

Abstract
Pharmacological ascorbate has been shown to induce toxicity in a wide range of cancer cell lines; using animal models pharmacological ascorbate has shown promise for use in cancer treatment. At pharmacological concentrations the oxidation of ascorbate produces a high flux of H2O2 via the formation of ascorbate radical (Asc•−). The rate of oxidation of ascorbate is principally a function of the level of catalytically active metals. Iron in cell culture media contributes significantly to the rate of H2O2 generation. We hypothesized that increasing intracellular iron would enhance ascorbate-induced cytotoxicity and that iron chelators could modulate the catalytic efficiency with respect to ascorbate oxidation. Treatment of cells with the iron-chelators deferoxamine (DFO) or dipyridyl (DPD) in the presence of 2 mM ascorbate decreased the flux of H2O2 generated by pharmacological ascorbate and reversed ascorbate-induced toxicity. Conversely, increasing the level of intracellular iron by pre-incubating cells with Fe-hydroxyquinoline (HQ) increased ascorbate toxicity and decreased clonogenic survival. These findings indicate that redox metal metals, e.g. Fe3+/Fe2+, have an important role in ascorbate-induced cytotoxicity. Approaches that increase catalytic iron could potentially enhance the cytotoxicity of pharmacological ascorbate in vivo.
Graphical abstract
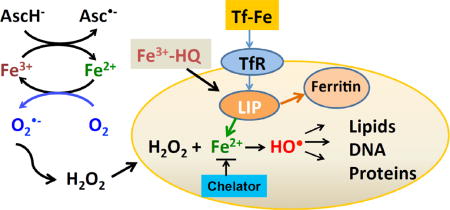
Introduction
Pharmacological ascorbate has been shown to induce toxicity in cancer cells both in vitro and in vivo [1, 2, 3]. Pharmacological concentrations of ascorbate produce hydrogen peroxide via the formation of ascorbate radical (Asc•−) [4]. In in vitro environments, the rate of ascorbate oxidation is principally a function of the level of catalytically active iron and copper [5, 6]. For example, catalytic iron in cell culture media containing ascorbate contributes significantly to the rate of H2O2 generation; Dulbecco’s modification of Eagle’s MEM (DMEM) generates more H2O2 than RPMI 1640 during a 6-hour incubation with increasing concentration of ascorbate [7] due to the fact that DMEM has an additional 0.25 μM Fe(NO3)3 in its formulation in addition to any adventitious iron.
Extracellular H2O2 readily diffuses into cells [8]; if not removed, it can lead to oxidative damage to proteins, lipids, and DNA [9]. These detrimental oxidations require that H2O2 be “activated” by appropriate redox-active transition metals, such as iron [10, 11]; labile iron, i.e. redox active iron associated with macromolecules, will lead to production of hydroxyl radical (HO•) causing site-specific damage [12, 13]. Intracellular labile iron can be coordinated by chelators such as deferoxamine (DFO) blunting their catalytically activity [14], thereby protecting cells from H2O2-induced DNA damage [15]. Conversely, increasing intracellular iron can enhance H2O2-induced injury as seen with cardiomyocytes [16]. Pre-incubation of the epithelial cell line CNCMI221 with Fe3+/8-hydroxyquinoline enhanced the observed cytotoxic effects of H2O2 [17]. It is well known that the primary oxidant generated by the Fenton reaction (H2O2 + Fe2+) is the hydroxyl radical, HO• [18]. HO• is very reactive and as such has an extremely limited diffusion distance, only about 6 nm in cells [19]. In cell culture experiments, introduction of Fe2+ to media containing H2O2 can actually protect cells [20, 21] because the H2O2 in the media is removed; HO• generated by the Fenton reaction in the media will have little consequence, as the majority will react with media components and not cells. Removal of extracellular H2O2 will minimize any possible site-specific oxidative damage to intracellular macromolecules, such as DNA.
We hypothesized that altering the concentration of iron would affect ascorbate-induced cytotoxicity: (1) by altering the rate of oxidation of ascorbate and thus the rate of production of H2O2; and (2) altering the rate of activation of H2O2 by intracellular labile iron, thereby altering the oxidative damage to cellular macromolecules, e.g. DNA, induced by site-specific production of HO•. We found that treatment of cells with iron-chelators decreased H2O2 generation and reversed ascorbate-induced toxicity while increasing intracellular iron enhanced ascorbate-induced cytotoxicity. Our study indicates that redox active metals, both inside and outside the cell, play an important role in ascorbate-induced cytotoxicity.
Materials and Methods
Chemicals
Iron (III) chloride (FeCl3), deferoxamine mesylate (DFO), 2, 2′-dipyridyl (DPD), 8-hydroxyquinoline (8-HQ), and ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Asc-2P) were from Sigma (St. Louis, MO). L-Ascorbic acid was purchased from Macron Chemicals (Center Valley, PA). Stock solutions of ascorbate (1.0 M) were made as previously described [3]. Phen Green™ SK diacetate (PG SK) was from Life Technologies (Grand Island, NY). Prior to use, DPD and PG SK were initially dissolved in a small amount of dimethylsulfoxide (DMSO) and then diluted with Hank’s balance salt solution (HBSS). To make the Fe3+(8-HQ)2 complex (Fe(HQ)2), ferric chloride (10 mM) and lipophilic iron ligand 8-hydroxyquinoline (20 mM) stock solutions were prepared in DMSO. Fe(HQ)2 was made using equal volumes of these stock solutions.
Cell lines
Human pancreatic cancer cells MIA PaCa-2 and Panc-1were cultured in DMEM high glucose supplemented with 10% FBS. Human pancreatic cancer cell line AsPC-1 was cultured in RPMI 1640 with 20% FBS and 1 mM pyruvate. Immortalized normal pancreatic ductal epithelial cells H6c7 were cultured in keratinocyte-serum free medium (KSFM) with supplements (2.5 μg human recombinant EGF and 25 mg bovine pituitary extract) [22]. All cells were maintained in a humidified atmosphere of 95% air/5% CO2 at 37 °C.
Measurement of OCR and H2O2 accumulation via Clark Electrode
The rate of oxygen consumption (OCR, -d[O2]/dt) by ascorbate was determined using a Clark electrode (YSI-5331A probe, YSI Inc., Yellow Springs, OH, USA) connected to an ESA Biostat multi-electrode system (ESA Products, Dionex Corp., Chelmsford, MA, USA) [4]. DMEM media (3.0 mL) was transferred to the reaction chamber, stirring for 2 min. After inserting the electrode, the baseline was recorded. Ascorbate was then added to the reaction chamber using a gas-tight Hamilton syringe.
Clonogenic assay
Cells (1 × 105) were seeded in 60 mm culture dishes and treated 48 h later. After treatment, cells were trypsinized and seeded into 6-well plates at 300 cell/well with 4.0 mL growth media. Colonies were allowed to form between 10 to 14 days at 37 °C. Colonies were fixed with 70% ethanol and stained with Coomasie blue. Colonies with more than 50 cells were counted. Plating efficiency was determined by the formula: (number of colonies formed /number of cells inoculated) ×100.
H6c7 cell lines do not form clones at low cell density, so exponentially growing MIA PaCa-2 cells were irradiated (30 Gy) and then detached by trypsinization. Cells were resuspended in KSFM at 5 × 104 cells/mL and then were seeded into 6-well plates as feeder cells for H6c7.
Measurement of cellular labile iron
The metal sensor Phen Green™ SK diacetate (PG SK) was used to detect intracellular labile iron. PG SK is a probe containing a fluorescein (dichlorofluoresceinamine) and a metal binding moiety (phenanthroline). The modification of carboxylic acids with acetate ester groups results in an uncharged molecule that can permeate cell membranes. Once inside the cell, the acetate ester groups are cleaved by nonspecific esterases, the resulting charged form of Phen Green™ SK chelates intracellular labile iron, which quenches the fluorescence of the fluorescein moiety.
Fluorescence Imaging
Cells (8 × 104) cultured in 35 mm glass-bottomed dishes were washed with PBS and loaded with 5 μM PG SK for 10 min in HBSS at 37 °C in the dark. After incubation, the cells were washed with HBSS to remove excess dye. The cells were imaged in an Olympus IX81 Inverted Microscope using a 60x oil lens. After the baseline fluorescence had been recorded, Fe(HQ)2 (a mixture of equal volumes of 8-hydroxyquinoline (20 mM) and ferric chloride (10 mM)) was added to the cells followed by the addition of DPD (500 μM).
Flow cytometry
Cells cultured in 60-mm dishes were washed with PBS and incubated with 5 μM PG SK for 10 min in HBSS at 37 °C in the dark. The cells were then washed and treated with Fe(HQ)2 (5 and 10 μM) or DPD (500 μM) for 30 min. Following a wash with PBS, cells were harvested by scraping and centrifuged at 1,000 rpm (200 g) for 5 min. The cell pellets were resuspended in 500 μL HBSS and transferred to FACS tubes. Fluorescence profile of the samples was analyzed by flow cytometry (LSR-UV, Becton Dickinson, Mountain View, CA).
To compare the intracellular iron concentration between H6c7, MIA PaCa-2, AsPC-1, and Panc-1 cells. Cells were grown in 60 mm tissue culture dishes until 80% confluence. Cells were washed twice with PBS and incubated with 5 μM PG SK for 10 min at 37 °C. Then the cells were washed twice and treated with DPD (500 μM) for 30 min or left untreated as control. Cells were collected and washed with PBS. Fluorescence of PG SK was measured at 488 nm excitation and 530 emission. The difference in the MFI of cells with and without DPD treatment was used to estimate LIP [23].
Measurement of Intracellular Ascorbate Concentration
Exponentially growing MIA PaCa-2 cells in 100 mm dishes were treated with 100 μM of ascorbate-2-phosphate (Asc-2P) for 24 h [24]. Then cells were washed with PBS and harvested by trypsinization. Cell pellets were stored at −80 °C until analysis. Cell pellets were thawed and extracted with 300 μL MeOH/H2O (60:40, v/v) containing DTPA. Ascorbate in the supernatant was oxidized to dehydroascorbic acid (DHA) using Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy) and then reacted with o-phenylenediamine (oPDA) to form the condensation product DHA-oPDA. The rate of appearance of DHA-oPDA was monitored over time using fluorescence as described [24].
Western Blotting, siUROD
siUROD (SI00008162) was purchased from QIAGEN (Germantown, MD); negative control was from Applied Biosystem (Foster City, CA). MIA PaCa-2 cells in 60 mm dishes were treated with siNegative or siUROD (10 nM) with Lipofectamine 2000 (Invitrogen) as specified by the manufacturer. Twenty-four hours after transfection, cell protein extracts were harvested and prepared for immunoblotting as previously described. PVDF membranes were probed with anti-UROD antibody (1:1000 dilution; Thermo Scientific), anti-GAPDH (1:3000 dilution, Millipore), followed by secondary antibodies conjugated to horseradish peroxidase (1:25000 dilution, Millipore).
Results
Iron chelators can protect cells from ascorbate-induced cytotoxicity
The oxidation of ascorbate in aqueous solution, cell culture media, or in extracellular fluid is primarily dependent on the presence of catalytic metal ions [5, 6, 25]. To investigate the role of iron in ascorbate-induced cytotoxicity, cells were exposed to metal chelators DFO (75 μM) or DPD (50 μM) in HBSS for 1 h followed by addition of 2 mM of ascorbate in DMEM-10% FBS for 1 h (Figure 1A). There was no difference in clonogenic survival for cells treated with either DFO (41 ± 3 %) or DPD (40 ± 1 %), compared to control (42 ± 6 %). However, upon exposure to ascorbate (2 mM) in the absence of chelators only 3 ± 1% survived as assessed by clonogenic assays; however, in cells treated with ascorbate in the presence of these chelators, the plating efficiency was around 12%, indicating that metal chelating agents can protect cells from ascorbate-induced cytotoxicity.
Figure 1A. Iron chelators can protect cells from the toxicity induced by ascorbate oxidation as seen by clonogenic survival.
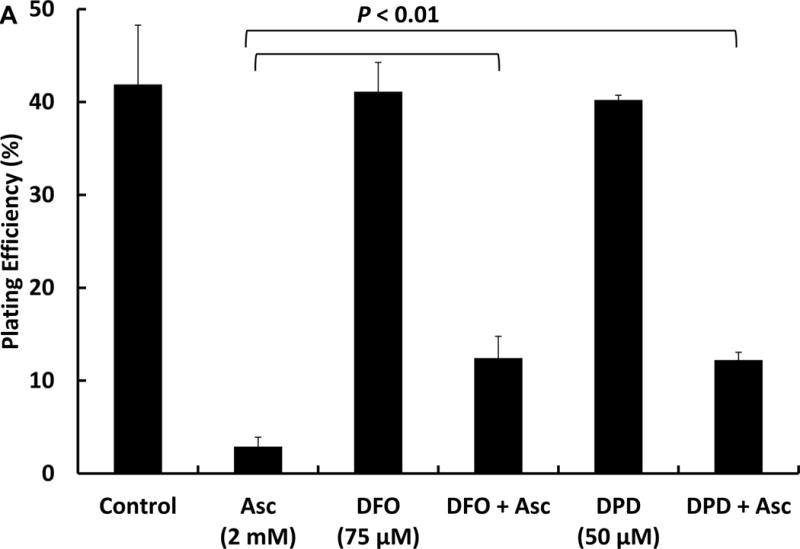
MIA PaCa-2 cells in 60 mm culture dishes were treated with 75 μM of DFO or 50 μM of DPD for 1 h in HBSS followed by ascorbate (2 mM) for 1 h in DMEM-10% FBS. After treatment, cells were trypsinized and seeded in 6-well plates. Colonies were allowed to form for 10–14 days at 37 °C. Colonies were fixed with 70% ethanol and stained with Coomasie blue. Colonies with more than 50 cells were counted.
Increasing intracellular iron enhanced extracellular ascorbate-induced cytotoxicity
Extracellular iron can increase the rate of ascorbate oxidation and thereby increase the flux of H2O2, which can damage cells. Paradoxically, extracellular iron can in some circumstances protect cells from H2O2-induced toxicity [20, 21, 26, 27] by initiating the Fenton reaction outside the cell; thus H2O2 is removed and the highly oxidizing hydroxyl radical reacts with media components, leading to minimal damage to cells. In order to investigate the role of intracellular labile iron, we used the iron chelator 8-hydroxyquinoline (HQ). This lipophilic ligand facilitates the entry of iron into cells, thereby increasing the labile iron pool without inducing the iron storage protein ferritin [28, 29]. 8-HQ alone (10 μM) did not cause cytotoxicity (n=3) to MIA PaCa-2 cells. Cells incubated with Fe(HQ)2 (5 μM in Fe3+ and 10 μM HQ, for 1 h in HBSS) had decreased clonogenic survival, 27 ± 2 % compared to controls (37 ± 3 %), Figure 1B. Ascorbate treatment alone decreased clonogenic survival to 13 ± 2 %; this decreased to 8 ± 1 % with the addition of Fe(HQ)2 (5 μM). Increasing the Fe(HQ)2 (10 μM) concentration further enhanced ascorbate induced cytotoxicity. These results suggest that transient increases in intracellular labile iron can enhance ascorbate-induced cytotoxicity.
Figure 1B. Iron supplementation increases the cytotoxicity of extracellular ascorbate.
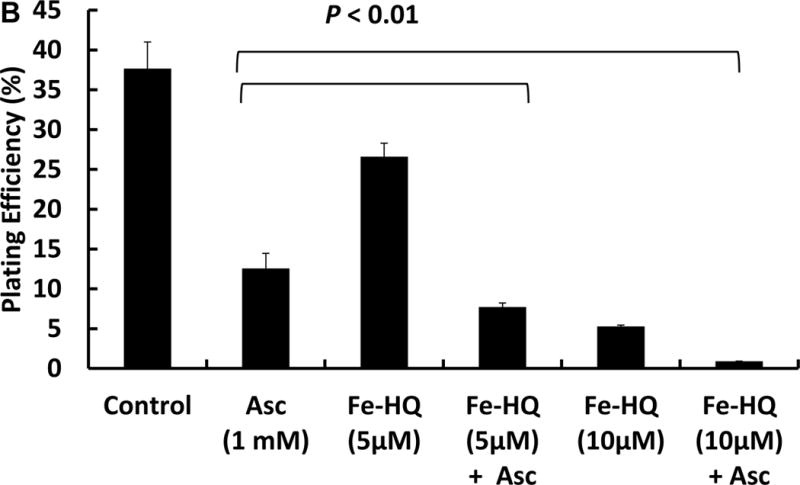
MIA PaCa-2 cells were treated with Fe(HQ)2 (5 and 10 μM) for 1 h in HBSS, followed by incubation with 1 mM ascorbate for 1 h in DMEM-10% FBS. Then the cells were trypsinized and seeded in 6-well plates. Clonogenic survival was determined 10–14 days later.
Increasing intracellular ascorbate and iron does not enhance cytotoxicity
Cultured cells generally lack ascorbate [24, 30]. To increase intracellular ascorbate in MIA PaCa-2 cells without exposing the cells to an extracellular flux of H2O2, 100 μM of ascorbate-2-phosphate (Asc-2P; this form of ascorbate does not oxidize to produce H2O2) was added to the media for 24 h, leading to intracellular ascorbate level of 0.5 mM, similar to observations in [24]. Cells were then exposed to 1, 2.5, or 5 μM of Fe(HQ)2 to increase intracellular iron. Although increasing iron alone decreased clonogenic survival of MIA PaCa-2 cells, the combination of increased intracellular ascorbate and iron did not further reduce clonogenic survival (Figure 1C). These results indicate that the flux of H2O2 from the oxidation of intracellular ascorbate is very low due to the small volume of the cytoplasm (≈2.0 pL [31]), compared to the flux achievable when ascorbate is in the cell culture media (for example, 3 × 105 cells (total intracellular volume ≈6 × 10−4 mL) in 3.0 mL media).
Figure 1C. Increasing intracellular ascorbate in combination with intracellular iron does not enhance cytotoxicity.
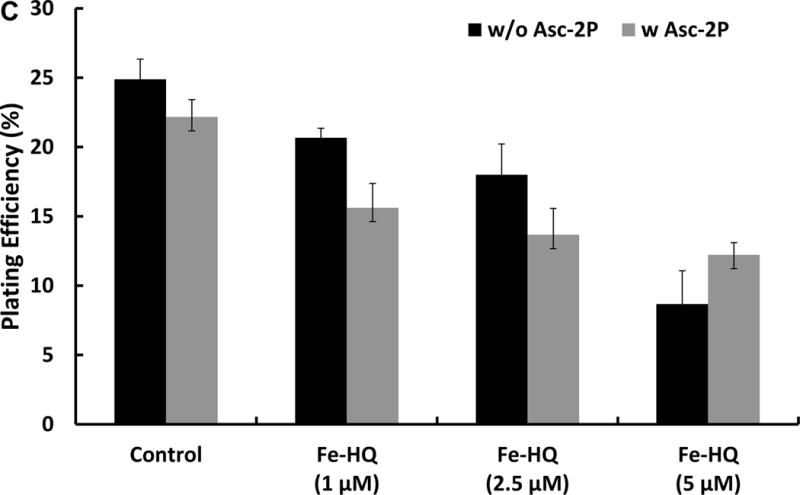
MIA PaCa-2 cells were treated with 100 μM of ascorbate-2-phosphate (Asc-2P) for 24 h, followed by Fe(HQ)2 for 30 min in HBSS. After treatment, cells were trypsinized and seeded into 6-well plates. There are no statistical differences in clonogenic survival when Asc-2P, which increases intracellular ascorbate, is combined with Fe(HQ)2.
Altering intracellular iron concentrations changes PG SK fluorescence
To verify that 8-hydroxyquinoline, a lipophilic iron ligand, facilitated the entry of iron into the cells, MIA PaCa-2 cells grown in 35 mm glass-bottomed dishes were loaded with 5 μM PG SK for 10 min in HBSS at 37 °C in the dark. Once inside the cell, the diacetate group of PG SK is cleaved by intracellular esterases, PG SK then chelates intracellular labile iron resulting in the quenching of its fluorescence. Real time changes in fluorescence from living cells stained with PG SK are shown in Figure 2. Figure 2A shows the baseline fluorescence of cells stained with PG SK; this fluorescence is quenched within minutes upon the addition of Fe(HQ)2 mixture, Figure 2B. The addition of the membrane permeable iron chelator DPD partially restored the quenched fluorescence, Figure 2C & 2D. Figure 2D shows the time frame for the changes in fluorescence intensity from individual cells.
Figure 2. Intracellular fluorescence of PG SK was quenched by Fe(HQ)2 and reversed by membrane permeable chelator DPD in MIA PaCa-2 cells.
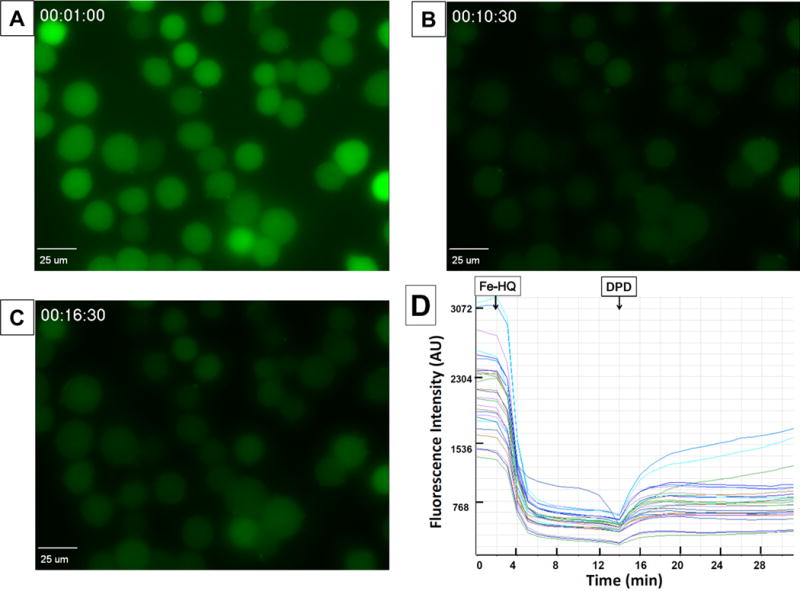
Cells were loaded with 5 μM PG SK for 10 min in HBSS at 37°C. (A) Baseline fluorescence image; (B) after the addition of 10 μM iron as Fe(HQ)2; (C) with the addition of 500 μM of the Fe2+ chelating agent DPD; (D) fluorescence intensity of individual cells.
Changes in intracellular iron content were confirmed by flow cytometry. MIA PaCa-2 cells in 60 mm dishes were loaded with 5 μM PG SK for 20 min in HBSS at 37 °C in the dark. Then cells were treated with either 5 or 10 μM Fe(HQ)2 or 500 μM DPD. After washing with HBSS to remove excess dye, cells were harvested by trypsinization and resuspended in 500 μL of HBSS. An overlay of the fluorescence signals showed that increasing intracellular labile iron with Fe(HQ)2 decreased the intensity of the fluorescence observed; DPD increased the fluorescence observed from PG SK indicating a decrease in the intracellular labile iron, Figure 3.
Figure 3. Flow cytometry measurement of intracellular iron in MIA PaCa-2 cells.
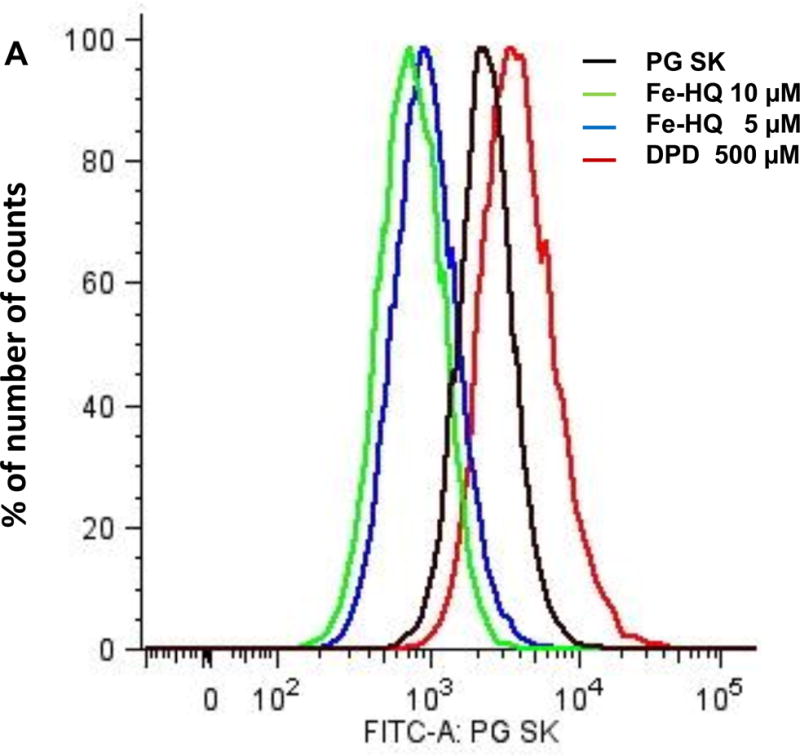
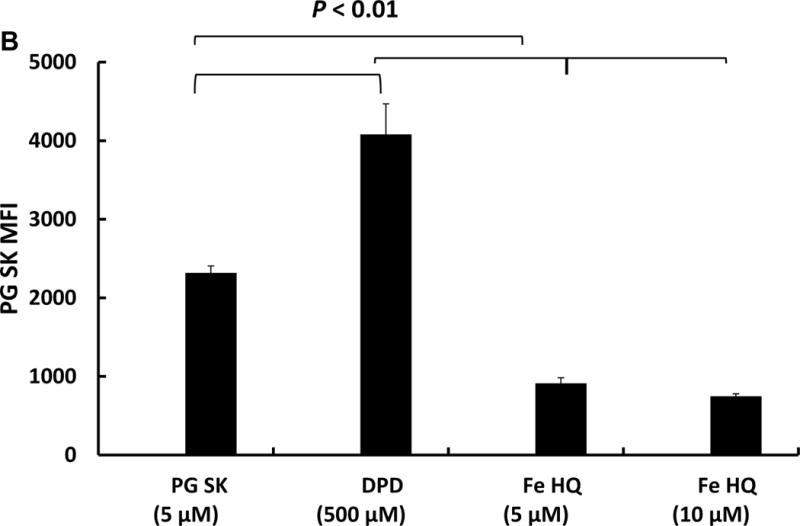
MIA PaCa-2 cells were treated with PG SK (5 μM) for 10 min in HBSS. After treatment, cells were washed with HBSS twice to remove excess dye. Then the cells were treated with Fe(HQ)2 (5 and 10 μM) or DPD (500 μM) for 30 min. Following a wash with PBS, cells were harvested and resuspended in 500 μL of HBSS. (A) Fluorescence profile of each sample; (B) Quantitive results of flow cytometry demonstrating decreases in intracellular labile iron with DPD and increases in intracellular iron with Fe(HQ)2.
Basal level of intracellular labile iron may contribute to the sensitivity of cells to ascorbate
H6c7, MIA PaCa-2, AsPC-1 and Panc-1 cells were treated with 1 mM ascorbate in DMEM-10% FBS for 1 h, then cells were trypsinized for clonogenic assays. Figure 4A shows the cancer cell lines MIA PaCa-2, AsPC-1 and Panc-1 are more sensitive to ascorbate treatment than H6c7 cells. However, when we compared the basal labile iron content of cancer cells to normal cell H6c7, only MIA PaCa-2 cells have significantly higher levels of labile iron (Figure 4B). There were no significant differences among H6c7, Panc-1and AsPC-1 cells. These results indicate that intracellular labile iron only partially contributes to the sensitivity to ascorbate.
Figure 4A. Effects of intracellular iron on cytotoxicity.
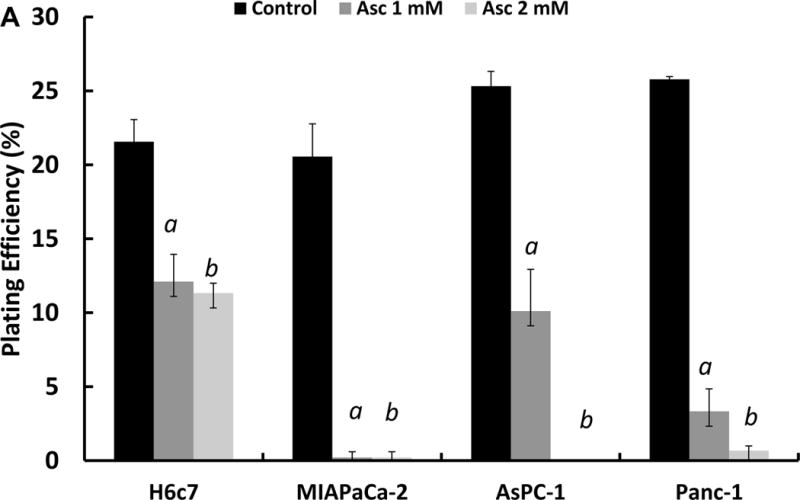
H6c7, MIA PaCa-2, AsPC-1 and Panc-1 cells were treated with 0 and 1 mM of ascorbate for 1 h in 3.0 mL DMEM containing 10% FBS. After treatment, cells were trypsinized and seeded into 6-well plates at 300 cells/well with 4.0 mL KSFM media, irradiated MIA PaCa-2 (30 Gy) were seeded at 5 × 104 /mL as feeder cells to help H6c7 cells form colonies. aP < 0.01 verses control.
Figure 4B.
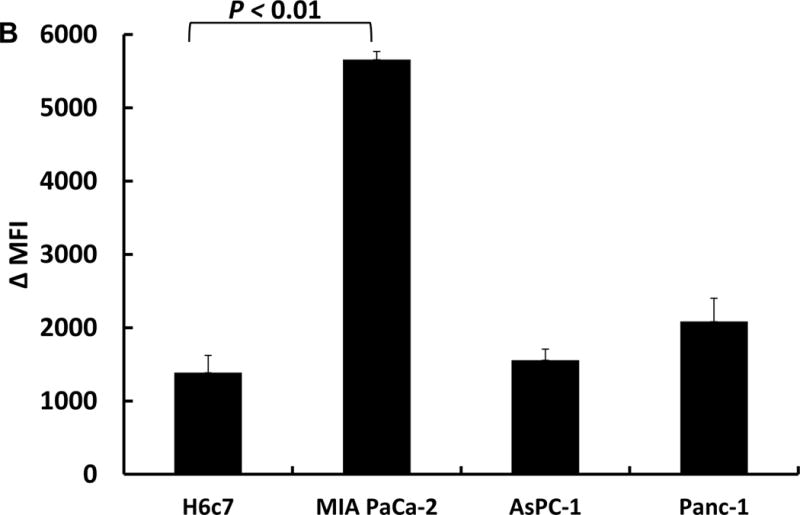
Flow cytometry measurement of intracellular iron in H6c7, MIA PaCa-2, AsPC-1 and Panc-1 cells. Cells (3 × 105) were seeded into 60 mm dishes for 48 h. Cells were treated with PG SK (5 μM) for 10 min in HBSS. After treatment, cells were washed with HBSS to remove excess dye. The cells were treated with DPD (500 μM) for 30 min or left untreated as control. Then cells were harvested and resuspended in 500 μL of HBSS. The difference in the MFI (ΔMFI) of cells with and without DPD treatment was used to estimate LIP.
siUROD enhances ascorbate-induced cytotoxicity
Uroporphyrinogen decarboxylase (UROD) is the fifth enzyme of the heme biosynthetic pathway. It catalyzes the decarboxylation of uroporphyrinogen to coproporphyrinogen. It has been reported that down regulation of UROD results in accumulation of intracellular iron; this iron sensitizes head and neck cancer to radiotherapy in vitro and in vivo [32]. The results presented in Figure 5A demonstrate a significant suppression of UROD expression 24 h after transfection of siRNA (siUROD). Clonogenic results in Figure 5B show that UROD knockdown enhances the cytotoxicity of 1 mM ascorbate. Pre-treatment of cells with δ-aminolevulinic acid (500 μM) for 4 h to induce porphyrin synthesis, followed by ascorbate (1 mM) did not enhance the cytotoxicity (data not shown), indicating that the combined effects of siUROD and ascorbate was not due to porphyrin accumulation. However, we were not able to detect the decrease in the LIP using PG SK since knockdown of UROD causes the accumulation of porphyrins (Ex ≈400 nm, Em ≈635 nm) that interferes with monitoring the fluorescence of PG SK (Ex 488 nm, Em 530 nm).
Figure 5. siUROD enhances ascorbate–induced cytotoxicity.
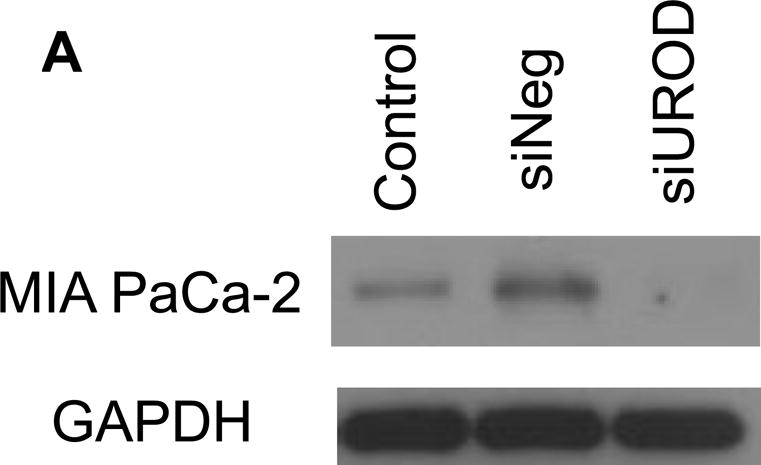
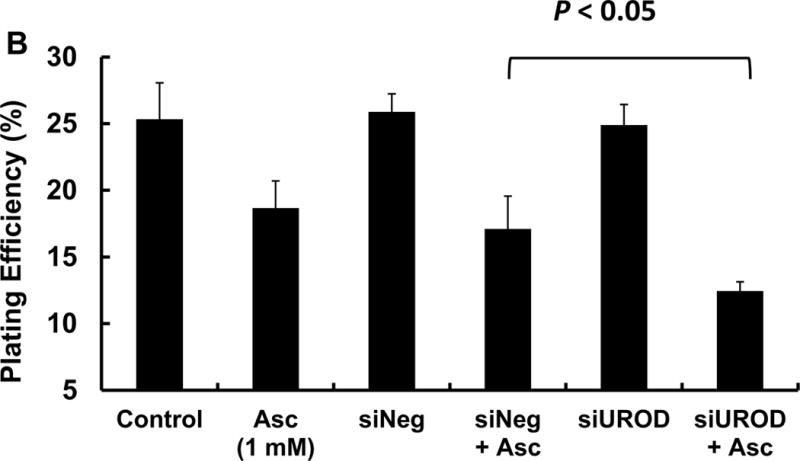
MIA PaCa-2 cells in 60 mm dishes were treated with siNeg or siUROD (40 pmol) for 24 h, followed by ascorbate (1 mM) for 1 h. After treatment, cells were trypsinized and seeded into 6-well plates at 300 cell/well with 4.0 mL growth media. (A) UROD knockdown was verified by immunoblotting; (B) Clonogenic assay results.
Discussion
The effects of iron on ascorbate oxidation-induced cytotoxicity are not straightforward. Chelating the adventitious catalytic metals with DTPA or DFO slows ascorbate oxidation in phosphate buffer at neutral pH [5]. However, pre-incubation of ascorbate (500 μM) with apo-Tf (50 g/mL) or 500 μM of DFO, ferrozine or DTPA then incubating human dermal fibroblasts (HDFs) with these solutions did not prevent DNA damage induced by ascorbate oxidation [33]. In our experimental settings, we found that pretreatment of human pancreatic cancer cells MIA PaCa-2 or AsPC-1 to DFO or DPD followed by exposure of these cells to ascorbate partially prevented cell death by ascorbate oxidation (Figure 1A). When adding chelators to ascorbate solution, chelators only slow the rate of ascorbate oxidation without significantly decreasing the corresponding accumulation of H2O2 after a fixed amount of time. For cell permeable chelators such as DPD, which permeate cells within minutes (Figure 2D) and chelates intracellular iron, we and others have seen the protective effects against ascorbate-induced cytotoxicity [37]. Although the ability of DFO to access cytosolic iron is poor with a short incubation time, it can readily chelate endosomal iron [34]; this maybe the reason it protects the cells from ascorbate oxidation after only a 1 h incubation.
Mammalian cells acquire iron through transferrin (Tf)-dependent and Tf-independent systems [35, 36]. Within the cell, iron can reside in endocytotic vesicles before being released into the cytosol. Most intracellular iron is bound to ferritin as storage iron or associated with proteins as prosthetic groups [37]. Only a small fraction (<5%) of total cellular iron exits as labile iron that can potentially participate redox cycling and can be scavenged by permeant chelators [38]. To increase intracellular labile iron, we loaded the cells with iron-hydroxyquinoline (Fe(HQ)2), which can increase intracellular iron within minutes due to the lipophilic property of HQ (Figure 2). The higher level of intracellular labile iron increases the toxicity of the H2O2 generated by the oxidation of extracellular ascorbate (Figure 1B), but did not appear to increase intracellular ascorbate oxidation-induced cytotoxicity (Figure 1C).
Calcein acetoxymethyl ester (calcein AM) and PG SK are the two most widely used iron sensors for detecting the labile iron pool [38, 39]. The cell permeable calcein AM is converted to a green-fluorescent calcein after acetoxymethyl ester hydrolysis by intracellular esterases, yielding a fluorescein conjugated with an EDTA-like moiety; the stoichiometry of calcein:iron is 1:1 or 1:2 [39, 40]. PG SK is a phenanthroline-based fluorescence probe for Fe2+; it binds iron with 3:1 stoichiometry. PG SK is more sensitive for detecting intracellular labile iron than calcein. Loading the cells using a final extracellular working concentration of 5 μM PG SK for 10 min is sufficient to give optimum signals without apparent toxic effects to cells. Changes in intracellular labile iron in pancreatic cancer cells were detected in real time using PG SK metal sensor (Figure 2 & 3).
Under normal physiological conditions, the majority of the iron in the circulation is tightly bound to transferrin [41], which is essentially catalytically inactive [6]. Extracellular catalytic iron, in general iron not bound to transferrin, can contribute to the oxidation of ascorbate. oxidation in the media, thereby modulating the flux of H2O2, i.e. mol cell s−1 that diffuses into cells. A constant flux of H2O2 causes long-lasting damage to cells compared to a bolus addition. It has been shown that the bolus addition of 200 μM of H2O2 to cells in DMEM-F12 for 80 min leads fewer DNA single strand breaks than shorter exposure [42], indicating that cells are able to repair DNA damage after the H2O2 is consumed. On the other hand, ascorbate oxidation by catalytic iron produces continuing flux of H2O2 that could compromise the ability to repair DNA, producing long-lasting DNA damage that can eventually lead to cell death. Our data support the proposal that increasing intracellular labile iron enhances extracellular ascorbate cytotoxicity by “activating” intracellular H2O2, producing powerful oxidants such as ferryl iron and HO•, that in turn damage biomolecules, e.g. DNA.
Studies have shown that breast cancer cells have higher concentrations of LIP than in normal breast epithelial cells [43]; an increase of the intracellular LIP was observed for cutaneous T-cell lymphoma or T cell from Sézary patients but not normal T cells [44]. Furthermore, cells transformed with oncogenic RAS have increased iron content relative to their normal cell counterparts [45]. To clarify how basal levels of LIP affect ascorbate cytotoxicity, we compared immortalized pancreatic epithelial cells H6c7 to MIA PaCa-2, Panc-1, and AsPC-1 cells. MIA PaCa-2, Panc-1, and AsPC-1 cells are more sensitive to ascorbate treatment than H6c7 cells. However, when we compared the intracellular labile iron levels among these cell lines, only MIA PaCa-2 cells appeared to have higher basal levels of labile iron than H6c7. There were no significant differences in the LIP among H6c7, Panc-1 and AsPC-1 cells. Thus, intracellular labile iron level is only one of many factors that affect cellular sensitivity to ascorbate-induced cytotoxicity [37, 46]; the cellular antioxidant defense system may also play a significant role [8, 47, 48]. However, increasing the level of extracellular as well as intracellular catalytically active labile iron in tumor tissue may enhance the effectiveness of pharmacological ascorbate as an adjuvant in cancer therapy.
Supplementary Material
AsPC-1 cells were treated with DFO (75 μM) or DPD (50 μM) for 1 h in HBSS, followed by ascorbate (2 mM) for 1 h in DMEM-10% FBS. After treatment, cells were trypsinized and seeded into 6-well plates at 300 cell/well with 4.0 mL growth media.
AsPC-1 cells were treated with Fe(HQ)2 (10 and 20 μM) for 1 h in HBSS, followed by ascorbate (0.5 mM) for 1 h in DMEM-10% FBS. Cells were trypsinized and seeded into 6-well plates at 300 cell/well with 4.0 mL growth media.
Highlights.
Catalytic metals are central in determining the rate of oxidation of ascorbate and production of H2O2
Extracellular catalytic iron is vital to the cellular toxicity of pharmacological ascorbate
Increasing intracellular labile iron enhances the toxicity of pharmacological ascorbate
Acknowledgments
Supported by NIH grants U01 CA166800, R01 CA169046, R01 GM073929, and the Medical Research Service, Department of Veterans Affairs 1I01BX001318-01A2. Core facilities were supported in part by NIH P30 CA086862.
The University of Iowa ESR Facility provided invaluable support. Data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine / Holden Comprehensive Cancer Center core research facility at the University of Iowa. The Facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. The content is solely the responsibility of the authors and does not represent views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M. Ascorbic acid at pharmacologic concentrations selectively kills cancer cells: ascorbic acid as a pro-drug for hydrogen peroxide delivery to tissues. Proc Natl Acad Sci U S A. 2005;102:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, Choyke PL, Pooput C, Kirk KL, Buettner GR, Levine M. Ascorbic acid in pharmacologic concentrations: a pro-drug for selective delivery of ascorbate radical and hydrogen peroxide to extracellular fluid in vivo. Proc Natl Acad Sci USA. 2007;104:8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, Taghiyev AF, Du C, Knudson CM, Cullen JJ. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16(2):509–520. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rawal M, Schroeder SS, Wagner BA, Cushing CM, Welsh J, Button AM, Du J, Sibenaller ZA, Buettner GR, Cullen JJ. Manganoporhyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013;73:1–10. doi: 10.1158/0008-5472.CAN-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buettner GR. In the absence of catalytic metals, ascorbate does not autoxidize at pH 7: Ascorbate as a test for catalytic metals. J Biochem Biophys Meth. 1988;16:27–40. doi: 10.1016/0165-022x(88)90100-5. [DOI] [PubMed] [Google Scholar]
- 6.Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate, and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- 7.Clément MV1, Ramalingam J, Long LH, Halliwell B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid Redox Signal. 2001;3(1):157–163. doi: 10.1089/152308601750100687. [DOI] [PubMed] [Google Scholar]
- 8.Wagner BA, Witmer JR, van ‘t Erve TJ, Buettner GR. An assay for the rate of removal of extracellular hydrogen peroxide by cells. Redox Biology. 2013;1:210–217. doi: 10.1016/j.redox.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 10.Blakely WF, Fuciarelli AF, Wegher BJ, Dizdaroglu M. Hydrogen peroxide-induced base damage in deoxyribonucleic acid. Radiat Res. 1990;121(3):338–343. [PubMed] [Google Scholar]
- 11.Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DA. Fluorescein diacetate is superior for detecting intracellular oxidants: comparison to 2′7′ dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′7′-dichlorodihydrofluorescein diacetate and dihydrorhodamine 123. Free Radic Biol Med. 1999;27:146–159. doi: 10.1016/s0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 12.Samuni A, Aronovitch J, Godinger D, Chevion M, Czapski G. On the cytotoxicity of vitamin C and metal ions. A site-specific Fenton mechanism. Eur J Biochem. 1983;1371–2:119–124. doi: 10.1111/j.1432-1033.1983.tb07804.x. [DOI] [PubMed] [Google Scholar]
- 13.Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. 1997;272(31):19095–19098. doi: 10.1074/jbc.272.31.19095. [DOI] [PubMed] [Google Scholar]
- 14.Buettner GR. In the absence of catalytic metals, ascorbate does not autoxidize at pH 7: Ascorbate as a test for catalytic metals. J Biochem Biophys Meth. 1988;16:27–40. doi: 10.1016/0165-022x(88)90100-5. [DOI] [PubMed] [Google Scholar]
- 15.Barbouti A, Doulias PT, Zhu BZ, Frei B, Galaris D. Intracellular iron, but not copper, plays a critical role in hydrogen peroxide-induced DNA damage. Free Radic Biol Med. 2001;31(4):490–498. doi: 10.1016/s0891-5849(01)00608-6. [DOI] [PubMed] [Google Scholar]
- 16.Chen YY, Ho KP, Xia Q, Qian ZM. Hydrogen peroxide enhances iron-induced injury in isolated heart and ventricular cardiomyocyte in rats. Mol Cell Biochem. 2002;231:61–68. doi: 10.1023/a:1014484907291. [DOI] [PubMed] [Google Scholar]
- 17.Jonas SK, Riley PA. Modification of the in vitro cytotoxicity of hydrogen peroxide by iron complexes. Free Radic Res Comm. 1992;17:407–419. doi: 10.3109/10715769209083145. [DOI] [PubMed] [Google Scholar]
- 18.Wardman P, Candeias LP. Fenton chemistry: an introduction. Radiat Res. 1996;145(5):523–531. [PubMed] [Google Scholar]
- 19.Roots R, Okada S. Estimation of life times and diffusion distances of radicals involved in X-ray induced strand breaks of killing of mammalian cells. Radiat Res. 1975;64:306–320. [PubMed] [Google Scholar]
- 20.Hempel SL, Buettner GR, Wessels DA, Galvan GM, O’Malley Y. Extracellular iron(II) can protect cells from hydrogen peroxide. Arch Biochem Biophys. 1996;330:401–408. doi: 10.1006/abbi.1996.0268. [DOI] [PubMed] [Google Scholar]
- 21.Qian SY, Buettner GR. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: An electron paramagnetic resonance spin trapping study. Free Radic Biol Med. 1999;26:1447–1456. doi: 10.1016/s0891-5849(99)00002-7. [DOI] [PubMed] [Google Scholar]
- 22.Qian J, Niu J, Li M, Chiao PJ, Tsao M-S. In vitro Modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–5053. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 23.Prus E, Fibach E. Flow cytometry measurement of the labile iron pool in human hematopoietic cells. Cytometry Part A. 2008;73A:22–27. doi: 10.1002/cyto.a.20491. [DOI] [PubMed] [Google Scholar]
- 24.Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Anal Biochem. 2007;365:31–39. doi: 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song Y, Buettner GR. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic Biol Med. 2010;49:919–962. doi: 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hata Y, Kawabe T, Hiraishi H, Ota S, Terano A, Ivey KJ. Hydrogen peroxide-mediated cytotoxicity to cultured colonic epithelial cells. Life Sci. 1997;60(24):2221–2230. doi: 10.1016/s0024-3205(97)00237-3. [DOI] [PubMed] [Google Scholar]
- 27.Mojić M, Bogdanović Pristov J, Maksimović-Ivanić D, Jones DR, Stanić M, Mijatović S, Spasojević I. Extracellular iron diminishes anticancer effects of vitamin C: an in vitro study. Sci Rep. 2014;4:5955. doi: 10.1038/srep05955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simpson RJ, Peters TJ. Studies of Fe3+ transport across isolated intestinal brush-border membrane of the mouse. Biochim Biophys Acta. 1984;772(2):220–226. doi: 10.1016/0005-2736(84)90047-6. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan J, Jordan I, Sturrock A. Regulation of the transferrin-independent iron transport system in cultural cells. J Biol Chem. 1991;266:2997–3004. [PubMed] [Google Scholar]
- 30.Chepda T, Cadau M, Girin P, Frey J, Chamson A. Monitoring of ascorbate at a constant rate in cell culture: effect on cell growth. In Vitro Cell Dev Biol Anim. 2001;37(1):26–30. doi: 10.1290/1071-2690(2001)037<0026:MOAAAC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 31.Wagner BA, Venkataraman S, Buettner GR. The rate of oxygen utilization by cells. Free Radic Biol Med. 2011;51:700–712. doi: 10.1016/j.freeradbiomed.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito E, Yue S, Moriyama EH, Hui AB, Kim I, Shi W, Alajez NM, Bhogal N, Li G, Datti A, Schimmer AD, Wilson BC, Liu PP, Durocher D, Neel BG, O’Sullivan B, Cummings B, Bristow R, Wrana J, Liu FF. Uroporphyrinogen decarboxylase is a radiosensitizing target for head and neck cancer. Sci Transl Med. 2011;3:67ra7. doi: 10.1126/scitranslmed.3001922. [DOI] [PubMed] [Google Scholar]
- 33.Duarte TL, Almeida GM, Johns GDD. Investigation of the role of extracellular H2O2 and transition metal ions in the genotoxic action of ascorbate in cell culture models. Toxicol Lett. 2007;170:57–65. doi: 10.1016/j.toxlet.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Glickstein H1, El RB, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106:3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 35.De Silva DM, Askwith CC, Kaplan J. Molecular mechanisms of iron uptake in eukaryotes. Physiol Rev. 1996;76:31–47. doi: 10.1152/physrev.1996.76.1.31. [DOI] [PubMed] [Google Scholar]
- 36.Lane DJ, Richardson DR. The active role of vitamin C in mammalian iron metabolism: much more than just enhanced iron absorption! Free Radic Biol Med. 2014;75:69–83. doi: 10.1016/j.freeradbiomed.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Brissot P, Ropert M, Le Lan C, Loréal O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820:403–410. doi: 10.1016/j.bbagen.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 38.Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement and participation in cellular process. Free Radic Biol Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 39.Petrat F, Rauen U, de Groot H. Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, Phen Green SK. Hepatology. 1999;29:1171–1179. doi: 10.1002/hep.510290435. [DOI] [PubMed] [Google Scholar]
- 40.Thomas F, Serratrice G, Béguin C, Aman ES, Pierre JL, Fontecave M, Laulhère JP. Calcein as a fluorescent probe for ferric iron. Application to iron nutrition in plant cells. J Biol Chem. 1999;274:13375–13383. doi: 10.1074/jbc.274.19.13375. [DOI] [PubMed] [Google Scholar]
- 41.De Domenico I, McVey Ward D, Kaplan J. Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Biol. 2008;9:72–81. doi: 10.1038/nrm2295. [DOI] [PubMed] [Google Scholar]
- 42.Giandomenico AR, Cerniglia GE, Biaglow JE, Stevens CW, Koch CJ. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic Biol Med. 1997;23:426–434. doi: 10.1016/s0891-5849(97)00113-5. [DOI] [PubMed] [Google Scholar]
- 43.Pinnix ZK, Miller LD, Wang W, D’Agostino R, Jr, Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, Torti FM. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43ra56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiessling MK, Klemke CD, Kaminski MM, Galani IE, Krammer PH, Gülow K. Inhibition of constitutively activated nuclear factor – κB induces reactive oxygen species-and iron-dependent cell death in cutanous T-cell lymphoma. Cancer Res. 2009;69:2365–2374. doi: 10.1158/0008-5472.CAN-08-3221. [DOI] [PubMed] [Google Scholar]
- 45.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, non-apoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic Biol Med. 2009;47:32–40. doi: 10.1016/j.freeradbiomed.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 47.Olney KE, Du J, van ‘t Erve TJ, Witmer JR, Sibenaller ZA, Wagner BA, Buettner GR, Cullen JJ. Inhibitors of hydroperoxide metabolism enhance ascorbate-induced cytotoxicity. Free Radic Res. 2013;47(3):154–163. doi: 10.3109/10715762.2012.755263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al-Qenaei A, Yiakouvaki A, Reelfs O, Santambrogio P, Levi S, Hall ND, Tyrrell RM, Pourzand C. Role of intracellular labile iron, ferritin, and antioxidant defence in resistance of chronically adapted Jurkat T cells to hydrogen peroxide. Free Radic Biol Med. 2014;68:87–100. doi: 10.1016/j.freeradbiomed.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AsPC-1 cells were treated with DFO (75 μM) or DPD (50 μM) for 1 h in HBSS, followed by ascorbate (2 mM) for 1 h in DMEM-10% FBS. After treatment, cells were trypsinized and seeded into 6-well plates at 300 cell/well with 4.0 mL growth media.
AsPC-1 cells were treated with Fe(HQ)2 (10 and 20 μM) for 1 h in HBSS, followed by ascorbate (0.5 mM) for 1 h in DMEM-10% FBS. Cells were trypsinized and seeded into 6-well plates at 300 cell/well with 4.0 mL growth media.