additional file 5). Even within the OsMIR156 family, specific members are differentially processed. For example, OsMIR156k and –l showed fewer PARE signatures when compared with the remaining MIR156 precursors. Likewise, OsMIR529a and b precursors have a smaller amount of signatures when compared with most OsMIR156 precursors (Additional file 5). It is possible that differential precursor processing also account for the distinct functions of OsmiR156 and OsmiR529 in development. It would be interesting in the future to address the question of whether miR529 regulation of SPLs is crucial for rice vegetative development. In contrast to miR156, the effects of over-expressing or down-regulating miR529 have yet to be examined in transgenic/mutant monocot plants.
Conserved miR529-responsive element is functional in AtSPL9 and AtSPL15 genes
As discussed above, miR529 has been lost in eudicots like A. thaliana, yet the closely related AtSPL9 and AtSPL15 genes [29] harbor a conserved miR529-responsive element, similarly to their homolog in A. lyrata (Fig. 1c). Given that miRNA::target gene interactions have been comprehensively identified in A. thaliana [15, 17, 42], we asked whether miR529 binding site is functional in this core eudicot. To test this hypothesis, we generated transgenic Arabidopsis plants harboring the well-annotated rice MIR529b precursor under the control of viral 35S promoter (plants were hereafter referred to as 35S::OsMIR529b lines). As AtSPL9 and AtSPL15 are involved in regulating leaf initiation and flowering time, we compared vegetative and flowering phenotypes among Col-0, 35S::OsMIR529b lines, transgenic plants overexpressing the AtMIR156a precursor, and the double mutant spl9;spl15 (Fig. 5a). At least three independent 35S::OsMIR529b lines displayed similar phenotypes as the double mutant spl9;spl15, such as an increased number of rosette leaves in combination with a slight delay in flowering, albeit these phenotypes are less pronounced than those from 35S::AtMIR156a plants (Fig. 5a).
Fig. 5.
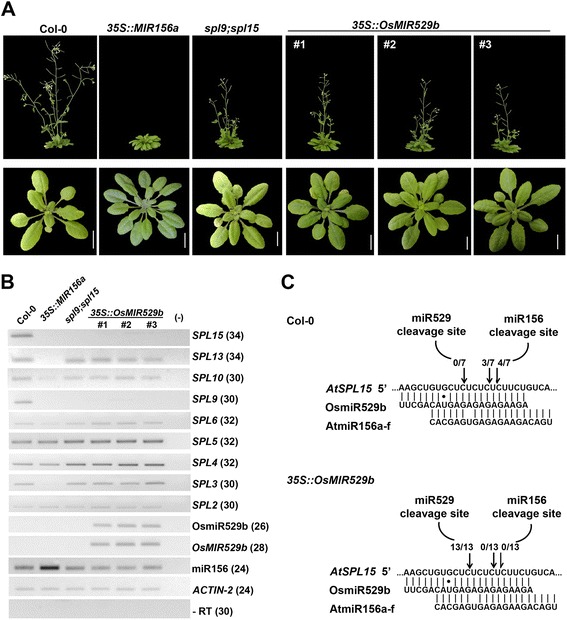
Phenotypic and expression analyses of 35S::OsMIR529b Arabidopsis plants. a Morphology of six-week-old (upper pannel) and 25-day-old (lowerpannel) plants (Col-0 or wild type, 35S::MIR156a, spl9;spl15 mutant, 35S::OsMIR529b). Scale bar represents 1 cm. b Stem–loop pulsed RT-PCR to detect OsMIR529b precursor, OsmiR529b, miR156, and AtSPL transcripts in Arabidopsis leaf tissues. Reactions without RT (−RT) and without cDNA (−) were used as negative controls. Numbers between brackets indicate PCR cycles. Actin-2 was used as an internal control. c Modified 5′-RACE analyses of cleaved SPL15 transcripts in Col-0 and 35S::OsMIR529b leaf tissues. The 5′-ends of the SPL15 cleavage products preferentially map to miR156 (in Col-0) and miR529b binding sites (in 35S::OsMIR529b)
To further confirm the phenotypic similarities between 35S::OsMIR529b and spl9;spl15 plants, we evaluated the average number of juvenile and rosette leaves (Table 1). Under our long-day (LD) growing conditions the 35S::MIR156a line produced 2.8 times more juvenile leaves than Col-0 (wild type), similarly to data previously reported [17], whereas 35S::OsMIR529b and spl9;spl15 plants produced, on average, only 1.4 times more. Likewise, the production of rosette leaves of 35S::OsMIR529b lines showed a stronger tendency toward the phenotype of the spl9;spl15 mutant (Table 1).
Table 1.
Phenotypic evaluation of 35S::OsMIR529b lines in comparison with Col-0, spl9;spl15, and 35S::MIR156a plants under LD growing conditions
| Juvenile leaves1 | Rosette leaves (DAG) | Bolting (DAG) | Anthesis2 (DAG) | ||||
|---|---|---|---|---|---|---|---|
| 15 | 20 | 25 | 30 | ||||
| Col-0 | 6.8 ± 0.5c | 7.7 ± 0.7c | 8.8 ± 0.9c | 13.3 ± 0.8c | 14.3 ± 0.9c | 16.5 ± 1.2d | 23.6 ± 1.6c |
| 35S::MIR156a | 19.4 ± 1.8a | 15.0 ± 0.9a | 19.5 ± 1.6a | 35.6 ± 4.6a | 52.9 ± 2.6a | 45.0 ± 1.8a | 54.0 ± 3.3a |
| spl9;spl15 | 10.3 ± 0.7b | 11.0 ± 1.2b | 13.3 ± 1.8b | 21.1 ± 2.3b | 29.4 ± 2.2b | 20.0 ± 0.7bc | 25.8 ± 1.9b |
| 35S::OsMIR529b #1 | 10.3 ± 0.5b | 12.3 ± 1.1b | 13.0 ± 0.6b | 20.3 ± 1.1b | 26.8 ± 2.0b | 19.4 ± 0.7c | 24.8 ± 1.5bc |
| 35S::OsMIR529b #2 | 10.0 ± 1.1b | 11.4 ± 1.0b | 12.6 ± 1.1b | 20.3 ± 2.7b | 27.5 ± 1.6b | 18.9 ± 0.9c | 23.4 ± 1.3c |
| 35S::OsMIR529b #3 | 9.8 ± 0.7b | 11.0 ± 1.3b | 13.2 ± 0.9b | 19.6 ± 1.7b | 26.5 ± 1.9b | 20.9 ± 1.2b | 26.8 ± 2.2b |
Fifteen plants per genotype were used for determination. DAG, days after germination. Average values followed by the same letter do not differ statistically (P < 0.01, Student’s t-test)
1Number of rosette leaves formed before the appearance of the first leaf with abaxial trichomes
2Opening of the first flower
In addition to estimating the number of leaves formed before the appearance of the first flowers, we also determined the time that 35S::OsMIR529b lines needed to bolt as well as to anthesis (Table 1). On average, transgenic plants overexpressing OsmiR529b showed a slight delay in the transition to flowering (3.3 days) when compared with Col-0 (wild type). In line with the observations of Schwarz and co-workers [29], we also observed that the double mutant spl9;spl15 showed an intermediate behavior between Col-0 and miR156 overexpressor (Table 1). Importantly, spl9;spl15 plants did not differ statistically from 35S::OsMIR529b lines in terms of transition to flowering and leaf development (Table 1). These data reinforced the observation that OsMIR529b overexpressors display similar vegetative and reproductive phenotypes as spl9;spl15 mutant, likely due to the low levels of SPL9 and SPL15 transcripts in both genotypes.
MiR156 targets, besides SPL9 and SPL15, exclusively other eight SPL family members [15] and these were shown to be down-regulated in AtMIR156b-overexpressing plants [17]. In comparison with spl9;spl15 double mutant and 35S::OsMIR529b lines, 35S::MIR156a line displays more severe, aberrant vegetative and reproductive phenotypes (Table 1; [35]), which is likely due to the fact that additional miR156-targeted SPL genes act redundantly to regulate leaf initiation and phase change [43]. Conversely, as in the spl9;spl15 double mutant, only AtSPL9 and AtSPL15 genes may be repressed in 35S::OsMIR529b lines, rendering them a less aberrant phenotype (Fig. 5a). To confirm this hypothesis, we evaluated the expression patterns of several SPL family members in leaf tissues of 35S::OsMIR529b lines, spl9;spl15 mutant, 35S::MIR156a line, and Col-0. We also evaluated the presence of transcripts from OsMIR529b precursor and the accumulation of its respective mature miRNA (Fig. 5b). As expected, several SPL genes were down-regulated in the 35S::MIR156a plants (Fig. 5b). However, in line with our phylogenetic (Fig. 1b) and phenotypic data (Table 1), only transcript levels of AtSPL9 and AtSPL15 showed a severe reduction in 35S::OsMIR529b plants. Moreover, levels of miR156 transcripts were similar in Col-0 and 35S::OsMIR529b plants (Fig. 5b), suggesting that the accentuated reduction in SPL9 and SPL15 expression in OsMIR529b overexpressors is most likely due to the accumulation of rice miR529b transcripts. Together, these results substantiated the fact that monocot MIR529 precursor is correctly processed in a core eudicot to generate mature miR529 transcripts and down-regulate specific SPL genes.
To confirm the post-transcriptional regulation of AtSPL genes by miR529b, we mapped cleavage sites in SPL15 transcripts in 35S::OsMIR529b plants employing the modified 5′-RACE approach [40]. RACE analyses showed that cleavage sites occurred between the base 10 and 11 of the miR529b (Fig. 5c), similarly to what has been described for monocots [44]. These results robustly showed that SPL15 is mainly regulated by OsmiR529b in Arabidopsis 35S::OsMIR529b plants, demonstrating a conserved function of miR529 in post-transcriptionally regulating specific SPL family members. Importantly, our data imply that the miR529-responsive element is conserved and functional in Arabidopsis SPL9 and SPL15 genes, likely due to the selective constraint on the amino acid or RNA secondary structure of the region surrounding miR156/529-responsive element.
Conclusions
We have shown that, although MIR529 genes have been lost in Arabidopsis and perhaps in all eudicot species, particular SPL genes in these species retained the miR529-responsive element, possibly due to the maintenance of synonymous codons for efficient miR156 binding and proper function [10]. More specifically, A. thaliana SPL9 and SPL15 genes retained a functional miR529-responsive element, even in the absence of a miR529-generated locus. Similarly to monocot SPLs, eudicot SPL genes containing the miR156/529-responsive element appear to be under evolutionary constraints distinct from those containing only the miR156-responsive element. Such tendency would be indicative of target evolution constrained by miRNA-mediated regulation.
It is possible that the interplay between miR156 and miR529 regulation of specific SPLs be important to fine-tune flower architecture development in monocots, particularly in grasses [26, 37]. As Arabidopsis does not have miR529, perhaps particular miR156 family members (such as miR156h.2, which is preferentially expressed in flower tissues; [45]) functionally replace miR529. It is conceivable that other core rosids and/or closely related species of A. thaliana share similar miR156/miR529/SPL evolutionary history, though such confirmation requires future studies.
Methods
Plant material and growth conditions
Arabidopsis thaliana plants (ecotype Columbia-0 or Col-0) were grown at 21 °C (day)/19 °C (night) under long-day conditions (16 h light/8 h dark). Transgenic plants 35S::MIR156a and the double mutant spl9-1;spl15-2 were described [35]. Transgenic plants were confirmed by PCR genotyping.
For transgenic Arabidopsis plants, the binary constructs 35S::OsMIR529b and 35S::AqcMIR529 were delivered into Agrobacterium tumefaciens GV3101 (pMP90) by the electroporation method. Transgenic plants were generated by the floral dipping method [46] and screened with 50 mg/mL kanamycin on half-strength MS plates. At least six independent kanamycin-resistant lines were selected for transgene integration by PCR and subsequently examined for transgene expression levels (data not shown). Further analyses were performed with selected lines in the T3 generation.
Rice seeds (Oryza sativa ssp japonica) were germinated on soil, and plants were grown under greenhouse conditions.
DNA constructs
Oligonucleotide primers for all constructs are given in the Additional file 6. A 1000-bp fragment encompassing the OsMIR529b precursor was amplified from genomic DNA of O. sativa. The PCR product was subcloned into pGEM (promega) and sequenced. The confirmed OsMIR529b precursor was digested with BamHI and SacI restriction enzymes and subsequently cloned into the binary vector pBI121 behind the CaMV35S promoter. For 35S::AqcMIR529 construction, a 125-bp fragment encompassing the annotated AqcMIR529 precursor [33] was amplified from genomic DNA of A. coerulea, sequenced, and further cloned into the plant binary destination vector pK7WG2 (Gateway System; [47]) behind the CaMV35S promoter.
RNA extraction and stem–loop pulsed reverse transcriptase (RT)-PCR
Total RNA from Arabidopsis (leaf tissues) and rice (vegetative apices, leaf, and panicle tissues) was extracted using Trizol reagent (Life Technologies, USA) according to manufacturer’s instructions and subsequently treated with DNAse I (Life Technologies, USA). For miRNA and mRNA detection, DNAse I-treated RNA (2.0 μg) was reverse-transcribed to generate the first-strand cDNA, according to Varkonyi-Gasic et al. [48]. Oligo(dT) primer was also added to the reaction for detecting target mRNAs and internal controls. cDNA dilutions were used for PCR reactions as follows: 1.0 μL cDNA, 1.5 mM Magnesium Sulfate, 0.25 mM each dNTP, 10 pmol each primer, and 1 U of Taq DNA Polymerase (Promega, USA). The reactions were done under the following conditions: 94 °C for two minutes and appropriate cycle numbers of 94 °C for 20 s, 60 °C for 30 s, and 72 °C for 45 s. All reactions were repeated at least twice with two biological samples. Primer sequences are described in Additional file 6.
Analysis of 5′-RACE
Five micrograms of total RNA from rosette leaves of Arabidopsis plants (Col-0, 35S::OsMIR529b and 35S::AqcMIR529) was ligated to a RNA adapter, in a reaction mixture containing 0.5 U/μL of T4 RNA Ligase, 4 U/μL RNAse inhibitor, and 1 mM ATP. The subsequent steps were performed according to the manufacturer’s guide of the GeneRacer kit (Invitrogen). The first PCR was done using the following AtSPL15 specific primer: 5′-AGCCATTGTAACCTTATCGGAGAATGAG. The PCR reaction was subsequently used as a template for a semi-NESTED PCR with an internal AtSPL15-specific primer (5′-TCATCGAGTCGAAACCAGAAGAT). After amplification, 5′-RACE products were gel-purified and cloned, and at least eight independent clones were randomly chosen and sequenced.
Phenotypic analysis
The number of rosette leaves was measured during several developmental stages (15, 20, 25, and 30 days after germination or DAG). Flowering and bolting time as well as the number of juvenile leaves were estimated as described. Data were subjected to statistical analyses by using the program ASSISTAT version 7.6 beta (t-student P <0.01).
In situ hybridization
Non-radioactive in situ hybridization was done as described [49]. Oryza sativa vegetative apices were collected from seedlings 25 days after germination. Locked nucleic acid probes with sequences complementary to miR56 and miR529 as described [38] and negative control Scramble-miR (5′-GTGTAACACGTCTATACGCCCA) were synthesized by Exiqon (USA) and digoxigenin-labeled with the DIG Oligonucleotide 3′-end Labeling kit (Roche Applied Science, USA). Ten picomoles of each probe was used for each slide. Hybridization and washing steps were performed at 55 °C.
Phylogenetic and sequence analysis
Sequences of MIR156 and MIR529 precursors (pre-miRNAs) were retrieved from miRBase v.21 (http://www.mirbase.org/) whereas SPL sequences were retrieved from PHYTOZOME v. 9.1 (http://www.phytozome.net/) and TAIR (https://www.arabidopsis.org/). For miRNA precursors, retrieved sequences were aligned using ClustalW [50] using default values. Phylogenetic analyses were performed in MEGA v. 5.05 [51] using default values. Phylogenetic inference was done using maximum-likelihood (ML) method with Bootstrap analysis (1000 trees).
The DNA sequences of SPLs were aligned using Muscle algorithm [52] and the well-aligned blocks were selected using Gblocks server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) by the most stringent option. Multiple sequence alignment is depicted in Additional file 7. The alignment was submitted to the estimation of proportion of invariant sites and substitution saturation test using the algorithm of Xia test implemented in DAMBE5 software [53]. The option for the best-fit evolutionary model was performed using Akaike information criterion implemented in jModelTest [54]. The phylogenetic reconstruction was determined by ML and Bayesian inference methods, using PhyML v3.0 [55] and Beast v1.8.0 [56], respectively, the latter being implemented in CIPRES Science Gateway (https://www.phylo.org/). The approximate likelihood ratio test or aLRT [57] was used for ML analysis. The posterior probability estimates were calculated for Bayesian inference. The software Tracer was applied to determine the burn-in (using the log likelihood scores) in Bayesian method generation and the TreeAnnotator [54] to summarize the data after the exclusion of the trees that appeared outside the convergence area. The proportion of invariable sites and gamma distribution (number of categories = 4) was estimated and random local clock model for Bayesian analysis was also used.
PARE signatures mapping to OsMIR156 and OsMIR529 precursors and RNA-seq and sRNA signatures were retrieved from Rice Next-Gen sequence DBs (http://mpss.udel.edu/). Sequence abundance was estimated by normalizing all samples to TP10M (transcript per 10 million reads).
Nonsynonymous (Ka) and synonymous (Ks) substitution calculation
The selective pressure analysis (Ka/Ks) was performed using best-aligned blocks for nucleotide sequences encompassing the SBP domain plus few nucleotides upstream/downstream (named “SBP domain”). Eudicot, monocot, and bryophyte SPL sequences were carefully selected based on the phylogenetic tree depicted in Fig. 1b and phylogenetic analyses reported previously [29]. Codons of each DNA sequence for each edited alignment were selected using an in-house Python script. Values of Ka/Ks were estimated by comparing sequences among and within eudicot, monocot, and bryophyte groups through the software KaKs_Calculator using the model selection (MS) method [58]. Selected “target site” sequences were aligned using the Muscle algorithm, and Logos were generated using Geneious tools (http://www.geneious.com).
Synteny analysis
Based on coordinates of neighbor genes of sites of pre-miR529 in O. sativa (OsMIR529a and OsMIR529b), A. coerulea, and pre-miR156h from V. vinifera and A. thaliana, the conservation of syntenic blocks among and within monocot and eudicot species was searched in Genomicus Plants v.16.03 [59]. For syntenic mapping of Aquilegia coerulea, we firstly used coordinates of pre-miR529 sites (scaffold_4:4,760,784..4,810,783) from Phytozome database to map flanking genes around aqc-MIR529 locus. Orthologous genes for those loci in selected eudicots and monocots were queried in Genomicus Plants v.16.03 [59]. Phytozome database was also used to search for homologs in A. coerulea of pre-miR529 and pre-miR156h neighbor genes from O. sativa, V. vinifera, and A. thaliana, respectively.
Availability of supporting data
All published datasets referred to in the manuscript are cited in the reference list. All the supporting data are included as additional files.
Accession numbers
AGI identifiers for Arabidopsis thaliana genes are as follows: SPL2, At5g43270; SPL3, At2g33810; SPL4, At1g53160; SPL5, At3g15270; SPL6, At1g69170; SPL9, At2g42200; SPL10, At1g27370; SPL13, At5g50570; SPL15, At3g57920; Actin-2, At3g18780.
Acknowledgments
We thank Dr. Scott Poethig for 35S::AtMIR156a seeds; Dr. Peter Huijser for spl9;spl15 seeds; Dr. Renato Vicentini for initial bioinformatic analyses and helpful discussions; and Dr. Luiz Del Bem for initial phylogenetic analyses. This work was supported by the State of Sao Paulo Research Foundation, FAPESP, Brazil (grants no. 07/58289-5 and 12/51146-2). EGOM was a recipient of a fellowship from Coordination for the Improvement of Higher Education Personnel (CAPES, Brazil). GFFS (from Centro de Energia Nuclear na Agricultura –CENA/USP) and EMS were recipients of a fellowship from the State of Sao Paulo Research Foundation, FAPESP, Brazil.
Additional files
Predicted SBP/SPL targets from eudicot, monocot, and bryophyte species that contain miR156/529-responsive element. (PDF 111 kb)
Estimated Ka/Ks ratios for SPL genes from distinct species. (XLS 33 kb)
Unrooted phylogenetic tree and sequence alignments depict the relationship between MIR156 and MIR529 precursors from representative species. (A) Available MIR156 and MIR529 precursor sequences were retrieved from miRbase v. 21 (http://www.mirbase.org/). Phylogenetic analysis was performed using maximum-likelihood with bootstrap analysis (1000 trees). The percentage of trees (above 50 %) in which the associated taxa clustered together is shown next to the branching sites. ath, Arabidopsis thaliana; aqc, Aquilegia coerulea; bdi, Brachypodium distachyon; osa, Oryza sativa; ppt, Physcomitrella patens; sbi, Sorghum bicolor; zma, Zea mays. (B) Sequence alignments of MIR529 and MIR156 precursors. Al, Arabidopsis lyrata. Alignments were done using ClustalW [50]. Harpin structure predictions of MIR529 and MIR156 precursors were estimated using MFOLD3.2 algorithm [60]. (PDF 569 kb)
Conservation of syntenic blocks. (A) Genomic regions containing homologs of rice pre-miR529a/b neighbor genes. (B) Genomic regions containing homologs of Aquilegia pre-miR529 neighbor genes. (C) Genomic regions containing homologs of Arabidopsis and grape pre-miR156h neighbor genes. Arrows sharing the same color in different species designate orthologous genes. Arrows with gray and black lines indicate paralogs if they share the same color, although not all black lined arrows indicated orthologs (please see Genomicus manual; http://www.dyogen.ens.fr/genomicus). The descriptions on top of some arrows indicate locus name and position in that particular genome; *, regions of pre-miRNAs; “:”, loci not shown. The quantity of this symbol indicates the number of loci; “//”, large genomic blocks not shown. Collinear arrows surrounded by a box indicate conserved syntenic blocks. (PDF 458 kb)
PARE and RNA-seq data for rice MIR156 and MIR529 precursors. (PDF 58 kb)
Oligonucleotide sequences used in this work. (PDF 84 kb)
Multiple sequence alignment of selected SPL nucleotide sequences. (PDF 86 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EGOM, EMS, GFFS, and CHBR carried out the molecular biology studies and analyzed the data; GTV and MV carried out the bioinformatic and phylogenetic analyzes; FTSN designed and coordinated the study, and FTSN and MV wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Edna Gicela Ortiz Morea, Email: omorea1010@gmail.com.
Eder Marques da Silva, Email: edermarques.silva@gmail.com.
Geraldo Felipe Ferreira e Silva, Email: geraldoffs@gmail.com.
Guilherme Targino Valente, Email: valentegt@gmail.com.
Carlos Hernan Barrera Rojas, Email: cahebaro@gmail.com.
Michel Vincentz, Email: mgavince@gmail.com.
Fabio Tebaldi Siveira Nogueira, Phone: (+55-19) 34294052, Email: ftsnogue@usp.br.
References
- 1.Axtell MJ, Westholm JO, Lai EC. Vive la differénce: biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011;2:221. doi: 10.1186/gb-2011-12-4-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen E, Xie Z, Gustafson AM, Sung GH, Spataford JW, Carrington JC. Evolution of microRNAs genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004;36:1282–90. doi: 10.1038/ng1478. [DOI] [PubMed] [Google Scholar]
- 3.Lu C, Tej SS, Luo S, Haudenschild CD, Meyers BC, Green PJ. Elucidation of the small RNA component of the transcriptome. Science. 2005;309:1567–9. doi: 10.1126/science.1114112. [DOI] [PubMed] [Google Scholar]
- 4.Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, et al. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;l5:e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Felippes FF, Schneeberger K, Dezulian T, Huson DH, Weigel D. Evolution of Arabidopsis thaliana microRNAs from random sequences. RNA. 2008;14:2455–9. doi: 10.1261/rna.1149408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piriyapongsa J, Jordan IK. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA. 2008;14:814–21. doi: 10.1261/rna.916708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20:3407–25. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Creasey KM, Zhai J, Borges F, Van Ex F, Regulski M, Meyers BC, et al. miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature. 2014;508:411–5. doi: 10.1038/nature13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao M, Meyers BC, Cai C, Xu W, Ma J. Evolutionary patterns and coevolutionary consequences of MIRNA genes and MicroRNA targets triggered by multiple mechanisms of genomic duplications in soybean. Plant Cell. 2015;27:546–62. doi: 10.1105/tpc.15.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu W, Wang X, Zhai C, Xie X, Zhou T. Selection on synonymous sites for increased accessibility around miRNA binding sites in plants. Mol Biol Evol. 2012;29(10):3037–44. doi: 10.1093/molbev/mss109. [DOI] [PubMed] [Google Scholar]
- 11.Axtell MJ, Bartel DP. Antiquity of microRNAs and their targets in land plants. Plant Cell. 2005;17:1658–73. doi: 10.1105/tpc.105.032185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuperus JT, Fahlgren N, Carrington C. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23:431–42. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montes RA, de Fátima Rosas-Cárdenas F, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, et al. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun. 2014;5:3722. doi: 10.1038/ncomms4722. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz-Morea FA, Vicentini R, Silva GF, Silva EM, Carrer H, Rodrigues AP, et al. Global analysis of the sugarcane microtranscriptome reveals a nique composition of small RNA associated with axillary bud outgrowth. J Exp Bot. 2013;64:2307–20. doi: 10.1093/jxb/ert089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Barte B, Barte DP. Prediction of plant microRNA targets. Cell. 2002;110:513–20. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- 16.Klein J, Saedler H, Huijser P. A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol Gen Genet. 1996;250:7–16. doi: 10.1007/BF02191820. [DOI] [PubMed] [Google Scholar]
- 17.Schwab R, Palatnik JF, Riester M, Schommer C, Schmid ME, Weigel D. Specific effects of MicroRNAs on the plant transcriptome. Dev Cell. 2005;8:517–27. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Chuck G, Cigan A, Saeteurn K, Hake S. The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat Genet. 2007;39:544–9. doi: 10.1038/ng2001. [DOI] [PubMed] [Google Scholar]
- 19.Wang JW, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138:738–49. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Nodine MD, Bartel DP. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010;24:2678–92. doi: 10.1101/gad.1986710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu N, Cai WJ, Wang S, Shan CM, Wang LJ, Chen XY. Temporal control of trichome distribution by microRNA 156-targeted SPL genes in Arabidopsis thaliana. Plant Cell. 2010;22:2322–35. doi: 10.1105/tpc.109.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gou JY, Felippes FF, Liu CJ, Weigel D, Wang JW. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell. 2011;23:1512–22. doi: 10.1105/tpc.111.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva GFF, Silva EM, Azevedo MS, Guivin MAC, Ramiro DA, Figueiredo CR, et al. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 2014;78:604–18. doi: 10.1111/tpj.12493. [DOI] [PubMed] [Google Scholar]
- 24.Ling LZ, Zhang SD. Exploring the evolutionary differences of SBP-box genes targeted by miR156 and miR529 in plants. Genetica. 2012;140:317–24. doi: 10.1007/s10709-012-9684-3. [DOI] [PubMed] [Google Scholar]
- 25.Zhang SD, Ling LZ, Zhang QF, Xu JD, Cheng L. Evolutionary comparison of two combinatorial regulators of SBP-Box genes, MiR156 and MiR529, in plants. PLoS One. 2015;10:e0124621. doi: 10.1371/journal.pone.0124621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, et al. Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell. 2011;23(12):4185–207. doi: 10.1105/tpc.111.089045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arif MA, Fattash I, Ma Z, Cho SH, Beike AK, Reski R, et al. DICER-LIKE3 activity in Physcomitrella patens DICER-LIKE4 mutants causes severe developmental dysfunction and sterility. Mol Plant. 2012;5:1281–94. doi: 10.1093/mp/sss036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39(Web Server issue):W155–9. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz S, Grande AV, Bujdoso N, Saedler H, Huijser P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol Biol. 2008;67((1-2)):183–95. doi: 10.1007/s11103-008-9310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preston JC, Hileman LC. Functional evolution in the plant SQUAMOSA-PROMOTER BINDING-LIKE (SPL) gene family. Front Plant Sci. 2013;4(80):1–13. doi: 10.3389/fpls.2013.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell. 2010;22(4):1074–89. doi: 10.1105/tpc.110.073999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 33.Taylor RS, Tarver JE, Hiscock SJ, Donoghue PCJ. Evolutionary history of plant microRNAs. Trends Plant Sci. 2014;19:175–82. doi: 10.1016/j.tplants.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Puzey JR, Kramer EM. Identification of conserved Aquilegia corulea microRNAs and their targets. Gene. 2009;448:46–56. doi: 10.1016/j.gene.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Odell JT, Nagy F, Chua N-H. Identification of DNA sequences required for activity of the cauliflower mosaic virus-35S promoter. Nature. 1985;313:810–2. doi: 10.1038/313810a0. [DOI] [PubMed] [Google Scholar]
- 36.Barakat A, Wall K, Leebens-Mack J, Wang YJ, Carlson JE, Depamphilis CW. Large-scale identification of microRNAs from a basal eudicot (Eschscholzia californica) and conservation in flowering plants. Plant J. 2007;51:991–1003. doi: 10.1111/j.1365-313X.2007.03197.x. [DOI] [PubMed] [Google Scholar]
- 37.Jeong DH, Szchmidt SA, Rymarquis LA, Park S, Ganssmann M, German MA, et al. Parallel analysis of RNA ends enhances global investigation of microRNAs and target RNAs of Brachypodium distachyon. Genome Biol. 2013;24:R145. doi: 10.1186/gb-2013-14-12-r145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie K, Shen J, Hou X, Yao J, Li X, Xiao J, et al. Gradual increase of miR156 regulates temporal expressin changes of numerous genes during leaf development in rice. Plant Physiol. 2012;158:1382–94. doi: 10.1104/pp.111.190488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang JW, Schwab R, Czech B, Mica E, Weigel D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell. 2008;20:1231–43. doi: 10.1105/tpc.108.058180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008;18:758–62. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng Y, Gou L, Chen D, Wu P, Chen M. High-throughput degradome sequencing can be used to gain insights into microRNA precursor metabolism. J Exp Bot. 2010;61:3833–7. doi: 10.1093/jxb/erq209. [DOI] [PubMed] [Google Scholar]
- 42.Jones-Rhoades M, Bartel D. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14:787–99. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 43.Poethig RS. Vegetative phase change and shoot maturation in plants. Curr Top Dev Biol. 2013;105:125–52. doi: 10.1016/B978-0-12-396968-2.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuck G, Whipple C, Jackson D, Hake S. The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development. 2010;137:1243–50. doi: 10.1242/dev.048348. [DOI] [PubMed] [Google Scholar]
- 45.Jeong DH, Thatcher SR, Brown RS, Zhai J, Park S, Rymarquis LA, et al. Comprehensive investigation of microRNAs enhanced by analysis of sequence variants, expression patterns, ARGONAUTE loading, and target cleavage. Plant Physiol. 2013;162:1225–45. doi: 10.1104/pp.113.219873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–43. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 47.Karimi M, Inzé D, Depicker A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002;7:193–5. doi: 10.1016/S1360-1385(02)02251-3. [DOI] [PubMed] [Google Scholar]
- 48.Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods. 2007;3:1–12. doi: 10.1186/1746-4811-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Javelle M, Timmermans MCP. In situ localization of small RNAs in plants by using LNA probes. Nat Protoc. 2012;7:533–41. doi: 10.1038/nprot.2012.006. [DOI] [PubMed] [Google Scholar]
- 50.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 51.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol. 2013;30:1720–8. doi: 10.1093/molbev/mst064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systs Biol. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 56.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Systs Biol. 2006;55:539–52. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Z, Li JL, Zhao X-Q, Wang J, Wong GK-S, Yu J. KaKs calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinfomatics. 2006;4:259–63. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louis A, Nguyen NTT, Muffato M, Crollius HR. Genomicus update 2015: KaryoView and MatrixView provide a genome-wide perspective to multispecies comparative genomics. Nucleic Acids Res. 2015;43:D682–9. doi: 10.1093/nar/gku1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]