

Abstract
Delayed neuronal death associated with stroke has been increasingly linked to the immune response to the injury. Splenectomy prior to middle cerebral artery occlusion (MCAO) is neuroprotective and significantly reduces neuroinflammation. The present study investigated whether splenic signaling occurs through interferon gamma (IFNγ). IFNγ was elevated early in spleens but later in the brains of rats following MCAO. Splenectomy decreased the amount of IFNγ in the infarct post-MCAO. Systemic administration of recombinant IFNγ abolished the protective effects of splenectomy with a concurrent increase in INFγ expression in the brain. These results suggest a role for spleen-derived IFNγ in stroke pathology.
Keywords: Brain ischemia, Cytokine, Microglia/macrophages, MCAO
Introduction
Current clinical and animal research has shown a complex interplay between the peripheral immune system and the progression of stroke-induced neurodegeneration. The brain communicates with the immune system largely via direct innervation of the lymphoid tissues and humoral control provided by the hypothalamic-pituitary-adrenal axis (Chrousos 1995).
The spleen is a mediator of the immune response to ischemic injury in all organ systems examined. Splenectomy reduces the ischemic-induced immune response in the liver (Okuaki et al. 1996), gastrointestinal system (Savas et al. 2003), kidney (Jiang et al. 2007) and brain (Ajmo et al. 2008). These reports indicate that the presence of the spleen is necessary for promotion of the inflammatory response to ischemic injury which is responsible for delayed cellular death. Splenectomy two weeks prior to middle cerebral artery occlusion (MCAO) in the rat significantly reduces infarct volume with a concomitant decrease in the number of immune cells within the infarct (Ajmo et al. 2008). The inflammatory signal from the spleen to the ischemic brain or other organs has yet to be identified.
Many studies have attempted to decipher the immune signature for an inflammatory response to stroke (Offner et al. 2006; Liesz et al. 2009a; Ren et al. 2010; Becker et al. 2005). Many different gene knockout models of inflammatory cytokines have been characterized in the field of stroke showing various degrees of increased neuronal death or protection (Lucas et al. 2006; Boutin et al. 2001). One study reports that the deletion of the interferon gamma (IFNγ) gene decreases brain damage after MCAO (Yilmaz et al. 2006). Moreover, when IFNγ neutralizing antibodies are infused intraventricularly three days post-MCAO this protects the brain from stroke induced injury (Liesz et al. 2009b). Also, mice with increased levels of brain IFNγ as a result of over-expression in oligodendrocytes (OL), have increased infarcts compared to wild-type mice (Lambertsen et al. 2004). IFNγ is associated with the Th1 inflammatory response by activating cells of the monocytic lineage, microglia and macrophages. Since activation of microglia/macrophages is partly responsible for the delayed cellular damage after ischemic insult, this cytokine could play a role in the splenic response by exacerbating the inflammation associated with ischemic injury.
In the present study, we examined the expression of IFNγ after MCAO. We discovered that splenectomy reduced IFNγ expression in the brain after MCAO and that systemic administration of IFNγ reversed the protective effects of splenectomy. These findings indicate that IFNγ may be one of the inflammatory signals originating from the spleen causing a delayed inflammatory response in the ischemic brain.
Materials and methods
Animal care
All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals with a protocol approved by the Institutional Animal Care and Use Committee at the University of South Florida. Male Sprague–Dawley rats (300–350 g) were used for the in vivo experiments. Postnatal day 3 (P3) rat pups from untimed pregnant female rats were used for in vitro primary oligodendrocyte cell culture experiments and prenatal day 18 (E18) rat embryos from timed pregnant females were used for in vitro primary neuron cell culture experiments. All rats were purchased from Harlan Labs (Indianapolis, IN), maintained on a 12 h light/dark cycle (6 am–6 pm) and given access to food and water ad libitum.
Splenectomy
Splenectomies were performed two weeks prior to MCAO by making a midline skin incision at the caudal terminus of the 13th rib on the anatomical left. The abdominal wall was opened along the midline and the spleen was externalized through the incision with blunt forceps. The splenic blood vessels were ligated and the spleen was removed. The incision was then closed with sutures, first closing the abdominal cavity and then the skin incision. Sham operations were also performed where the spleen was exteriorized and then reinserted into the cavity.
Laser doppler blood flow measurement
Laser Doppler was used to monitor blood perfusion (Moor Instruments Ltd, Devon, England). A hole was drilled into the right parietal bone (1 mm posterior and 4 mm lateral from Bregma), and a guide screw was set. The probe was inserted into the guide screw, and the tip of the probe was placed against the pial surface of the brain. Rats that did not show ≥60% reduction in perfusion during MCAO were excluded from the study (Ajmo et al. 2006; Ajmo et al. 2008; Hall et al. 2009a)
Permanent middle cerebral artery occlusion
MCAO surgery was performed using the intraluminal method originally described by Longa et al. (Longa et al. 1989) and previously reported (Ajmo et al. 2006; Ajmo et al. 2008; Hall et al. 2009a). Briefly, rats were anesthetized, the common carotid artery was separated from the vagus nerve, and blunt dissection was performed to isolate the internal carotid artery (ICA), and the external carotid artery (ECA). A 40 mm monofilament was introduced into the ECA, fed distally into the ICA, and advanced approximately 25 mm through the Circle of Willis to the origin of the middle cerebral artery. The filament was tied off at the internal/external carotid junction to produce permanent occlusion. The incision was then sutured closed and the rat was allowed to wake in a fresh cage. Following recovery, animals were randomly assigned into treatment groups.
Recombinant IFNγ administration
Naïve rats were given increasing doses of rIFNγ until an observable physiological response occurred to determine the optimal rIFNγ dosage. A physiological response to the rIFNγ was determined by the presence of several characteristics: pilo erection, excessive porphrin production, lethargy, and chills or fever. The rats were monitored every 15 min for 2 h following intravenous (i.v.) injections. The dosage of 20 μg was the lowest dosage which elicited a physiological response and was used to determine the effects of IFNγ on neural injury in splenectomized and sham-splenectomized rats. The animals were injected i.v., via the tail vein, at 48 and 72 h post-MCAO with 0.21 ml of either 20 μg (in ddH2O) of recombinant IFNγ (rIFNγ) (Prospec, Rehovot, Israel) or 0.21 ml ddH2O.
Brain extraction and sectioning
The animals were euthanatized with ketamine/xylazine mix, 75 mg/kg and 7.5 mg/kg respectively, intraperitoneal (i.p.) at 3, 24, 48, 51, 72 and 96 h post-MCAO, and perfused transcardially with 0.9% saline followed by 4% paraformaldehyde in phosphate buffer. The brains were harvested, post fixed in 4% paraformaldehyde, and immersed in 20% followed by 30% sucrose in phosphate buffered saline (PBS). Brains were frozen and sliced into 30 μm sections with a cryostat. Coronal brain sections were taken at six points from 1.7 to −3.3 mm from Bregma. Sections were either thaw mounted on glass slides or placed in Walter’s Anti-freeze cryopreservative and stored at −20°C.
Fluoro-Jade staining
Slides were stained with Fluoro-Jade, which labels degenerating neurons. This method was adapted from that originally developed by Schmued et al. (Schmued et al. 1997) and has been described by Duckworth et al. (Duckworth et al. 2005). Slides were dried, placed in 100% ethanol for 3 min, 70% ethanol for 1 min, and then ddH2O for 1 min. Slides were oxidized using a 0.06% KMnO4 solution for 15 min followed by three 1 min rinses with ddH2O. Slides were stained in a 0.001% solution of Fluoro-Jade (Histochem, Jefferson, AR) in 0.1% acetic acid for 30 min. Slides were rinsed 4 times with ddH2O for 3 min, allowed to dry at 45°C for 20 min, cleared with xylene and then cover slipped with DPX mounting medium (Electron Microscopy Sciences, Ft. Washington, PA).
Infarct volume quantification
Fluoro-Jade stained tissue was digitally photographed with Zeiss Axioskop2 (Carl Zeiss INC, Thornwood, NY) microscope controlled by Openlab software (Improvision, Waltham, MA) at a magnification of 1x. Area of neurodegeneration was measured using the NIH ImageJ software. The area of the contralateral side of the brain was also measured and used to compensate for possible edema in the ipsilateral hemisphere. Infarct volumes were then calculated by the total area of ipsilateral staining divided by the total contralateral area for a given animal. Infarct quantification was only done at 96 h post-MCAO because this has been shown to be the time point at which the infarct is stable (Newcomb et al. 2006).
Immunohistochemistry in the brain
The slides were dried at 45°C for 1 h then rinsed with PBS pH 7.4. Endogenous peroxidase activity was extinguished by incubating the slides for 20 min in 3% hydrogen peroxide. Slides were placed in permeabilization buffer containing 10% serum, 3% 1 M lysine, and 0.3% Triton X-100 in PBS for 1 h at room temperature. Next, sections were incubated overnight at 4°C in a primary antibody solution (PBS with 2% serum and 0.3% Triton X-100) in a humidified chamber. Slides were subsequently washed with PBS and incubated with a secondary antibody solution (PBS, 2% serum, 0.3% Triton X-100) for 1 h. For staining with metal-enhanced 3, 3′-diaminobenzidine (DAB) visualization sections were washed in PBS (3×5 min) following secondary antibody solution and incubated in an avidin/biotin/horseradish peroxidase complex (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA) for 1 h at room temperature. Sections were washed in PBS, and DAB (Pierce, Rockford, IL) was used for color development. Slides were washed thoroughly with PBS and dried for 1 h at 45°C then dehydrated, rinsed with xylene and cover slipped using DPX (Electron Microscopy Sciences).
For fluorescence staining, the same procedure was followed up to the incubation with the secondary antibody, though sections were not incubated in hydrogen peroxide. Slides were washed with PBS after secondary incubation and then cover slipped using Vectashield hard set mounting media with DAPI (Vector Laboratories). Slides were protected from light during these steps. Double-labeled immunohistochemistry, for IFNγ and immune cell surface markers, was achieved by co-incubating the slides with primary antibodies raised in two distinct species, followed by co-incubation with secondary antibodies conjugated to distinct fluorophores.
The following primary antibodies were used: goat anti-rat IFNγ (1:200; R&D Systems, Minneapolis, MN), mouse anti-rat CD3 for T cells (1:2,000; BD Biosciences, San Jose, CA), mouse anti-rat CD161 for NK cells (1:1,000; Serotec, Raleigh, NC), mouse anti-rat CD45R for B cells (1:5,000; BD Biosciences), and mouse anti-rat CD11b for microglia/macrophages (1:3,000; Serotec). Horse anti-goat biotinylated antibody (1:300; Vector Laboratories) and Alexa-Fluor® 594 rabbit anti-goat (1:300; Invitrogen, Carlsbad, CA) secondary antibodies were used with the IFNγ antibody. Alexa-Fluor® 488 rabbit anti-mouse (1:300; Invitrogen) secondary was used in conjunction with all other antibodies noted above.
IFNγ Immunohistochemistry in the spleen
Spleens were fixed in 4% paraformaldehyde overnight. The spleens were then placed in a solution of 20% glycerol and 2% dimethyl sulfoxide (DMSO) and embedded in a gelatin matrix using MultiBrain Technology© (NeuroScience Associates, Knoxville, TN). The block of spleens was rapidly frozen in isopentane with crushed dry ice (−70°C). Using a microtome the block was sliced into 25 μm sections. Six consecutive sections were taken and collected in Antigen Preservation solution (50% ethylene glycol, 49% PBS pH 7.0, 1% polyvinyl pyrrolidone). The spleen sections were stained free floating in Tris-buffered saline (TBS) solutions. Endogenous peroxide activity was extinguished by treatment with 3% hydrogen peroxide for 15 min. After washing with TBS sections were incubated for 30 min in permeabilization buffer (TBS with 0.3% TritonX-100 and 10% rabbit serum). Following permeabilization, slides were incubated overnight at room temperature with primary antibody in TBS with 2% rabbit serum. The sections were rinsed with TBS and incubated in secondary biotinylated antibody in TBS with 2% rabbit serum for 1 h. After being rinsed with TBS, sections were incubated with an avidin/biotin/horseradish peroxidase complex (Vectastain Elite ABC kit) for 1 h. Staining was visualized with DAB (Sigma-Aldrich, St. Louis, MO). The sections were then mounted on gelatinized slides, dried, dehydrated, cleared with xylene, and cover slipped with Permount (Fischer Scientific, Pittsburg, PA). The primary antibody used was goat anti-rat IFNγ (1:1,500; R&D Systems,) and the secondary antibody was biotinylated rabbit anti-goat (1:256; Vector Laboratories,).
IFNγ immunohistochemistry quantification
IFNγ stained tissue sections were digitally photographed with Zeiss Axioskop2 (Carl Zeiss INC, Thornwood, NY) microscope controlled by Openlab software (Improvision, Waltham, MA) at a 10× magnification. One image from each Bregma point was taken for a total of six images per brain. The area selected for quantification was from the peri-infarct region of the ipsilateral hemisphere for all animals. The images were analyzed for percent of immunostaining per area with ImageJ software. These six values were then averaged for each brain.
Splenic images were taken with a Nikon 90i microscope using a 20× objective and NIS Elements BR 2.30 software at a high resolution. The images were processed and analyzed with Photoshop CS5 (Adobe Systems Inc., San Jose, CA). The intensity of the staining was measured in the histogram for the entire image and the amount of staining per image was analyzed. Six sections per spleen were analyzed for each rat.
Neuronal cultures
Cortices from E18 rat embryos were dissociated with a solution of 0.25% trypsin/2.21 mM EDTA for 10 min at 37°C. The solution was triturated to obtain a uniform single cell suspension. Then 40 ml of DMEM (Mediatech, Manassas, VA) was added and the solution was allowed to settle. The supernatant was transferred to a fresh conical tube and centrifuged at 1000 rpm for 10 min. The supernatant was aspirated off, the pellet was re-suspended in DMEM and the solution was allowed to settle. The debris from the bottom was removed with a pipette and the solution was centrifuged for 10 min at 1000 rpm. The supernatant was aspirated off and the cells were re-suspended in DMEM. Trypan blue exclusion was used to count viable cells and 3×105 cells in a final volume of 1 ml were plated in 24 well poly-L-lysine treated culture plates. Twenty-four hours later the media was changed to neurobasal complete (neurobasal media (Invitrogen), B-27 (Invitrogen), 0.05 mM L-glutamine (Mediatech)) for seven days. After a media change, the cells were used for oxygen glucose deprivation (OGD) experiments.
Mixed glial cultures
A 2.21 mM EDTA solution containing 0.25% trypsin was used to dissociate cortices from P3 rat pups. The suspension was triturated and pelleted. The pellet was re-suspended in DMEM+, which consisted of DMEM (Mediatech), 2.5% fetal bovine serum, 10% horse serum, and 1% antibiotic/antimycotic (Mediatech). After Trypan Blue exclusion to assess cell viability, cells were seeded at a concentration of 1.5×107 cells in 75 cm2 poly-L-lysine treated tissue culture flasks. The following day the media was changed to fresh DMEM+and the cultures were incubated at 37°C for 8 days (Gottschall et al. 1995; Rowe et al. 2010).
Oligodendrocyte purification
To separate the microglia cell fraction from the OL/astrocyte monolayer, flasks were mechanically shaken for 1 h and the media was discarded. Fresh DMEM+was added to the flasks and incubated for 2 days at 37°C. Following the 2 day incubation, the flasks were mechanically shaken for 18 h to remove the astrocytes from the OLs and microglia. The media was collected and cells were pelleted and re-suspended in fresh DMEM+. Trypan Blue exclusion was used to count the viable cells. The media containing OLs and microglia was added to 10 cm plastic tissue culture dishes at a concentration of 107 cells/dish and incubated for 15 min at 37°C. This was repeated three times to assure microglial adherence to the plastic. Following the final incubation the dishes were gently agitated and the media was removed. The cells were pelleted, re-suspended in DMEM+, and plated on poly-L-lysine treated glass coverslips at a concentration of 3×105 cells/coverslip (McCarthy and de Vellis 1980). After 24 h the media was changed to neurobasal complete with 10 ng/ml of platelet derived growth factor-AA (PDGF-AA) (Barres et al. 1993; Yang et al. 2005) and the OLs were allowed to proliferate for 7 days. Afterwards, the PDGF-AA was withdrawn for 5 days allowing the OLs to progress to the mature phenotype (Yang et al. 2005). All experiments were conducted on cultures following the 5 day PDGF-AA withdrawal and all cultures used for experiments were 95% pure OLs (Hall et al. 2009b; Rowe et al. 2010).
Oxygen glucose deprivation and rIFNγ administration
Mature OLs that were seeded on glass coverslips in 6 well culture plates were subjected to 24 h of OGD. Neurons that were seeded in 24 well culture plates were subjected to either OGD or normoxia for 24 h. OGD conditions were induced using DMEM without glucose and placing the cultures in an air tight chamber that was flushed with hypoxic gas (95% N2, 4% CO2, and 1% O2; Airgas, Tampa, Fl) for 15 min and sealed for 24 h at 37°C. Cultures exposed to normoxia were incubated in DMEM with glucose in a standard tissue culture incubator for 24 h at 37°C. The two groups were further divided into cultures that received 20 ng/ml of rIFNγ or vehicle just prior to the 24 h OGD or normoxic conditions. The concentration of 20 ng/ml of rIFNγ was previously shown to kill immature oligodendrocytes but not mature OLs (Horiuchi et al. 2006).
Lactate dehydrogenase assay
The amount of neuronal and OL cell death was determined using a lactate dehydrogenase (LDH) assay (Takara Bio, Inc, Madison, WI). Following 24 h of OGD or normoxia, the culture media was removed and centrifuged. Then 100 μl of media was added to 100 μl of LDH reagent and incubated in a 96 well plate for 30 min at room temperature protected from light. The plate was then read at 548 nm on the μQuant platereader (Bio-tek, Winooski, VT).
Statistical analysis
All data are expressed as group mean± SEM. Significance of the data was determined by ANOVA with a Dunnet’s post hoc test for IFNγ immunostaining in the brain and neuronal culture survival. Following ANOVA, a Bonferroni’s post hoc test was used for the rIFNγ splenectomy treatment groups. ATukey’s post-hoc test was used following ANOVA to determine significance for the splenic IFNγ protein levels. A two tailed t-test was used to for OL survival in culture. A value of p<0.05 was considered significant. All sections were blinded prior being analyzed by an investigator.
Results
IFNγ levels are increased in the brain following MCAO
To determine if IFNγ is present in the brain following MCAO, its expression in the infarct was characterized over time. To quantify IFNγ levels, immunohistochemistry for IFNγ was performed on brain sections from sham operated animals and from animals euthanized at 3, 24, 48, 72, and 96 h following MCAO. IFNγ protein levels were significantly increased at 72 h (p<0.01) and remained elevated at 96 h (p<0.05) compared to sham operated rats 96 h after surgery (Fig. 1). Immunohistochemistry was also performed on brain sections from rats that received splenectomy two weeks prior to MCAO and were euthanized at 72 and 96 h post-MCAO. Splenectomy decreased IFNγ protein levels down to those not significantly different from sham MCAO at both 72 and 96 h post-MCAO. Additionally splenectomy reduces IFNγ protein levels significantly at 72 h compared to 72 h post-MCAO only and as well as at 96 h when compared to 96 h post-MCAO only.
Fig. 1.
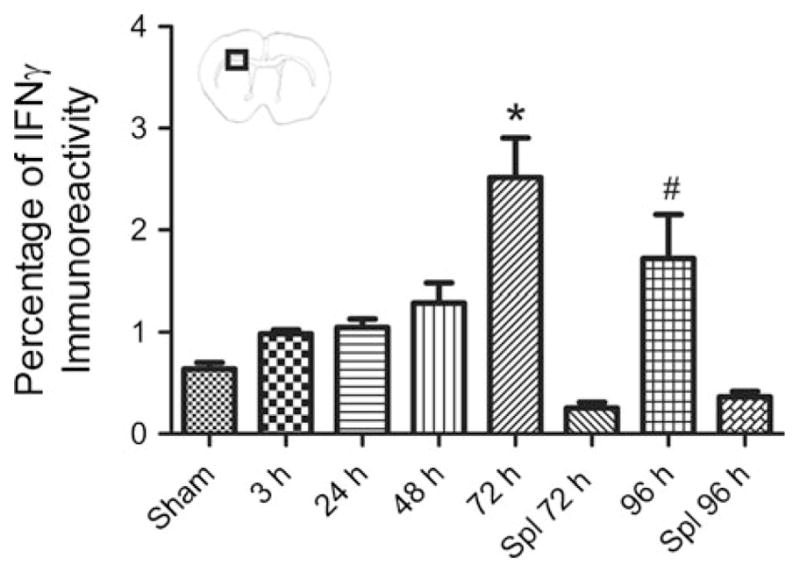
IFNγ levels increase in the injured brain post-MCAO. IFNγ immunohistochemistry of brain tissue from sham operated animals, animals that received splenectomies two weeks prior to MCAO and were euthanized at 72 and 96 h post-MCAO, and animals 3, 24, 48, 72, and 96 h post-MCAO. IFNγ protein levels were significantly higher at 72 and 96 h compared to sham operated animals and animals that received splenectomy prior to MCAO at 72 and 96 h post-MCAO (* p<0.01; # p<0.05). For each group n≥3. Box in brain graphic depicts area used for quantification of IFNγ levels. Sham denotes a sham MCAO and Spl denotes rats that underwent splenectomy prior to MCAO
IFNγ protein levels in the spleen are elevated at 24 h following MCAO
Splenic production of IFNγ was measured by immunohistochemical analysis of the spleen. IFNγ protein levels were significantly elevated at 24 h post-MCAO compared to 48, 72, and 96 h post-MCAO, and also elevated compared to the sham-operated rats at 48 and 96 h after surgery (p<0.0002). Naïve spleens showed very low levels of IFNγ protein expression (Fig. 2).
Fig. 2.
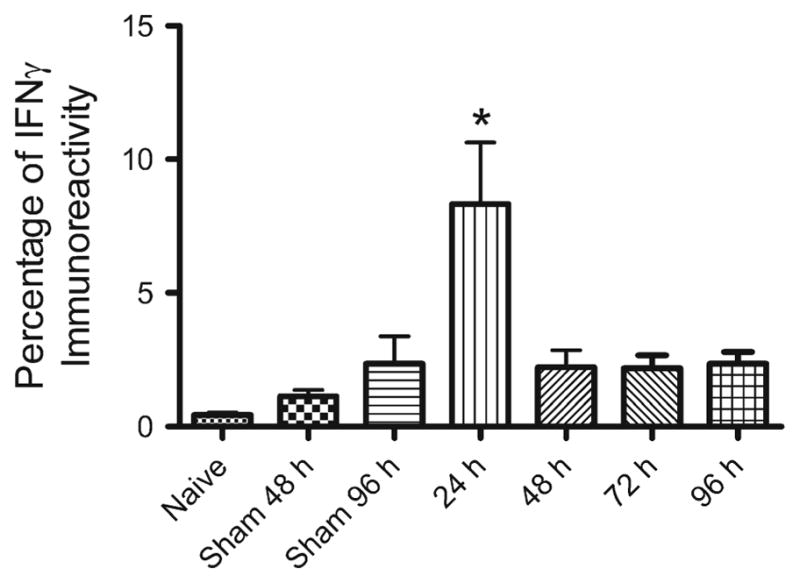
Splenic IFNγ production is elevated at 24 h post-MCAO. Spleens from animals 24, 48, 72, 96 h post-MCAO along with naïve, 48 and 96 h sham-MCAO were assayed using immunohistochemistry for IFNγ. IFNγ protein levels were found to be significantly elevated 24 h post-MCAO (* p<0.0002) compared to the other groups
IFNγ expression by T cells, NK cells, and B cells in and around the infarct
Immunostaining for IFNγ was abundant in the infarct of rat brains at 96 h post-MCAO (Fig. 3a). Double staining with antibodies against immune cell markers and IFNγ showed co-localization of CD3 (T cells) (Fig. 3b), CD161 (NK cells) (Fig. 3c), and CD45R (B cells) (Fig. 3d) with IFNγ. These results indicate T cells, NK cells and B cells were producing IFNγ in and around the infarct. CD11b positive cells did not co-localize with IFNγ staining cells, indicating that microglia/macrophages were not producing IFNγ (Fig. 3e).
Fig. 3.

Representative brain sections from rats 96 h post-MCAO were stained with IFNγ and immune cell surface markers to identify what types of cells are expressing IFNγ in the infarct and peri-infarct. Micrographs show IFNγ (red) (a), and double staining merged images of IFNγ (red) with CD3 (green) for T cells (b), CD161 (green) for natural killer cells (c), and CD45R (green) for B cells (d); yellow cells with white arrows indicate areas of co-localization. A micrograph of staining with CD11b (green) for microglia/macrophages and IFNγ (red) (e) demonstrate a lack of co-localization of CD11b and IFNγ. In figure e, arrow heads indicate IFNγ positive cells and yellow arrows indicate CD11b positive cells. Scale bars=20 μm. Box in brain graphics depicts the regions where images were taken for a given micrograph
T cells, B cells, NK cells, and microglia/macrophages are present in the ipsilateral hemisphere following MCAO
Antibodies directed against immune cell surface markers showed that T cells (CD3), NK cells (CD161), B cells (CD45R), and microglia/macrophages (CD11b) are localized in the infarcted area of the ipsilateral hemisphere 96 h following MCAO (Fig. 4a–d). In splenectomized rats, there was a decrease in the immunostaining for T cells, NK cells, and B cells in the injured hemisphere (Fig. 4e–g). Microglia/macrophages in the infarct declined in the ipsilateral hemisphere of splenectomized rats (Fig. 4h). In splenectomized rats the predominant form of microglia/macrophages appear with an amoeboid morphology but these cells still display evident ramifications in the damaged area. Only microglia in the resting, ramified morphology were present in the contralateral hemispheres (Fig. 4i–l).
Fig. 4.

T cells, B cells, NK cells, and microglia/macrophages are present in the ipsilateral hemisphere following MCAO. At 96 h post-MCAO immunohistochemistry for immune cell surface markers shows peripheral immune cells are present in the ipsilateral hemisphere. Micrographs show CD3 positive cells (T cells) (a), CD161 positive cells (NK cells) (b), CD45R positive cells (B cells) (c), and CD11b positive cells (microglia/macrophages) (d) in the infarcted hemisphere. Micrographs from splenectomized rats demonstrate a decrease in immunostaining for T cells (e), NK cells (f), B cells (g), and microglia/macrophages (h) in the ipsilateral hemisphere. However in the contralateral hemisphere there is an absence of staining for T cells (i), NK cells (j), and B cells (k). Only microglia/macrophages were detected in the contralateral hemispheres (l). Inserts provide representative images of the morphological states of the microglia/macrophages present in each group and show an amoeboid cell (d), an amoeboid cell with evident ramifications (h), and a ramified cell (l). Scale bars=100 μm. The scale bar of the inserts=20 μm. Box in brain graphics depicts the regions where images were taken for a given micrograph
Administration of rIFNγ following MCAO abolishes the protective effect of splenectomy
IFNγ production originating from the spleen could contribute to delayed neural death and explain why splenectomy prior to MCAO is neuroprotective. To test this, rats underwent splenectomy or sham-splenectomy two weeks prior to MCAO. Animals were then administered rIFNγ (20 μg/rat i.v.) or ddH2O at 48 and 72 h post-MCAO. Infarct volumes, as measured by Fluoro-Jade staining, at 96 h post-MCAO showed splenectomized rats that received systemic rIFNγ had infarcts that were significantly greater than splenectomized vehicle rats (p<0.0001). The splenectomized rIFNγ rats had infarcts that were not significantly different from either of the sham-splenectomized rat groups (Fig. 5e). There was an average of 5% infarct in the splenectomy vehicle brain sections (Fig. 5c), compared to the average infarcts of (50–70%) for all other treatment groups (Figs. a, 5b, and 5d).
Fig. 5.

Recombinant IFNγ increases neural injury following MCAO in splenectomized rats. Recombinant IFNγ increases infarct volume in splenectomized rats at 96 h post-MCAO to levels not different from sham-splenectomized rats. Infarct volumes were measured as a percentage of the contralateral hemisphere with Fluoro-Jade staining. Graph depicts average infarct volumes for each group at 96 h post-MCAO (e). The splenectomy-vehicle treated rats had significantly lower infarcts than the other treatment groups (* p<0.0001). The splenectomy-IFNγ treated rats had infarcts that were not significantly different from the sham-splenectomy groups. Representative images for each treatment group at 96 h post-MCAO: sham-splenectomy-vehicle (SS-V) n=4 (a), sham-splenectomy-rIFNγ (SS-IFNγ) n=6 (b), splenectomy-vehicle (S-V) n= 4 (c), and splenectomy-rIFNγ (S-IFNγ) n=6 (d). Scale bars=2 mm
Recombinant IFNγ increases IFNγ expression in the infarct of splenectomized rats
Immunohistochemical analysis for IFNγ in the brain was performed to determine the effect of rIFNγ administration on levels of this cytokine in the infarct. IFNγ expression was significantly decreased in the infarct of splenectomized-vehicle rats (Fig. 6c) (p<0.02) compared to all other groups (Fig. 6e). The addition of rIFNγ to splenectomized rats (Fig. 6d) increased IFNγ protein levels in the infarct to levels found in rats which underwent sham-splenectomy prior to MCAO (Fig. 6a and b).
Fig. 6.

Recombinant IFNγ increases IFNγ expression in the infarct of splenectomized rats. The graph shows splenectomy results in a significant decrease in IFNγ protein expression at 96 h post-MCAO (* p< 0.02) (e). However rats that received splenectomy and rIFNγ had IFNγ protein levels not significantly different than the rats which underwent sham-splenectomy prior to MCAO. Representative images from each treatment group at 96 h following MCAO: sham-splenectomy-vehicle (SS-V) (a), sham-splenectomy-rIFNγ (SS-IFNγ) (b), splenectomy-vehicle (S-V) (c), and splenectomy-rIFNγ (S-IFNγ) (d). Box in brain graphics depicts the regions where images were taken for a given micrograph
Recombinant IFNγ is not cytotoxic to cultured primary neurons or OLs
To determine if rIFNγ is directly toxic to neural cells, cultured neurons and OLs were treated with rIFNγ prior to OGD. Cell death as measured with LDH assays show that treatment with rIFNγ does not directly enhance death of neurons (Fig. 7a) in culture under nor-moxic or OGD conditions. Recombinant IFNγ does not increase the death of cultured OLs (Fig. 7b) exposed OGD conditions. Representative images of primary neuronal (Fig. 7c) and primary OL cultures (Fig. 7d) prior to experimentation are provided.
Fig. 7.

Recombinant IFNγ is not cytotoxic to cultured primary neurons or OLs. Primary neuronal and OL cultures were treated with 20 ng/ml of rIFNγ under normoxic and OGD for 24 h. Recombinant IFNγ does not increase the amount of cell death, as measured by LDH, in neuronal cultures under normoxic or OGD conditions for 24 h (a). Oligodendrocytes subjected to 24 h of OGD and rIFNγ did not have significantly different survival rates (b). Representative images depict neuronal (c) and OL (d) cultures prior to treatment. Scale bars=30 μm
Discussion
The spleen is a key component in the immune response to ischemic injury of the brain and other organs (Okuaki et al. 1996; Savas et al. 2003; Jiang et al. 2007). Splenectomy is protective in models of ischemic (Ajmo et al. 2008), hemorrhagic (Lee et al. 2008), and severe traumatic brain injury (Li et al. 2011). Together, these studies suggest there is a splenic response that exacerbates neural injury by initiating a delayed inflammatory response.
Notably, IFNγ perpetuates the pro-inflammatory response by promoting Th1 cell differentiation while inhibiting Th2 cell differentiation. Additionally, it is known to activate numerous immune cell types including microglia/macrophages, NK cells, B cells, and T cells, as well as vascular endothelial cells. Furthermore, this pro-inflammatory cytokine also influences antibody isotype production, up regulates both major histocompatibility complexes (MHC I and MHC II), induces changes in vascular endothelial cell adhesion, and increases the production of reactive oxygen species (Boehm et al. 1997). These actions are detrimental to the survival of compromised neural cells. In particular the enhanced Th1 response seen with IFNγ has been found to be detrimental in ischemic brain injuries. A Th1 response to brain antigens has been shown in animal studies to result in a more severe injury (Becker et al. 2005) and is a poor prognostic factor regardless of stroke severity in people (Becker et al. 2011).
Experimental data suggest that IFNγ plays an important role in exacerbating neural injury, as IFNγ knockout mice show reduced infarct volume following transient MCAO (Yilmaz et al. 2006). In contrast, a different study demonstrated that there was no difference in infarct volume between IFNγ knockout and wild type mice following MCAO (Lambertsen et al. 2004). However, this latter study used a different model of MCAO from the one used in the Yilmaz study. Increased serum levels of IFNγ have been detected in mice 24 h following MCAO (Liesz et al. 2009a).
In stroke patients IFNγ production was reduced 6 h following symptom onset. However, IFNγ expression returns to levels not significantly different than healthy controls 72 h following symptom onset. In these patients, IFNγ was being produced by the innate cells of the immune system, specifically γδT cells, NK cells, and natural killer T (NKT) cells (Peterfalvi et al. 2009). Both animal and human studies provide strong support for IFNγ and the innate immune system response in the progression of tissue damage in ischemic brain injury.
Splenic IFNγ protein levels were elevated at 24 h post-MCAO and decrease by 48 h. The spike of IFNγ found in the spleen of the rats suggests it is being produced by innate immune cells, in particular NK cells as they are a major source innate IFNγ (Boehm et al. 1997). This transient spike in IFNγ protein expression in the spleen at 24 h is consistent with rises in IFNγ mRNA in the spleens of mice 22 h following transient MCAO (Hurn et al. 2007).
Our results indicated an increase in IFNγ protein expression in the injured brain at 72 h post-MCAO, with expression remaining elevated at 96 h. These results are consistent with studies examining IFNγ mRNA levels in the brain following MCAO. For example, Li et al. (2001) demonstrated that IFNγ mRNA increased in the infarct at 2 days post-MCAO and remained elevated 6 days following MCAO (Li et al. 2001). Other reports have shown that IFNγ mRNA levels were decreased in the mouse brain at 22 h following MCAO (Offner et al. 2006). An experiment by Liesz et al. (2011) in which mice were administered an antibody directed against CD49d (VLA-4) 24 h prior to transient MCAO provides further support for delayed IFNγ production in the brain. Trafficking of T and NK cells into the brain was decreased in the CD49d antibody treated mice compared to control mice. In the same experiment there was also a decrease in the amount of IFNγ mRNA at 72 h post-MCAO in the antibody treated mice compared to the control mice (Liesz et al. 2011). This experiment suggests that T cells and NK cells are a source of IFNγ in the brain at later time points following MCAO which is also consistent with our findings.
The increase in IFNγ expression in the brain at 72 h post-MCAO coincides with the time point at which microglia/macrophages become maximally activated in the brain (Leonardo et al. 2010). As IFNγ is a potent activator of microglia/macrophages, the delay in the activation of these cells in the brain suggests that splenic IFNγ is acting through other immune cells to elicit this delayed effect to the infarct. A direct systemic IFNγ response from the spleen would be expected to cause a more immediate response. Therefore, it is more likely that IFNγ production in the spleen acts on target cells within the spleen and these cells then migrate to other immune organs to interact with other cell types. These cells could then infiltrate the brain stimulating the microglia/macrophages to degrade the infarcted area in the brain. As T cells and NK cells have been found in the peri-infarct region producing IFNγ 96 h following MCAO, the likely sequence of events starts with an initial increase in IFNγ in the spleen leading to delayed neural injury.
Our results suggest the neuroprotection resulting from splenectomy is caused by the loss of IFNγ. Systemic administration of rIFNγ to splenectomized rats resulted in infarct volumes that were not different from sham-splenectomized rats, suggesting that spleen derived IFNγ is responsible for the delayed expansion of the penumbra. Interestingly, sham-splenectomized rats that received rIFNγ did not have larger infarcts than sham-splenectomized rat that received vehicle. This finding suggests the endogenous IFNγ response from the spleen is enough to cause maximal delayed neural damage following a stroke.
Splenectomy reduced the amount of IFNγ protein in the brain following MCAO and administration of rIFNγ restores IFNγ production in the brains of splenectomized animals to levels seen in sham-splenectomized rats. Additionally IFNγ expression was not significantly higher in the brains of sham-splenectomy rats that received rIFNγ compared to rats that received vehicle treatment. This observation provides evidence that the IFNγ from the spleen has a relationship to the IFNγ produced in the brain following MCAO. Whether this is a direct (systemic) or indirect (cellular) relationship is yet to be determined.
As previously reported, splenectomy reduced the number of Isolectin IB4 and myeloperoxidase (MPO) positive cells, activated microglia/macrophages and neutrophils respectively, in the infarcted hemisphere 96 h post-MCAO (Ajmo et al. 2008). Splenectomy reduces the number of peripheral immune cells, specifically T cells, B cells, and NK cells, in the ipsilateral hemisphere and alters the morphology of microglia/macrophages responding to the injury at 96 h following MCAO. A majority of the microglia in the splenectomized rats appear in transitional state with amoeboid-like cell body with ramifications, not the completely amoeboid morphology observed in MCAO only rats at 96 h. As IFNγ activates microglia/macrophages, the lack of this cytokine would maintain these cells towards a resting state. Therefore, blocking splenic IFNγ could prove to be a therapeutic option in modulating the immune response following ischemic stroke.
Experiments with rIFNγ were performed on cell cultures to ensure that the increase in infarct volume in the splenectomy-rIFNγ group was due to activation of the immune system and not the result of the rIFNγ being directly cytotoxic to neural cells. A previous study demonstrated that IFNγ is not cytotoxic to primary mature OLs at 20 ng/ml (Horiuchi et al. 2006). This concentration was used to treat primary neural cell cultures under normoxic and OGD conditions. Recombinant IFNγ is not directly cytotoxic to cultured neurons or OLs demonstrating that other cells through activation by IFNγ, like microglia, are eliciting their cytotoxic effect. This contention is further supported by Bal-Price and Brown (2001) who showed that IFNγ added to mixed brain cell cultures results in neuronal cell death. The neurotoxic effects of IFNγ appear to be mediated through the activation of microglia/macrophages.
From these various experiments, blocking IFNγ from facilitating a pro-inflammatory response to ischemic stroke is a potential way to reduce injury. Selectively blocking IFNγ signaling will allow for targeting one facet of the immune response, leaving the anti-inflammatory or pro-regenerative facets able to respond to the injury.
Acknowledgments
We would like to thank Dr. Chris Katnik for his help obtaining neuronal cultures and Dr. Thomas Klein for his insights into immunology. This work was supported by the National Institutes Health grant RO1 NS052839.
Funding
NIH grant RO1 NS052839.
Abbreviations
- MCAO
Middle cerebral artery occlusion
- IFNγ
Interferon gamma
- OL
Oligodendrocyte
- P3
Postnatal day 3
- E18
Prenatal day 18
- ICA
Internal carotid artery
- ECA
External carotid artery
- rIFNγ
Recombinant interferon gamma
- i.v
Intravenous
- i.p
Intraperitoneal
- PBS
Phosphate buffered saline
- DAB
3 3′-diaminobenzidine
- DMSO
Dimethyl sulfoxide
- TBS
Tris-buffered saline
- OGD
Oxygen glucose deprivation
- PDGF-AA
Platelet derived growth factor-AA
- LDH
Lactate dehydrogenase
- MHC
Major histocompatibility complex
- NKT cell
Natural killer T cell
- MPO
Myeloperoxidase
Footnotes
Conflicts of Interest
The authors have no conflicts of interest.
Contributor Information
Hilary A. Seifert, Department of Molecular Pharmacology and Physiology, School of Basic Biomedical Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Christopher C. Leonardo, Department of Molecular Pharmacology and Physiology, School of Basic Biomedical Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Aaron A. Hall, Department of Molecular Pharmacology and Physiology, School of Basic Biomedical Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Derrick D. Rowe, Department of Molecular Pharmacology and Physiology, School of Basic Biomedical Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Lisa A. Collier, Department of Molecular Pharmacology and Physiology, School of Basic Biomedical Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Stanley A. Benkovic, NeuroScience Associates, Knoxville, TN 37934, USA
Alison E. Willing, Center for Excellence in Aging and Brain Repair, Department of Neurosurgery and Brain Repair, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Keith R. Pennypacker, Email: kpennypa@health.usf.edu, Department of Molecular Pharmacology and Physiology, School of Basic Biomedical Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
References
- Ajmo CT, Jr, Vernon DO, Collier L, Pennypacker KR, Cuevas J. Sigma receptor activation reduces infarct size at 24 hours after permanent middle cerebral artery occlusion in rats. Curr Neurovasc Res. 2006;3(2):89–98. doi: 10.2174/156720206776875849. [DOI] [PubMed] [Google Scholar]
- Ajmo CT, Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. 2008;86:2227–2234. doi: 10.1002/jnr.21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21(17):6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118(1):283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- Becker KJ, Kindrick DL, Lester MP, Shea C, Ye ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab. 2005;25(12):1634–1644. doi: 10.1038/sj.jcbfm.9600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KJ, Kalil AJ, Tanzi P, Zierath DK, Savos AV, Gee JM, Hadwin J, Carter KT, Shibata D, Cain KC. Autoimmune Responses to the Brain After Stroke Are Associated With Worse Outcome. Stroke. 2011 doi: 10.1161/STROKEAHA.111.619593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21(15):5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332(20):1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR. Temporary focal ischemia in the mouse: technical aspects and patterns of Fluoro-Jade evident neurodegeneration. Brain Res. 2005;1042(1):29–36. doi: 10.1016/j.brainres.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Yu X, Bing B. Increased production of gelatinase B (matrix metalloproteinase-9) and interleukin-6 by activated rat microglia in culture. J Neurosci Res. 1995;42(3):335–342. doi: 10.1002/jnr.490420307. [DOI] [PubMed] [Google Scholar]
- Hall AA, Guyer AG, Leonardo CC, Ajmo CT, Jr, Collier LA, Willing AE, Pennypacker KR. Human umbilical cord blood cells directly suppress ischemic oligodendrocyte cell death. J Neurosci Res. 2009a;87(2):333–341. doi: 10.1002/jnr.21857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AA, Leonardo CC, Collier LA, Rowe DD, Willing AE, Pennypacker KR. Delayed treatments for stroke influence neuronal death in rat organotypic slice cultures subjected to oxygen glucose deprivation. Neuroscience. 2009b;164(2):470–477. doi: 10.1016/j.neuroscience.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Itoh A, Pleasure D, Itoh T. MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendroglial progenitor cells. J Biol Chem. 2006;281(29):20095–20106. doi: 10.1074/jbc.M603179200. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27(11):1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Meng F, Li W, Tong L, Qiao H, Sun X. Splenectomy ameliorates acute multiple organ damage induced by liver warm ischemia reperfusion in rats. Surgery. 2007;141(1):32–40. doi: 10.1016/j.surg.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Gregersen R, Meldgaard M, Clausen BH, Heibol EK, Ladeby R, Knudsen J, Frandsen A, Owens T, Finsen B. A role for interferon-gamma in focal cerebral ischemia in mice. J Neuropathol Exp Neurol. 2004;63(9):942–955. doi: 10.1093/jnen/63.9.942. [DOI] [PubMed] [Google Scholar]
- Lee ST, Chu K, Jung KH, Kim SJ, Kim DH, Kang KM, Hong NH, Kim JH, Ban JJ, Park HK, Kim SU, Park CG, Lee SK, Kim M, Roh JK. Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain. 2008;131(Pt 3):616–629. doi: 10.1093/brain/awm306. [DOI] [PubMed] [Google Scholar]
- Leonardo CC, Hall AA, Collier LA, Ajmo CTJ, Willing AE, Pennypacker KR. Human umbilical cord blood cell therapy blocks the morphological change and recruitment of CD-11b-expressing isolectin-binding proinflammatory cells after middle cerebral artery occlusion. J Neurosci Res. 2010;88(6):1213–1222. doi: 10.1002/jnr.22306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Kostulas N, Huang YM, Xiao BG, van der Meide P, Kostulas V, Giedraitas V, Link H. IL-17 and IFN-gamma mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol. 2001;116(1):5–14. doi: 10.1016/s0165-5728(01)00264-8. [DOI] [PubMed] [Google Scholar]
- Li M, Li F, Luo C, Shan Y, Zhang L, Qian Z, Zhu G, Lin J, Feng H. Immediate Splenectomy Decreases Mortality and Improves Cognitive Function of Rats After Severe Traumatic Brain Injury. J Trauma. 2011 doi: 10.1097/TA.0b013e3181f30fc9. [DOI] [PubMed] [Google Scholar]
- Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, Hug A, Zorn M, Dalpke A, Nawroth P, Veltkamp R. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009a;40(8):2849–2858. doi: 10.1161/STROKEAHA.109.549618. [DOI] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebro-protective immunomodulators in acute experimental stroke. Nat Med. 2009b;15(2):192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, Sommer C, Dalpke AH, Veltkamp R. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134(Pt 3):704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Longa E, Weinstein P, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb JD, Ajmo CT, Jr, Sanberg CD, Sanberg PR, Pennypacker KR, Willing AE. Timing of cord blood treatment after experimental stroke determines therapeutic efficacy. Cell Transplant. 2006;15(3):213–223. doi: 10.3727/000000006783982043. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26(5):654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Okuaki Y, Miyazaki H, Zeniya M, Ishikawa T, Ohkawa Y, Tsuno S, Sakaguchi M, Hara M, Takahashi H, Toda G. Splenectomy-reduced hepatic injury induced by ischemia/reperfusion in the rat. Liver. 1996;16(3):188–194. doi: 10.1111/j.1600-0676.1996.tb00726.x. [DOI] [PubMed] [Google Scholar]
- Peterfalvi A, Molnar T, Banati M, Pusch G, Miko E, Bogar L, Pal J, Szereday L, Illes Z. Impaired function of innate T lymphocytes and NK cells in the acute phase of ischemic stroke. Cerebrovasc Dis. 2009;28(5):490–498. doi: 10.1159/000236527. [DOI] [PubMed] [Google Scholar]
- Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. CD4 +FoxP3+ regulatory T-cells in cerebral ischemic stroke. Metab Brain Dis. 2010;26(1):87–90. doi: 10.1007/s11011-010-9226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe DD, Leonardo CC, Hall AA, Shahaduzzaman MD, Collier LA, Willing AE, Pennypacker KR. Cord blood administration induces oligodendrocyte survival through alterations in gene expression. Brain Res. 2010;1366:172–188. doi: 10.1016/j.brainres.2010.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas MC, Ozguner M, Ozguner IF, Delibas N. Splenectomy attenuates intestinal ischemia-reperfusion-induced acute lung injury. J Pediatr Surg. 2003;38(10):1465–1470. doi: 10.1016/s0022-3468(03)00497-4. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751(1):37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Yang Z, Watanabe M, Nishiyama A. Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. J Neurosci Meth. 2005;149(1):50–56. doi: 10.1016/j.jneumeth.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113(17):2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]