

Abstract
This work focuses on improving the efficacy of photoactivatable Ru complexes for photodynamic therapy by employing cross-linked nanoassemblies (CNAs) as a delivery approach. The effects of complex photoactivation, hydrophobicity, and solution ionic strength and pH on complex loading and release from CNAs were analyzed. The cell cytotoxicity of CNA formulations was similar to free Ru complexes despite reduced or eliminated DNA interactions. The release rate and the amount of each Ru complex released (%) varied inversely with complex hydrophobicity, while the effect of solution ionic strength was dependent on complex hydrophobicity. Premature release of two photoactivatable prodrugs prior to irradiation was believed to account for higher activity in cells studies compared to DNA interaction studies; however, for photostable 1O2 generator-loaded CNAs this cannot explain the high cytotoxicity and lack of DNA interactions because release was incomplete after 48 hrs. The cause remains unclear, but among other possibilities, accelerated release in a cell culture environment may be responsible.
1 Introduction
Following the unprecedented clinical success of cisplatin as an anticancer chemotherapeutic in the late 20th century, there has been an impetus to formulate other metal-containing therapeutics to improve anticancer efficacy and to address the limitations of platinum (Pt)-based drugs, which include harmful side effects and drug resistance. Polypyridyl ruthenium (Ru) complexes have been investigated extensively in recent years as potential anticancer agents.2 In particular, photoactivatable Ru complex prodrugs have been developed as promising agents for cancer therapy.1, 3 Integrating strain into the structure to create distorted octahedral Ru complexes is one approach to preparing photoactivated systems, as it facilitates ligand ejection when exposed to visible (λ>400 nm) and near IR (λ>650 nm) light.1, 3b This generates a ligand deficient and highly reactive metal center that readily forms bonds with biomolecules, such as DNA. The cytotoxic activity of the photoactivatable Ru complexes increases upon ligand ejection due in large part to DNA adduct formation.1, 3b, 3c, 4 In this manner, polypyridyl Ru complexes are similar to current Pt chemotherapeutics; however, while cisplatin primarily forms intrastrand DNA cross-links, Ru complexes similar to the ones analyzed here are believed to preferentially form interstrand DNA cross-links.2a, 2b, 5 Based on this, the potential of photoactivatable Ru complexes is hypothesized to be greater than Pt-based therapies, as the difficulty of repairing interstrand cross-links is much higher compared to intrastrand cross-links, and it also may be possible to form cross-links between a strand of DNA and a protein, further complicating potential repair.5b, 6 By exploiting these features, it is possible to develop light-activated Ru prodrugs for localized cancer therapy. A number of photoactivatable polypyridyl Ru complexes have been developed in our laboratory that bind to DNA and displayed cytotoxicity similar to cisplatin after photoactivation; however, while preliminary in vitro results have been promising, these therapeutics have not been characterized in vivo.1, 3b, 4c Many free polypyridyl Ru complexes can be quite hydrophilic; this, in addition to their small size, can result in rapid clearance from the blood stream, which would limit their efficacy in vivo. These issues were made apparent in the 1950’s by Dwyer and coworkers, where structurally related polypyridyl Ru complexes were analyzed in vivo in mice and found to be rapidly excreted in the urine.7 To address this concern, polymeric cross-linked nanoassembly (CNA) drug carriers were employed as a means to extend circulation time and improve tumor accumulation of promising Ru complexes. Moreover, enhanced targeting of Ru complexes to tumor tissue may limit potential side effects such as those observed by Dwyer in mice.8 Three Ru complexes were prepared with varying levels of hydrophobicity and differing mechanisms of action to analyze the impact of these parameters on CNA loading and release in addition to in vitro anticancer potential. It was hypothesized that by increasing hydrophobicity it may be possible to slow Ru complex release from CNAs. Two of the complexes were developed as photoactivatable prodrugs ([Ru(bpy)2(dmbpy)]Cl2 (bpy = 2,2′-bipyridine and dmbpy = 6,6′-dimethyl-2,2′-bipyridine) (1) and [Ru(dmbpy)2(dip)]Cl2 (dip = 4,7-diphenyl-1,10-phenanthroline) (2)) while the third complex ([Ru(dip)3]Cl2 (3)) was photostable and was selected as a potential traditional photodynamic therapy (PDT) agent1b (Figure 1). Complexes 1 and 2 undergo ligand ejection upon light exposure, generating biologically active aqua complexes ([Ru(bpy)2(H2O)2]Cl2 for 1 and initially [Ru(dmbpy)(dip)(H2O)2]Cl2 for 2 with some [Ru(dip)(H2O)4]Cl2 upon prolonged light exposure), which preferentially bind with DNA. Complex 3 has been shown to generate singlet oxygen (1O2) when exposed to light in air-equilibrated D2O at room temperature (ΦΔ = 0.42),9 which can be used to locally eliminate cancer cells in a manner similar to other PDT agents; however, it possesses significant cytotoxicity in the dark.1b The three selected Ru complexes (Figure 1) were synthesized and characterized along with a poly(ethylene)glycol-poly(aspartate) (PEG-ASP) (PEG = 5,000 MW) CNA delivery system (Figure 2). The Ru complexes were physically entrapped in PEG-ASP CNAs and the photochemical, photobiological, and physiochemical properties compared to free Ru complexes. The release of Ru complexes from PEG-ASP CNAs was characterized when protected from light and after irradiation, to mimic conditions used in phototherapy. Finally, the in vitro anticancer potential of free Ru complexes and Ru complex-loaded PEG-ASP CNA formulations was evaluated through the analysis of DNA binding interactions and A549 human lung adenocarcinoma cell cytotoxicity.
Figure 1.
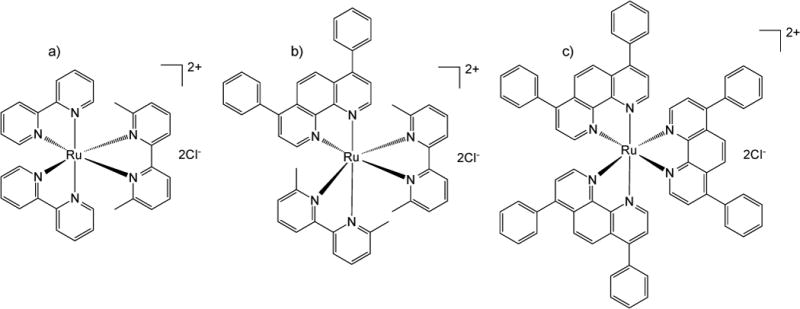
Structures of Ru complexes a) [Ru(bpy)2(dmbpy)]Cl2 (1), b) [Ru(dmbpy)2(dip)]Cl2 (2), and c) [Ru(dip)3]Cl2
Figure 2.
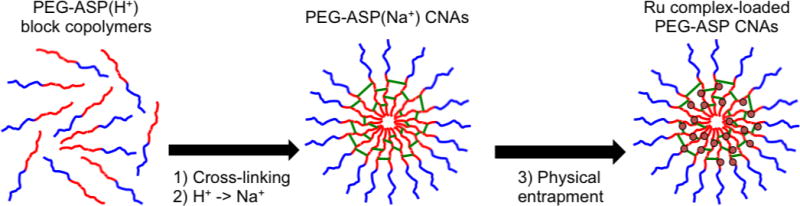
PEG (5k) (blue lines)-ASP (35 units) (red lines) CNA preparation. 1) PEG-ASP(H+) block copolymers were cross-linked (green lines) with diaminooctane (DAO) activated with diisopropylcarbodiimide (DIC)/ N-hydroxysuccinimide (NHS)/dimethylaminopyridine (DMAP); 2) ASP(H+) groups of PEG-ASP CNAs converted to ASP(Na+); 3) positively charged Ru complexes (red circles) were physically entrapped inside PEG-ASP CNAs through electrostatic interactions with ASP(Na+) groups.
2 Materials and methods
2.1. Materials
Solvents were obtained from commercial sources and used without further purification. All solvents were not anhydrous unless stated otherwise. Ru(Cl)3•xH2O was purchased from Johnson Matthey. Bathophenanthroline (dip) was purchased from Alfa Aesar. EcoRI restriction enzyme and EcoR1 buffer were purchased from New England BioLabs. 6,6′-dimethyl-2,2′-bipyridine (dmbpy), β-benzyl L-aspartate (BLA), triphosgene, hydrochloric acid (HCl), sodium hydroxide (NaOH), and sodium chloride (NaCl) were purchased from Sigma Aldrich. Ru(bpy)2(Cl)2•2H2O was purchased from Strem Chemicals Inc. α-Methoxy-ω-amino poly(ethylene glycol) (PEG, MW = 5,000) was purchased from NOF. Potassium hexafluorophosphate (KPF6), phosphate buffered saline (PBS) (pH 7.4), potassium phosphate buffer (PB), 6,000–8,000 molecular weight cut-off (MWCO) Fisherbrand regenerated cellulose membranes, and 50,000 MWCO Spectra/Por regenerated cellulose membranes were purchased from Fisher Scientific. Slide-A-Lyzer dialysis cassettes (10 kDa MWCO, G2) were purchased from Thermo Scientific. Dulbecco’s modified eagle media (DMEM, 4.5 g/L D-glucose with L-glutamine), opti-MEM I reduced serum media (opti-MEM), penicillin-streptomycin (pen-strep) solution, and heat inactivated fetal bovine serum (FBS) were purchase from Invitrogen. Serum supreme was purchased from Lonza.
2.2. Ruthenium complex synthesis
[Ru(bpy)2(dmbpy)]Cl2 (1) and [Ru(dip)3]Cl2 (3) were synthesized using established methods.1, 9 Complex 3 was purified using flash chromatography (silica gel, 100% acetonitrile (MeCN) ramped to 9% deionized H2O (diH2O) and 1.5% KNO3 in MeCN). [Ru(dmbpy)2(dip)]Cl2 (2) was synthesized as follows: [Ru(dip)Cl4] was prepared as described in literature.10 Briefly, Ru(Cl)3•xH2O (1.91 mmol) was added to a 1.5:1 mixture of ethanol and diH2O (25 mL) and the solution was refluxed for 3 hrs under a N2 atmosphere. The reaction mixture was then chilled in an ice bath. Dip (2.10 mmol) was suspended in a mixture of ethanol (10 mL) and hydrochloric acid (2 mL) and added to the Ru solution. The transfer vessel was rinsed with additional ethanol (5 mL), which was added to the reaction solution. The reaction mixture was stirred overnight at room temperature and the resulting precipitate was recovered by vacuum filtration, and washed with ethanol (100 mL) and ethyl ether (200 mL). The product was purified using flash chromatography (silica gel with a methanol mobile phase) to give the product in 27% yield. [Ru(dip)Cl4] (0.11 mmol) was combined with dmbpy (0.91 mmol) in a 4:1 mixture of ethanol and diH2O (5 mL) in a pressure tube and heated to 115 °C for 48 h. The volume of the reaction mixture was then reduced to 20% under reduced pressure, washed three times with dichloromethane (DCM) (50 mL), and the complex precipitated through the addition of saturated KPF6 (aq) (1 mL). The product was extracted into DCM (100 mL) and dried under reduced pressure, dissolved in minimal DCM, precipitated from ethyl ether (200 mL), collected by vacuum filtration, and washed twice with ethyl ether (25 mL) to give the product in 51% yield. 1H-NMR (CD3CN, 400 MHz) δ: 8.57 (d, J=8.2 Hz, 2H), 8.37 (d, J=8.2 Hz, 2H), 8.27 (d, J=5.8 Hz, 2H), 8.20 (t, J=8.0 Hz, 2H), 8.01 (s, 2H), 7.70 (t, J=8.0 Hz, 2H), 7.64 (d, J=5.9 Hz, 2H), 7.59 (m, 8H), 7.52 (m, 4H), 6.95 (d, J=7.8 Hz, 2H), 1.91 (s, 6H), 1.61 (s, 6H). 13C-NMR (CD3CN, 400 MHz) δ: 167.93, 166.41, 160.54, 159.28, 153.80, 150.32, 149.51, 139.53, 138.12, 136.27, 130.73, 130.70, 130.05, 129.02, 128.95, 127.62, 126.84, 126.55, 124.16, 123.62, 26.73, 25.44. ESI MS calculated for C48H40N6PF6Ru [M]+ 947.2; found 947.3 [M]+; C48H40N6Ru [M]2+ 401.1; found 401.2 [M]2+. UV/Vis (diH2O): λmax nm (ε M−1 cm−1): 280 (63,900), 295 (63,700), 465 (18,600).
2.3. PEG-ASP CNA synthesis
PEG-BLA block copolymers were prepared as previously described.11 β-Benzyl-L-aspartate N-carboxyanhydride (BLA-NCA) was prepared by reacting β-benzyl L-aspartate with triphosgene in anhydrous THF. The reaction was carried out under a N2 atmosphere at 45 °C until the solution became clear. BLA-NCA was recrystallized from hexane at −20 °C. PEG-BLA block copolymers were prepared by dissolving PEG and BLA-NCA separately in anhydrous DMSO (100 mg/mL) under N2 then transferring BLA-NCA to PEG and reacting at 45 °C for 72 hrs under N2. PEG-BLA was precipitated from solution three times using diethyl ether and lyophilized. Successful synthesis was verified by 1H-NMR (Figure S1). The number of BLA units in the BLA block was estimated using the ratio of the BLA and PEG peaks and determined to be 35 units. PEG-BLA block copolymers were suspended in diH2O and the benzyl ester groups deprotected with a 10:1 molar ratio of NaOH to BLA to form PEG-poly(aspartate) (PEG-ASP(Na+)) block copolymer. Excess NaOH was removed by dialysis in diH2O (replaced every two hrs over a period of six hrs). Dialysis was carried out using a 6,000 – 8,000 MWCO regenerated cellulose membrane. PEG-ASP was protonated to form PEG-ASP(H+) by adding a 2:1 molar ratio of HCl to ASP then excess HCl and NaCl was removed by dialysis in diH2O (replaced every two hrs over a period of six hrs). After dialysis, PEG-ASP was lyophilized and stored at −20 °C. PEG-ASP CNAs were prepared based on a previously established protocol.11b PEG-ASP(H+) (0.072 mmol, MW = 9100 (estimated based on known PEG molecular weight and the average number of aspartate units determined using NMR)) was dissolved in anhydrous DMSO (5 mL). In a separate vessel, 1,8-diaminooctane (DAO) (1.26 mmol) was combined with diisopropylcarbodiimide (DIC) (10.04 mmol), n-hydroxysuccinimide (NHS) (10.04 mmol), and 4-dimethylaminopyridine (DMAP) (0.50 mmol) in anhydrous DMSO (5 mL) for 15 min at room temperature to form a reactive ester cross-linker. The reaction mixture was slowly transferred to the PEG-ASP(H+) solution and reacted overnight at room temperature. Following CNA formation, DMSO and any remaining unreacted materials were removed by dialysis in diH2O (replaced twice a day for three days). Dialysis was carried out using a 50,000 MWCO Spectra/Por regenerated cellulose membrane. After purification, PEG-ASP(H+) CNAs were lyophilized and stored at −20 °C. The average molecular weight of the PEG-ASP CNAs was determined using gel permeation chromotography (GPC) with a superose 12 10/300 GL column by detecting UV/Vis absorbance at 280 nm as a function of time. This study was conducted with PBS as the running buffer. A PEG standard curve was employed to estimate the molecular weight of PEG-ASP CNAs.11b Particle diameter, surface charge, and polydispersity index (PDI) were determined in triplicate using dynamic light scattering (DLS) techniques with a Zetasizer Nano ZS (Malvern).
2.4. Ru complex PEG-ASP CNA loading and stability
The ASP(H+) groups of PEG-ASP(H+) CNAs were converted to ASP(Na+) as described above, then the PEG-ASP(Na+) CNAs were lyophilized and stored at −20 °C. PEG-ASP(Na+) CNAs were dissolved in anhydrous DMSO along with Ru complexes with a theoretical maximum Ru complex loading of 25% and incubated overnight at room temperature. Reaction mixtures were protected from light to prevent premature complex photoactivation. Unbound Ru complexes were then removed by dialysis in diH2O (replaced twice a day for three days). Dialysis was carried out using 6,000 – 8,000 MWCO regenerated cellulose membranes. After purification, Ru complex-loaded CNAs were lyophilized and stored at −20 °C protected from light. Ru complex loading was determined based on UV/Vis absorption determined using a Cary 60 spectrometer (Agilent) at the wavelengths associated with the metal-ligand charge transfer (MLCT) peaks for each complex.
PEG-ASP CNAs loaded with 1 and 2 were dissolved in PBS (6 μM Ru complex) and the particle diameter determined as a function of time (t = 0, 24, 48, 72 hrs) using a Zetasizer Nano ZS. The photoluminescence of 3-loaded PEG-ASP CNAs prevented them from being analyzed in this manner. The size of 3-loaded PEG-ASP CNAs relative to empty PEG-ASP CNAs was analyzed at 280 nm and at 450 nm (complex 3 MLCT wavelength) using GPC in a manner identical to the protocol detailed above for empty PEG-ASP CNAs.
2.5. Ru complex photoactivation kinetics
The photoactivation kinetics of free 1 and 2 and 1- and 2-loaded PEG-ASP CNAs were analyzed in PBS and carried out using a Cary 60 spectrometer. Each solution was irradiated with a Dell 1410× 200 W projector fitted with an NT43-941 optical filter (λ>400 nm) (Edmund Optics) and the change in UV/Vis absorption plotted as a function of time. Experiments were conducted in a 1 cm pathlength quartz cuvette located 18 in from the light source. Based on absorption values, photoactivation t1/2 values were calculated using GraphPad Prism 5.0a using a single-phase non-linear exponential regression (indicating a one-step process). However, in the case of free 2, a two-phase non-linear exponential regression produced a better fit, suggesting a two-step ligand exchange process. The photoactivation t1/2 was defined as the time required for half the chemical reaction (as indicated by spectral change) for one process.
2.6. Ru complex release from PEG-ASP CNAs
The rates of release of the Ru complexes from PEG-ASP CNAs were determined in triplicate using dialysis under sink conditions. Ru complex-loaded PEG-ASP CNAs were prepared at a concentration of 100 μg/mL of each Ru complex in diH2O. To determine the effect of irradiation on photoactivatable Ru complex release, 1- and 2-loaded PEG-ASP CNAs were irradiated (200 W, λ>400 nm) for 3 hrs (120 J/cm2) prior to analysis and compared to identical samples protected from light. Each solution was irradiated with a Dell 1410× 200 W projector fitted with an NT43-941 optical filter (λ>400 nm) (Edmund Optics); the projector was measured for the power output generated by the lamp, using a 1918-R meter (Newport Corporation) in the presence of the cutoff filter. The samples were then placed in 3 mL 10,000 MWCO Slide-A-Lyzer dialysis cassettes and dialysis carried out in 5 L of PBS at 37 °C protected from light. The release of the free complexes (not encapsulated) was also carried out to illustrate that once released from the PEG-ASP CNA carrier, the complexes rapidly diffuse out of the dialysis cassettes into the buffer solution. Identical dialysis studies were conducted in 5 L of 10 mM phosphate buffer without NaCl (pH 6.0 and 7.4, 37 °C) to examine the effect of ionic strength and pH on the release rate of Ru complexes from PEG-ASP CNAs. The percentage of Ru complexes remaining inside the CNAs was determined by sampling the Ru complex-loaded PEG-ASP CNA (or free Ru complex) solution inside the dialysis cassettes as a function of time (between 0 and 48 hrs) and measuring the UV/Vis absorption at the wavelength associated with the Ru complex MLCT peak using a Spectroflur Plus microplate reader (Tecan). Based on absorption values, release parameters were calculated using GraphPad Prism 5.0a using a one-phase non-linear exponential regression, except in the case of 3-loaded PEG-ASP CNAs in phosphate buffer with 0 mM NaCl where two-phase non-linear exponential regression produced a better fit.
2.7. Ruthenium complex DNA agarose gel electrophoresis
DNA damage initiated by Ru complexes and Ru complex-loaded PEG-ASP CNAs in the dark and after irradiation (200 W, λ>400 nm) for 1 hr (40 J/cm2) were analyzed using DNA agarose gel electrophoresis using a previously established protocol.1 Briefly, serial dilutions of each Ru complex (0 – 500 μM) were mixed with 40 μg/mL pUC19 plasmid DNA in 10 mM potassium phosphate buffer (PB, pH 7.4), then exposed to light for 60 min (40 J/cm2) or kept in the dark, followed by incubation in the dark for 24 hrs at room temperature. Controls were prepared to create single and double strand DNA breaks. Single strand breaks were induced with 8 μM copper phenanthroline (Cu(Phen)2) mixed with 40 μg/ml pUC19 in 10 mM PB. The reaction was initiated by mixing with DTT and H2O2 (final concentration 1 mM for both) at room temperature for 40 min and used immediately. Double strand breaks were induced with EcoRI using 40 μg/ml pUC19 in EcoR1 buffer. The reaction was performed at 37 °C for 90 min, and stored at −20 °C.
Samples were resolved on 1% agarose gels prepared in tris-acetate (TA) buffer. Each lane was loaded with 0.3 μg of pUC19 and the samples were run for 90 min at 100 mV. The gels were stained with 500 ng/mL ethidium bromide in TA buffer at room temperature for 40 min, washed with TA buffer for 40 min, and imaged using a ChemiDoc™ MP System (Bio-Rad).
2.8 Effect of oxygen on the photoluminescence of 3-loaded PEG-ASP CNAs
The effect of oxygen on the photoluminescence of free 3 and 3-loaded PEG-ASP CNAs was evaluated as an indicator of the 1O2 mediated quenching of 3 entrapped inside PEG-ASP CNAs. As photoluminescence and 1O2 generation are competing pathways, it was expected that when oxygen was removed from the buffer solution the emission of free 3 would increase. On the other hand, the emission of 3-loaded PEG-ASP CNAs should not change if 1O2 generation was already impeded in oxygenated buffer solution due to an inability of oxygen to reach 3 in the core of the PEG-ASP CNA carriers. Deoxygenated diH2O was prepared by purging 20 mL of diH2O with argon (Ar) gas for 1 hr prior to use. Free 3 and 3-loaded PEG-ASP CNAs were prepared at concentrations of 10 μM (500 μL) in air-equilibrated diH2O and Ar gas purged diH2O and the photoluminescence spectra measured using a Jobin Yvon Fluorolog-3 spectrofluorometer (Horiba). The absorbance spectra of free 3 and 3-loaded PEG-ASP CNAs were also obtained using a Cary 60 spectrometer in air-equilibrated diH2O and Ar gas purged diH2O.
2.9. Analysis of Ru complex hydrophobicity
The hydrophobicity of the Ru complexes was quantified by measuring their log P values in triplicate by shake flask method.12 Because the densities of diH2O and n-octanol are quite different, both solvents were saturated with the other prior to testing to prevent volume changes resulting from phase equilibration that would affect the accuracy of log P measurements.13 Prior to testing, 40 mL of n-octanol was mixed with 10 mL of diH2O for 24 hrs to produce a saturated n-octanol solution. Similarly, 40 mL of diH2O was mixed with 10 mL of n-octanol to generate a saturated diH2O solution. While protected from light, 1 and 2 were prepared at 200 μM in diH2O (3 was prepared at 100 μM due to lower solubility in diH2O), diluted 1:1 with n-octanol solution to a total final volume of 1 mL, shaken vigorously for 10 min, and the phases separated by centrifugation at 17,000 × g for 30 sec. The absorbance of both solutions were analyzed using a Cary 60 spectrometer, and log P calculated based on the absorbance values at the wavelength associated with the MLCT peak of each Ru complex. Samples of 1 and 2 were also irradiated with a Dell 1410× 200 W projector fitted with an NT43-941 optical filter for 30 min (20 J/cm2) to form photoactivated 1 ([Ru(bpy)2(H2O)2]Cl2) or photoactivated 2 (initially [Ru(dmbpy)(dip)(H2O)2]Cl2) and analyzed in an identical manner.
2.10. Cell culture
A549 cells were grown in 75 cm2 culture flasks in DMEM supplemented with 10% FBS and 1% pen-strep (stock = 5,000 units/mL pen and 5,000 μg/mL strep) at 37 °C, 5% CO2. Cells were maintained below 70% confluence.
2.11. Ruthenium complex A549 cell cytotoxicity
The anticancer potentials of Ru complexes and Ru complex-loaded PEG-ASP CNAs were evaluated in vitro in A549 human lung adenocarcinoma cells in the dark and after irradiation in triplicate. Cells were placed in opti-MEM supplemented with 1% serum supreme and 1% pen-strep and added to 96-well plates at a density of 1,500 cells/well. The cells were incubated for 6 hrs at 37 °C, 5% CO2 followed by the addition of serial dilutions of 1 or 2 (0 – 300 μM) or 3 (0 – 20 μM) in opti-MEM while protected from light, and incubated for 18 h at 37 °C, 5% CO2. The cells were then irradiated with a 410 Watt 955 Model 900 AJH projector (3M) fitted with an NT43-941 optical filter for 5 min in 30 sec pulses with 30 sec between pulses for a total irradiation time of 5 min, or protected from light, before incubation for 72 hrs at 37 °C, 5% CO2. Resazurin was added at a concentration of 73.3 μM and the cells were incubated for 4 hrs at 37 °C, 5% CO2. Sample fluorescence was measured using a SpectraFluor Plus microplate reader with an excitation wavelength of 560 nm and an emission wavelength of 590 nm. Cells dosed with free Ru complexes were compared with cells dosed with Ru complex-loaded CNAs and to untreated A549 cells.
3 Results and discussion
3.1. Ru complex and PEG-ASP CNA synthesis
Ru complexes (Figure 1) were prepared and characterized as described above. Two of the prepared complexes were photoactivatable (1 and 2), ejecting a bidentate ligand upon exposure to visible light; the third (3) was photostable and capable of generating 1O2. The ligands coordinated with the Ru center were varied to modify the hydrophobicity of the synthesized Ru complexes. 1 was the most hydrophilic of the three selected Ru complexes. For 2, the addition of a single dip ligand produced a Ru complex with moderately higher hydrophobicity than 1, and the replacement of all three coordinating groups with dip ligands in 3 generated a highly hydrophobic Ru complex. Based on literature reports and our previous studies, increased cellular uptake,14 DNA binding,15 and toxicity in the absence of light (dark toxicity)12, 1b were expected for Ru complexes containing one or more dip ligands; however, it was hypothesized that it may be possible to slow the release of Ru complexes from PEG-ASP CNAs into solution by increasing the hydrophobicity of the complex, thereby attenuating toxic effects prior to CNA accumulation at the targeted site.
PEG-BLA was prepared, verified by 1H-NMR, and the benzyl ester groups deprotected to form PEG-ASP. The molecular weight of the block copolymer was estimated based on the size of the BLA peak with respect to the PEG (5k) peak (Figure S1, MW = 9060). PEG-ASP CNAs (Figure 2) were subsequently synthesized as described above, and their physiochemical properties determined (Figure 3). The synthesized PEG-ASP CNAs were confirmed to be nanometer scale (diameter = 19.3 ± 0.8 nm, MW = 340 kDa), were relatively monodisperse (PDI = 0.24 ± 0.08), and possessed a neutral surface charge (−4.1 ± 1.0 mV), making them suitable for Ru complex entrapment.
Figure 3.
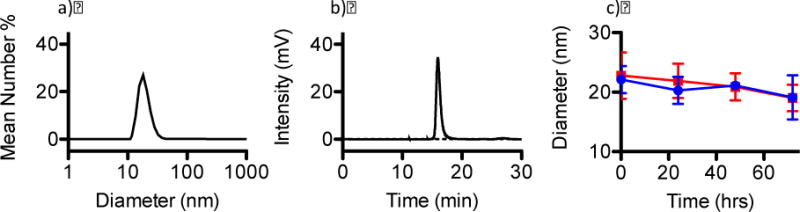
a) Empty PEG-ASP CNA diameter determined by DLS, b) empty PEG-ASP CNA molecular weight estimated based on GPC, and c) 1- and 2-loaded CNA stability was determined by DLS (1-loaded PEG-ASP CNAs (
 ) and 2-loaded PEG-ASP CNAs (
) and 2-loaded PEG-ASP CNAs (
 )).
)).
3.2. Ru complex-loaded CNA preparation and characterization
The PEG-ASP CNA system was selected for entrapment of Ru complexes due to the convenience of physical entrapment and attractive ionic interactions between positively charged Ru complexes and negatively charged aspartate (ASP) groups. To ensure maximum Ru complex loading, ASP groups were converted to their sodium salt form, which enabled Ru complexes to interact with the PEG-ASP CNAs more readily. In addition to the advantage of straightforward Ru complex loading, CNA systems should remain stable in vivo due to the cross-linking of the CNA core.11b After Ru complex-loaded PEG-ASP CNAs were purified by dialysis, the loading efficiency and total amount of entrapped Ru complexes were determined based on absorbance at the MLCT peak of each Ru complex. The wavelengths and MLCT extinction coefficients (ε) used for loading determination were 452 nm (ε = 14,000 M−1cm−1)1, 465 nm (ε = 18,600 M−1cm−1), and 450 nm (ε = 29,500 M−1cm−1)9 for 1, 2, and 3 respectively. Satisfactory loading efficiencies and weight % of the Ru complex-loaded PEG-ASP CNAs were observed: 84% (21 wt %), 68% (17 wt %), and 88% (22 wt %) for 1, 2, and 3 respectively with a maximum theoretical loading of 25 wt %. High loading (a maximum of approximately 20 wt %), in addition to suitable physiochemical properties noted above (nanometer scale, monodisperse, and neutral surface charge), demonstrate that PEG-ASP CNAs can act as a carrier for polypyridyl Ru complexes.
Often, drug entrapment in a nanoparticle delivery vehicle alters the physiochemical properties of the system, which in turn changes its pharmacological properties. For example, it is common for the size or surface charge of a nanoparticle carrier to be altered, leading to significantly different cellular interactions, circulation time, and in vivo biodistribution.16 Because of this, it was important to analyze the effect that Ru complex loading has on PEG-ASP CNAs. The stabilities of Ru complex-loaded PEG-ASP CNAs were analyzed in solution using DLS to ensure that no significant aggregation occurred over time, which could negatively influence the pharmacological properties of the PEG-ASP CNA delivery system. Only 1- and 2-loaded PEG-ASP CNAs could be studied in this fashion because the photoluminescence of 3 interfered with DLS particle size determination, making 3-loaded PEG-ASP CNAs appear much larger than their actual size. GPC analysis of 3-loaded PEG-ASP CNAs revealed an elution time of 15.7 min, which was very close to the elution time of empty PEG-ASP CNAs (16.0 min) (Figure S6). This indicates that the size of 3-loaded PEG-ASP CNAs was similar to empty PEG-ASP CNAs. In addition, the fact that 3-loaded PEG-ASP CNAs displayed absorbance at 450 nm confirms the presence of 3 because empty PEG-ASP CNAs do not absorb at this wavelength.
Figure 3 shows that the entrapment of 1 and 2 inside PEG-ASP CNAs did not alter the diameter of PEG-ASP CNAs (20 nm) and that the formulations were stable in PBS at room temperature for up to 72 hrs. The surface charge of 1-loaded PEG-ASP CNAs was also confirmed to be neutral (−7.4 ± 0.6 mV), comparable to empty PEG-ASP CNAs (−4.1 ± 1.0 mV), illustrating that positively charged Ru complexes are located primarily in the core of PEG-ASP CNAs. This suggests that positively charged Ru complexes can be effectively shielded from solution inside PEG-ASP CNAs. These qualities indicate that Ru complex-loaded PEG-ASP CNA formulations are generally stable in solution and that Ru complex entrapment does not adversely affect the physiochemical properties of the PEG-ASP CNA system. In addition, the small size of the PEG-ASP CNAs (diameter < 20 nm) compared to other nanoparticle platforms may potentially improve cell internalization and enable better diffusion through tumor tissue.
3.3. Photoactivation kinetics of free Ru complexes and Ru complex-loaded PEG-ASP CNAs
The rate of Ru complex photoactivation is a critical variable in successful therapeutic application. If a Ru complex is extremely sensitive to light it may undergo premature reaction prior to administration, and if the Ru complex is exceptionally photostable it may not experience sufficient photoactivation when irradiated with light, resulting in poor activity. Ligand photoejection from free 1 and 2 and 1- and 2-loaded PEG-ASP CNAs to form photoactivated 1 ([Ru(bpy)2(H2O)2]Cl2) or 2 (initially [Ru(dmbpy)(dip)(H2O)2]Cl2) was analyzed and the photoactivation kinetics modeled to compare the photosensitivity of the two complexes and to elucidate effects that PEG-ASP CNA entrapment may have on complex photoactivation. Both 1 and 2 formulations were stable in low light at room temperature and ejected rapidly when exposed to a light source (200 W, λ>400 nm). The photo-stability of 3 was confirmed by verifying that its UV/Vis absorbance spectrum did not change after a 30 min irradiation time (200 W, λ>400 nm). Free 1 and 1-loaded CNA photoactivation were modelled based on a single-phase non-linear exponential regression (consistent with a single step process), and displayed comparable photoactivation t1/2 values of 15.1 ± 0.7 sec and 21.1 ± 0.2 sec (Figure 4a–b). In contrast, the photoactivation kinetics of free 2 were more complex than 2-loaded PEG-ASP CNAs (Figure 4c–d). While 2-loaded PEG-ASP CNAs exhibited a single-phase process with a clear isosbestic point, free 2 was characterized by a primary phase that closely matched 2-loaded PEG-ASP CNA photoactivation, followed by a secondary phase exhibiting a second isosbestic point. As a result, free 2 photoactivation was modelled based on a two-phase non-linear exponential regression (consistent with a two step process) while 2-loaded PEG-ASP CNA photoejection followed a single-phase non-linear exponential regression similar to free 1 and 1-loaded PEG-ASP CNAs. The primary phase photoactivation t1/2 values of free 2 and 2-loaded PEG-ASP CNAs were 35.7 ± 1.5 and 51.5 ± 0.7 sec and the secondary phase photoactivation t1/2 value of free 2 was 577.4 ± 47.6 sec. The identity of the photoejected ligand in all cases was also confirmed to be dmbpy by HPLC (Figure S2–S4).
Figure 4.
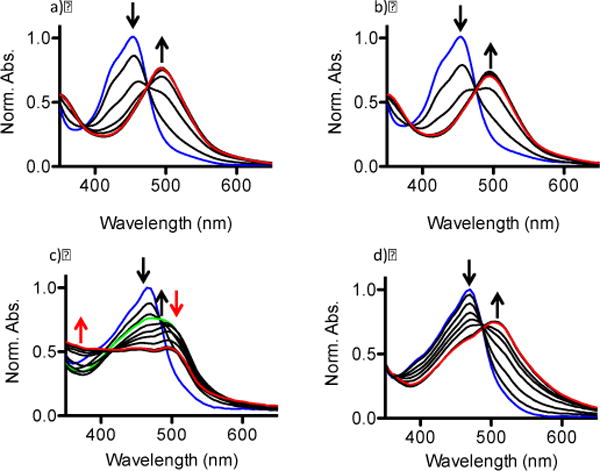
Photoejection kinetics of a) free 1, b) 1-loaded PEG-ASP CNAs, c) free 2, and d) 2-loaded PEG-ASP CNAs. The blue lines indicate the spectra of initial Ru complex prodrugs, red lines indicate final photoactivated product(s), and the green line in c) indicates a transition between the primary and secondary phases (t = 90 sec).
The more complex photochemistry of 2 implies that when entrapped in PEG-ASP CNAs, 2 was converted from a single starting material to a single product, but when it was free in solution it was further converted to a third product. This may result from the ejection of the second dmbpy ligand or from a rearrangement of the remaining chelated ligands. In contrast, interactions between 2 and PEG-ASP CNAs seemed to stabilize photoactivated 2, preventing further alteration of its chemical structure, possibly resulting from interactions between ASP groups and photoactivated 2. This outcome indicates that entrapment of Ru complexes inside PEG-ASP CNAs does not significantly affect photoactivation kinetics; however, if a particular complex is prone to ligand rearrangement or secondary ligand photoactivation, entrapment in PEG-ASP CNAs may offer increased complex stability, likely due to interactions with PEG-ASP CNA ASP groups.
3.4. Release of Ru complexes from PEG-ASP CNAs
There are a number of competing forces involved in the entrapment and retention of polypyridyl Ru complexes inside PEG-ASP CNAs, including hydrophobic effects and electrostatic interactions. Photoactivation of 1 and 2 further complicated the situation by introducing the possibility of secondary coordination with ASP groups inside the PEG-ASP CNAs. As the goal of this system is retention of Ru complexes inside PEG-ASP CNAs in the dark followed by quick release after light activation of the complex, it was important to understand the effect of Ru complex hydrophobicity, buffer ionic strength, and buffer pH on the release rate and maximum amount of Ru complex released from PEG-ASP CNAs. Ru complex-loaded PEG-ASP CNAs were irradiated and the release of Ru complexes from the PEG-ASP CNAs compared to their prodrug forms in PBS at 37 °C and pH 7.4. The release profiles (Figure 5) and kinetics (Table 1) for Ru complex-loaded PEG-ASP CNAs were modelled based on a one-phase non-linear exponential regression. The hydrophobicity of the selected complexes was used as a predictor of Ru complex release rate.
Figure 5.
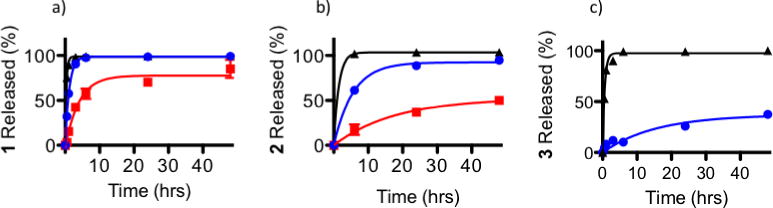
Percent of a) 1, b) 2, and c) 3 PEG-ASP CNA-entrapped Ru complexes released from 10K MWCO dialysis cassettes in PBS (pH 7.4) at 37 °C (PEG-ASP CNAs protected from light (
) and light irradiated PEG-ASP CNAs (
)) compared to free Ru complexes released from 10K MWCO dialysis cassettes (
 ). Because 3 was a photostable Ru complex, release was only evaluated when protected from light. The MLCT peaks of the entrapped Ru complexes were monitored in order to determine the amount of each Ru complex remaining inside the PEG-ASP CNAs, and this was used to calculate the amount of Ru complexes released as a function of time (N = 3).
). Because 3 was a photostable Ru complex, release was only evaluated when protected from light. The MLCT peaks of the entrapped Ru complexes were monitored in order to determine the amount of each Ru complex remaining inside the PEG-ASP CNAs, and this was used to calculate the amount of Ru complexes released as a function of time (N = 3).
Table 1.
Summary of the release kinetics of Ru complexes from PEG-ASP CNAs when protected from light and after light exposure (photoactivated) in PBS (pH 7.4) Uncertainty expressed as standard error (N = 3).
| 1 | 2 | 3 | 1 | 2 | |
|---|---|---|---|---|---|
|
|
|||||
| Dark | Photoactivated | ||||
|
|
|||||
| log P | −1.85 ± 0.04 | −1.27 ± 0.03 | 1.80 ± 0.02 | −1.84 ± 0.02 | −0.39 ± 0.11 |
| t1/2 (hrs) | 0.8 ± 0.03 | 3.9 ± 0.3 | 10.7 ± 2.4 | 3.0 ± 0.4 | 12.3 ± 2.7 |
| Release (%) | 98.7 | 92.4 | 37.0 | 77.7 | 52.6 |
In general, hydrophobic molecules are internalized well by cells but require additional formulation steps to improve their water solubility and enable successful delivery in vivo. On the other hand, while hydrophilic molecules are more straightforward to deliver due to their high water solubility, they are taken up poorly by cells and are often rapidly cleared from circulation.17 Entrapment inside PEG-ASP CNAs offers advantages to both hydrophobic and hydrophilic Ru complexes as the solubility of hydrophobic Ru complexes could be enhanced without the addition of potentially toxic excipients, while circulation time and cell internalization of hydrophilic complexes could be improved. However, it was expected that the release rate of Ru complexes would decrease with increasing complex hydrophobicity, making the relatively hydrophobic complexes, 2 and 3, more attractive delivery targets than 1, which is highly hydrophilic. This was supported by release studies where both the release rate and the maximum amount of Ru complex released were related to complex hydrophobicity and coordination state (prodrug or photoactivated form).
In PBS at 37 °C, when protected from light, the t1/2 values of Ru complex release increased with increasing hydrophobicity of the complex. The t1/2 of 1 release was 0.8 ± 0.03 hrs, the t1/2 of 2 release was 3.9 ± 0.3 hrs, and the t1/2 of 3 release was 10.7 ± 2.4 hrs (Table 1). This correlated well with log P values, which were −1.85 ± 0.04, −1.27 ± 0.03, and 1.80 ± 0.02 for 1, 2, and 3 respectively. Interestingly, irradiation caused the release rate of 1 and 2 from PEG-ASP CNAs to slowed significantly (p < 0.05). After photoactivation, the t1/2 of Ru complex release increased approximately 3× to 3.0 ± 0.4 hrs for photoactivated 1 and to 12.3 ± 2.7 hrs for photoactivated 2. The amount of Ru complex released decreased from 98.7% to 77.7% for photoactivated 1 and from 92.4% to 52.6% for photoactivated 2. Photoactivation did not change the hydrophobicity of 1, but the hydrophobicity of photoactivated 2 was significantly higher than its prodrug form (−0.39 ± 0.11 (p < 0.05)). This suggests that complex hydrophobicity alone is not sufficient to explain the decelerated release of photoactivated complexes. It seems likely that additional factors contributed to slowed release of photoactivated 1 and 2 from PEG-ASP CNAs, including secondary coordination with PEG-ASP CNA ASP groups after the photoejection of a dmbpy ligand (similar to the interactions noted above that are believed to stabilize photoactivated 2).
In order to ascertain the effect of ionic strength and pH on Ru complex release from PEG-ASP CNAs, dialysis release experiments were also conducted in 10 mM phosphate buffer without NaCl at pH 6.0 and at 7.4 (37 °C) (Tables 2 and 3 and Figures 6 and 7). As the Ru complexes were loaded into PEG-ASP CNAs utilizing ionic interactions between the positively charged complexes and the negatively charged ASP groups of the PEG-ASP CNAs, t1/2 values for the release of Ru complexes from PEG-ASP CNAs were expected to vary inversely with the ionic strength of the buffer due to competitive binding of Ru complexes and ASP groups by ions in solution. This was the case for 1- and 2-loaded PEG-ASP CNAs in the dark, where the decrease in buffer ionic strength slowed the release of 1 (t1/2 of release increased from 0.8 ± 0.03 hrs to 2.1 ± 0.2 hrs) and 2 (t1/2 of release increased from 3.9 ± 0.3 hrs to 8.8 ± 0.6 hrs) (p < 0.05). Similarly, the amount of 1 and 2 released decreased with decreasing buffer ionic strength, going from 98.7% to 90.0% for 1 and from 92.4% to 77.0% for 2 when the concentration of NaCl was lowered from 100 mM to 0 mM. These results indicate that the rate of release and the amount of complex released vary directly with buffer ionic strength for hydrophilic Ru complexes.
Table 2.
Summary of the effect of buffer ionic strength on the release kinetics of 1, 2, and 3 from PEG-ASP CNAs when protected from light and after light exposure (photoactivated) in phosphate buffer (0 mM NaCl) and PBS (100 mM NaCl) (pH 7.4). Uncertainty expressed as standard error (N = 3).
| 1 | 2 | 3a | 1 | 2 | ||
|---|---|---|---|---|---|---|
| Dark | Photoactivated | |||||
| 0 mM NaCl | t1/2 (hrs) | 2.1 ± 0.2 | 8.8 ± 0.6 | 0.2 ± 0.07 (13.5 ± 4.8) | 9.9 ± 1.1 | 13.9 ± 1.8 |
| Release (%) | 90.0 | 77.0 | 45.2 (51.9) | 65.8 | 55.9 | |
|
| ||||||
| 100 mM NaCl | t1/2 (hrs) | 0.8 ± 0.03 | 3.9 ± 0.3 | 10.7 ± 2.4 | 3.0 ± 0.4 | 12.3 ± 2.7 |
| Release (%) | 98.7 | 92.4 | 37.0 | 77.7 | 52.6 | |
The release of 3 in solution with 0 mM NaCl was fit to a two-phase non-linear exponential regression. The first t1/2 value corresponds to release in the primary phase while the t1/2 value in parenthesis corresponds to release in the secondary phase. The first value for release indicates the percentage of 3 released in the primary phase while the value in parenthesis indicates the total percentage of 3 released in both phases.
Table 3.
Summary of the effect of buffer pH on the release kinetics of 1, 2, and 3 when protected from light and after light exposure (photoactivated) in phosphate buffer. Uncertainty expressed as standard error (N = 3).
| 1 | 2 | 3a | 1 | 2 | ||
|---|---|---|---|---|---|---|
| Dark | Photoactivated | |||||
| pH 7.4 | t1/2 (hrs) | 2.1 ± 0.2 | 8.8 ± 0.6 | 0.2 ± 0.07 (13.5 ± 4.8) | 9.9 ± 1.1 | 13.9 ± 1.8 |
| Release (%) | 90.0 | 77.0 | 45.2 (51.9) | 65.8 | 55.9 | |
|
| ||||||
| pH 6.0 | t1/2 (hrs) | 1.9 ± 0.2 | 7.7 ± 0.6 | 0.2 ± 0.06 (9.0 ± 2.1) | 7.5 ± 0.7 | 10.1 ± 1.1 |
| Release (%) | 95.3 | 72.5 | 45.0 (46.4) | 70.0 | 50.3 | |
The release of 3 in solution with 0 mM NaCl was fit to a two-phase non-linear exponential regression. The first t1/2 value corresponds to release in the primary phase while the t1/2 value in parenthesis corresponds to release in the secondary phase. The first value for release indicates the percentage of 3 released in the primary phase while the value in parenthesis indicates the total percentage of 3 released in both phases.
Figure 6.
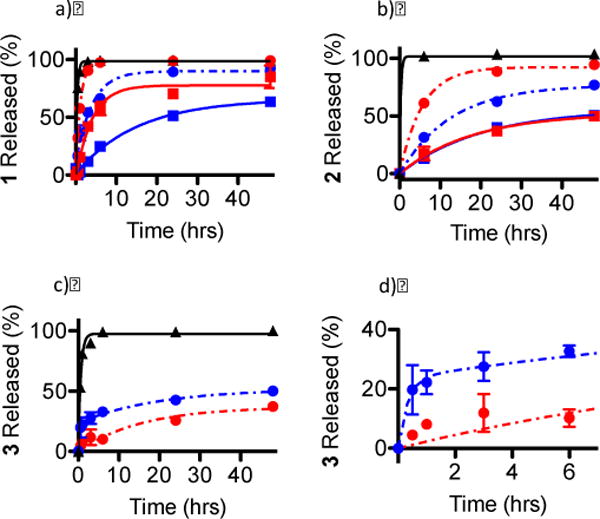
The effect of buffer ionic strength on release from PEG-ASP CNAs for a) 1, b) 2, and c) 3 (free complex (
), dark phosphate buffer (
 ), dark PBS (
), dark PBS (
 ), light phosphate buffer (
), light phosphate buffer (
 ), and light PBS (
)); d) shows a magnification of the rapid release of 3 from PEG-ASP CNAs when placed in a solution with low ionic strength (0 mM NaCl) (N = 3). Because 3 was a photostable Ru complex, release was only evaluated when protected from light.
), and light PBS (
)); d) shows a magnification of the rapid release of 3 from PEG-ASP CNAs when placed in a solution with low ionic strength (0 mM NaCl) (N = 3). Because 3 was a photostable Ru complex, release was only evaluated when protected from light.
Figure 7.
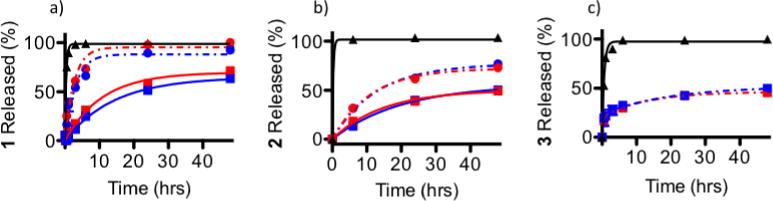
The effect of phosphate buffer pH on complex release from PEG-ASP CNAs through 10K MWCO dialysis cassettes for a) 1, b) 2, and c) 3 (dark pH 6.0 (
), dark pH 7.4 (
), light pH 6.0 (
), and light pH 7.4 (
)) compared to the release of free complexes from 10K MWCO dialysis cassettes (
) (N = 3). Because 3 was a photostable Ru complex, release was only evaluated when protected from light (N = 3).
The decrease in buffer ionic strength slowed the release of 1 after irradiation, (t1/2 of release increased from 3.0 ± 0.4 hrs to 9.9 ± 1.1 hrs for photoactivated 1) (p < 0.05) but did not affect the release of 2 (t1/2 values of release were 12.3 ± 2.7 in the dark and 13.9 ± 1.8 hrs for photoactivated 2). The amount of 1 released was slightly diminished with decreasing buffer ionic strength, going from 77.7% to 65.8% for photoactivated 1, while the amount of 2 released increased slightly, going from 52.6% to 55.9% for photoactivated 2. The fact that the t1/2 for the release of photoactivated 1 increased 3× at 0 mM NaCl compared to 100 mM NaCl while the t1/2 for the release of photoactivated 2 was not statistically different is informative. This likely indicates that the strength of the interaction between photoactivated 2 and the PEG-ASP CNAs was stronger than between photoactivated 1 and PEG-ASP CNAs, possibly due to secondary coordination between photoactivated 2 and ASP groups rather than purely electrostatic interactions between anionic ASP groups and the cationic Ru2+ complexes. It is also possible that any acceleration in the release of photoactivated 2 resulting from higher ionic strength of the solution was negated by additional hydrophobic interactions between complexes due to an increase in the hydrophobicity of 2 after photoactivation. The fact that the decrease in ionic strength led to the release of a slightly greater amount of 2 may point toward the involvement of hydrophobic interactions. Once again, for a hydrophilic Ru complex (photoactivated 1) the release rate and amount of complex released were directly related to ionic strength, whereas for the more hydrophobic complex (photoactivated 2) the release rate and amount of complex released were inversely related to buffer ionic strength.
Remarkably, buffer ionic strength had a large impact on the release behavior of 3. The most significant effect of the decrease in ionic strength on the release of 3 was a pronounced shift from one step (at 100 mM NaCl the release t1/2 was 10.7 ± 2.4 hrs) to two-step kinetics (at 0 mM NaCl the release t1/2 values were 0.2 ± 0.07 hrs (fast) and 13.5 ± 4.8 hrs (slow) with 45.2% of release occurring during the fast phase; see Table 2). The total amount of 3 released also increased when the ionic strength was decreased, going from 37.0% to 51.9%. Due to the combination of high hydrophobicity and positive charge, considerable hydrophobic effects and electrostatic interactions exist between 3 and PEG-ASP CNAs. The two-step release behavior of 3 in the absence of NaCl was likely due to the combined influence of these two effects; however, when the ionic strength of the solution was increased, the electrostatic interactions were eliminated, leaving only the hydrophobic interactions; hence the transition from two-phase to one-phase release kinetics. The release of nearly half of the maximum amount of 3 within 12 min in the absence of NaCl may suggest that hydrophobic interactions were weak compared to electrostatic repulsion between complexes, which led to rapid release; however, when the buffer ionic strength was increased by adding 100 mM NaCl, the solvation layer around 3 was compressed, decreasing the repulsion between complexes and promoting hydrophobic interactions. Once again, an inverse relationship was observed between release rate and the amount of complex released and buffer ionic strength for a hydrophobic complex (3). Based on these results, it seems that hydrophobic interactions resulting from the strong hydrophobic character of 3 were the primary factor controlling the interactions between the complex and the PEG-ASP CNAs.
The effect of buffer pH on Ru complex release was also investigated. It was suspected that partial protonation of ASP groups in the PEG-ASP CNAs may accelerate the release of complexes by reducing ionic interactions with the cationic Ru complexes. However, somewhat surprisingly, the effect of pH on the release of the Ru complexes from PEG-ASP CNAs was found to be negligible both in the dark and after irradiation. From this, it appeared that protonation of ASP groups was insufficient at pH 6.0 to promote more rapid release of polypyridyl Ru complexes.
It was theorized that if secondary coordination between charged photoactivated Ru complexes and PEG-ASP CNA ASP groups (which may be observable with a shift in the MLCT peak for the Ru complex) accounted for the slowed release of 2, that the interaction could be disrupted by increasing buffer ionic strength. The MLCT peaks of 1 and 2 were compared in diH2O and in PBS to test this hypothesis (Figure S5). When protected from light, 1-loaded PEG-ASP CNAs were indistinguishable from free 1, while a small red-shift was observed for 2-loaded PEG-ASP CNAs compared to free 2. After photoactivation, the MLCT peak of 1-loaded PEG-ASP CNAs was red-shifted in diH2O; however in PBS it was indistinguishable from free 1 after photoactivation. This hints that inside PEG-ASP CNAs 1 interacts with ASP groups after photoactivation and that increasing buffer ionic strength disrupts this interaction. Conversely, the MLCT peak of photoactivated 2 inside PEG-ASP CNAs was identical in diH2O and PBS, which adds support to the theory that the interaction between photoactivated 2 and PEG-ASP CNAs is stronger than for 1, as it was not affected by an increase in buffer ionic strength. Indeed, this was reinforced by the greater effect of buffer ionic strength on the release rate of photoactivated 1 compared to 2 detailed previously (when the NaCl concentration was increased from 0 mM to 100 mM photoactivated 1 was released 3× faster while photoactivated 2 was not released significantly faster).
Based on these results, it appears that by varying the relative hydrophobicity of the Ru complexes it is possible to control the release rate and the amount of Ru complexes released from PEG-ASP CNAs. While the observed decelerated release of photoactivated Ru complexes is undesirable for a light triggered system, an analogous approach could be readily applied to improve the pharmacological properties of any metal-based drugs with weak coordinating ligands, including many Pt-based drugs as well as Ru therapeutics in clinical trials (e.g., KP1019 and NAMI-A). Coordination of such compounds inside PEG-ASP CNAs could offer improved circulation time and enhanced tumor accumulation. The roles that hydrophobicity and charge played in Ru complex release also offer insight into general drug carrier development. The release rate of the more hydrophobic 2 (both intact and after photoactivation), as well as 3 compared favorably to many liposomal formulations with the added benefit that PEG-ASP CNAs are stable in vivo due to core cross-linking.18 While the use of ionic interactions was effective for loading positively charged complexes inside negatively charged PEG-ASP CNAs, they were quickly disrupted at physiologically relevant salt concentrations, leading to rapid release from PEG-ASP CNAs. Modulation of the hydrophobic character of Ru complexes seemed to be a more effective strategy than charge-based attractions to affect the release rate. Based on these observations, it seems that ideal drug candidates for entrapment in PEG-ASP CNAs should possess a positive charge (for high loading) and have high hydrophobicity (to slow release). Moreover, it is straightforward to alter the core chemistry of CNAs to allow the loading of negatively charged drugs as well. As many drugs are hydrophobic and bear a charge under certain conditions, there are many possible applications for such CNA systems. With proper CNA core design, sustained release can likely be achieved in vivo.
3.5. Interactions of Ru complex-loaded PEG-ASP CNAs with plasmid DNA
The mechanism of action of a number of Ru complexes has been linked to interactions with DNA.2e, 3c, 3d, 4a, 4b, 5b, 5c Whether a result of DNA strand cleavage due to 1O2 generation or from DNA cross-linking, these interactions are often thought to play a role in determining the cytotoxicity of Ru complexes. For this reason, it was important to assess the covalent DNA binding (with complexes 1 and 2) or DNA cleavage (with complex 3) of the selected free Ru complexes and Ru complex-loaded PEG-ASP CNAs.
The DNA interactions of free Ru complexes and Ru complex-loaded PEG-ASP CNAs were investigated by agarose gel electrophoresis using supercoiled plasmid DNA (Figure 8). In general, reduction in DNA migration was used as an indicator of DNA binding while strand cleavage was assessed by comparison of supercoiled to relaxed circle or linear DNA. In both cases, a DNA interaction EC50 was defined as the concentration at which one half of the maximum effect of a particular Ru complex was observed. Free 1 did not interact with DNA when protected from light, while free 2 and 3 led to DNA precipitation at high concentrations (EC50 = 62.5 – 125 μM and EC50 = 31.3 – 62.5 μM, Figure S7). It was also noted that 1-loaded PEG-ASP CNAs did not interact with DNA when protected from light indicating that PEG ASP CNAs alone do not have an effect on DNA. After photoactivation, free 1 displayed significant DNA binding (EC50 = 7.8 – 15.6 μM), 2 bound DNA at very low concentrations (EC50 < 7.8 μM), and 3 generated single strand DNA breaks (EC50 = 15.6 – 31.3 μM) analogous to a single strand break control (Cu(Phen)2). When 1 was entrapped in PEG-ASP CNAs, the DNA binding by the photoactivated complex decreased (EC50 = 31.3 – 62.5 μM), likely due to delayed complex release over the course of the experiment and competitive binding of photoactivated Ru complexes by the PEG-ASP CNA ASP groups. At the same time, when 2- and 3-loaded PEG-ASP CNAs were irradiated, they experienced a complete suppression of DNA binding and strand cleavage. For 2, this can be correlated with the significantly slower release of the photoactivated complex and once again may be related to the combined effects of the increase in complex hydrophobicity after photoactivation and secondary coordination with ASP groups. Similar to 2-loaded PEG-ASP CNAs, 3-loaded PEG-ASP CNAs likely failed to interact with DNA due to slow complex release. Based on agarose gel electrophoresis experiments, it is once again apparent that the release rate of Ru complexes is inversely proportional to complex hydrophobicity and that under these experimental conditions PEG-ASP CNAs can shield Ru complex-DNA interactions, including those mediated by the sensitization of 1O2, as oxygen may not readily penetrate into the CNA, preventing generation of 1O2.19
Figure 8.
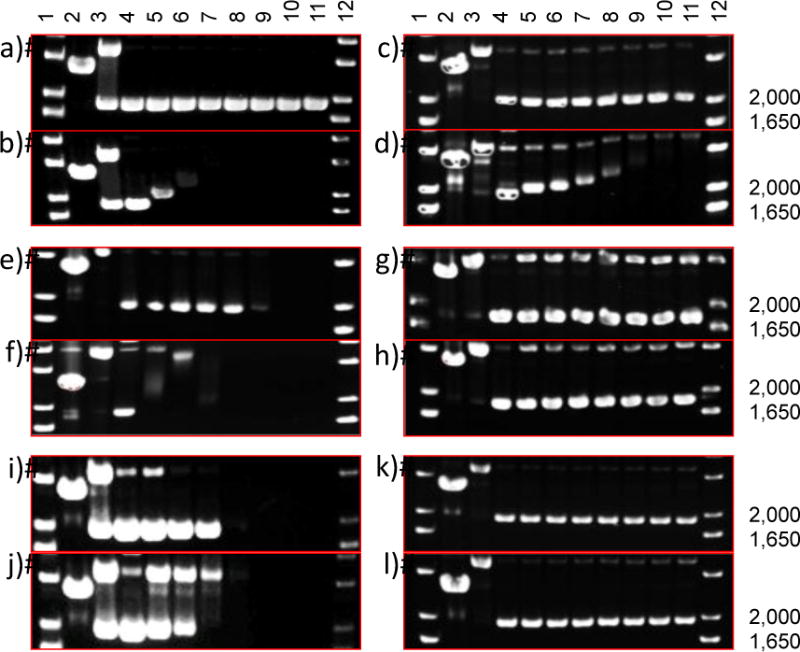
Agarose gel electrophoresis of 40 μg/mL pUC19 plasmid (10 mM phosphate buffer, pH 7.4) with light-sensitive Ru complexes. The supercoiled plasmid form migrates at 2000 bp, relaxed circle form is ~4000 bp, and linear form is just below 3000 bp. Dose response profiles: a) free 1 in the dark and b) photoactivated (40 J/cm2; 200 W source (λ> 400 nm)), c) 1-loaded PEG-ASP CNAs in the dark and d) photoactivated, e) free 2 in the dark and f) photoactivated, g) 2-loaded PEG-ASP CNAs in the dark and h) photoactivated, i) free 3 in the dark and j) photoactivated, and k) 3-loaded PEG-ASP CNAs in the dark and l) photoactivated. Lanes 1 and 12, DNA molecular weight standard; lane 2, linear pUC19; lane 3, relaxed circle (Cu(phen)2 reaction with pUC19); lanes 4–11, 0, 7.5, 15, 30, 60, 120, 240, and 500 μM complexes.
3.6 Effect of oxygen on the photoluminescence of 3-loaded PEG-ASP CNAs
It was hypothesized that oxygen may not readily penetrate into the cores of the PEG-ASP CNA carriers, resulting in a signfiicant reduction or complete elimination of 1O2 generation from 3-loaded PEG-ASP CNAs. In order to test this idea, free 3 and 3-loaded PEG-ASP CNAs were prepared in air-equilibrated and deoxygenated (Ar gas purged) diH2O and their emission spectra compared (Figure 9). The UV/Vis absorbance spectra were also collected and are included in Figure 9 to demonstrate that differences in photoluminescence are not a result of differences in complex concentration.
Figure 9.
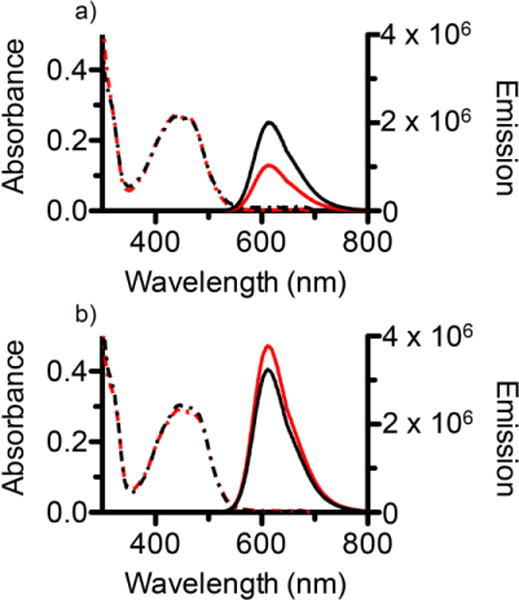
UV/Vis absorbance spectra (hatched lines) and emission spectra (solid lines) in air-equilibrated diH2O (red) and deoxygenated (Ar purged) diH2O (black) for a) free 3, and b) 3-loaded PEG-ASP CNAs.
In air-equilibrated diH2O the emission of free 3 (500 μL, 10 μM) was 1.0 × 106 counts per second (CPS). This value doubled to 2.0 × 106 CPS when the solution was deoxygenated with Ar. Compared to these values, the emission of 3-loaded PEG-ASP CNAs was noticeably higher in air-equilibrated diH2O (3.8 × 106 CPS) and in deoxygenated diH2O (3.2 × 106 CPS). In all cases, the maximum emission was at a wavelength of approximately 615 nm. As expected, the photoluminescence of free 3 increased substantially when the buffer solution was deoxygenated due to a reduction in 1O2 generation. In contrast, this trend was not observed for 3-loaded PEG-ASP CNAs where there was actually a slight decrease in photoluminescence after deoxygenation. These results support the theory that oxygen is not able to penetrate into the core of PEG-ASP CNAs, and as a result the removal of oxygen from the buffer solution had no effect on photoluminescence. In addition, the fact that the emission of 3-loaded PEG-ASP CNAs was higher than for an equal amount of free 3 in deoxygenated buffer solution further suggests that little to no 1O2 is being generated. Instead, the majority of incident light is utilized to generate photoluminescence.
3.7. In vitro cell cytotoxicity of free Ru complex and Ru complex-loaded PEG-ASP CNAs
While the binding or cleavage of DNA is generally regarded as central to the mechanism of action for many Ru complexes, DNA binding studies do not fully replicate the multifaceted interactions that occur when a complex enters a cell. In order to gain a more complete understanding of the manner in which the physiochemical and photodynamic properties of Ru complexes and their encapsulation in PEG-ASP CNAs affects cellular activity, the in vitro cytotoxicity of selected Ru complexes was assessed. Ru complex-loaded PEG-ASP CNAs were expected to display light-sensitive activity comparable to free Ru complexes; however, it was hypothesized based on the results of DNA agarose gel electrophoresis experiments that entrapment in PEG-ASP CNAs may alleviate the toxicity of 2 and 3 in the dark by shielding the Ru complexes from cellular interactions.
The in vitro cytotoxicity of free Ru complexes and Ru complex-loaded PEG-ASP CNAs was analyzed in A549 human lung adenocarcinoma cells. Briefly, A549 cells were incubated overnight with free or PEG-CNA entrapped Ru complexes, activated with light (λ>400 nm) in 30 sec pulses with 30 sec between pulses for a total irradiation time of 5 min, then cytotoxicity was assessed after 72 hrs. Cell cytotoxicity curves are shown in Figure S8 and Ru complex and cisplatin light and dark EC50 values and phototoxicity indices (PIs) are included in Table 4. PEG-ASP CNAs have been found to be relatively nontoxic (EC50>10 mg/mL20; Figure S18), and dmbpy has an EC50 of 80 μM; thus the cytotoxicity was attributed to Ru complex activity. Additionally, no adverse effects (body weight loss or hepatotoxicity) have been observed in mice administered PEG-ASP CNAs doses up to 100 mg/kg (unpublished data). The activity of free Ru complexes and Ru complex-loaded PEG-ASP CNAs was similar for all formulations. The EC50 values were greater than 300 μM for free 1 and 1-loaded PEG-ASP CNAs when protected from light. After photoactivation, free 1 was slightly more active than 1-loaded PEG-ASP CNAs (8.3 ± 1.2 μM and 12.9 ± 1.1 μM) (p < 0.05). The EC50 values of free 2 and 2-loaded PEG-ASP CNAs were indistinguishable when protected from light (9.7 ± 1.3 μM and 9.6 ± 1.4 μM) and after irradiation (3.9 ± 1.1 μM and 2.6 ± 1.1 μM). For free 3 and 3-loaded PEG-ASP CNAs, the EC50 values were also indistinguishable when protected from light (0.6 ± 1.1 μM and 0.6 ± 1.2 μM) and after irradiation (0.1 ± 1.0 μM and 0.1 ± 1.1 μM). Free 1 and 1-loaded PEG-ASP CNAs displayed the largest PI (36.1 and 23.3), while the PIs of free 2, 2-loaded PEG-ASP CNAs, free 3, and 3-loaded PEG-ASP CNAs were similar (2.5, 3.7, 6.0, and 6.0 respectively). These values were comparable to photostable cisplatin, which as expected had the same EC50 when protected from light and after irradiation (2.5 ± 0.6 μM) and a PI of 1.
Table 4.
A549 cell cytotoxicity (EC50) and phototoxicity indices for Ru complexes (N = 3) after 5 min light irradiation (λ>400 nm).
| Complex | Formulation | EC50(μM)
|
Phototoxicity Index, Pia | |
|---|---|---|---|---|
| Dark | Light | |||
| 1 | Free | >300 | 8.3 ± 1.2 | > 36.1 |
| CNA | >300 | 12.9 ± 1.1 | >23.3 | |
| 2 | Free | 9.7 ±1.3 | 3.9 ± 1.1 | >2.5 |
| CNA | 9.6 ± 1.4 | 2.6 ±1.1 | >3.7 | |
| 3 | Free | 0.6 ± 1.1 | 0.1 ±1.0 | >6 |
| CNA | 0.6 ±1.2 | 0.1 ±1.1 | >6 | |
| cisplatin | 2.5 ± 0.6 | 2.5 ±0.4 | 1 | |
The Phototoxicity Index (PI) is a ratio of the dark and light A549 EC50 values (see Figures S8 and S18 for cell viability curves).
It is important to highlight that cells were dosed with Ru complex-loaded PEG-ASP CNA formulations and incubated in the dark for 18 hrs before irradiation. Based on experimental data, near complete release of 1 and 2 was expected prior to photoactivation, which accounts for the similarities in the cytotoxicity of free and PEG-ASP CNA formulations. On the other hand, these results ran counter to the slow release kinetics of 3 (approximately 20% released after 18 hrs) and the previous DNA binding study in which the DNA interactions of 3 were almost completely inhibited after the complex was entrapped in PEG-ASP CNAs. To further examine the effect of PEG-ASP CNA entrapment on Ru complex efficacy a study was conducted that examined the potency of PEG-ASP CNA formulations of 1 and 3 compared to the complexes free in solution when exposure time was limited to 8 hrs (Figure S9). Free and PEG-ASP CNA entrapped 1 and 3 were incubated with A549 cells for 8 hrs, extracellular free or CNA entrapped Ru complexes were washed away using fresh media, the cells were irradiated with light (λ>400 nm), incubated for 72 hrs, and the cell viability determined as described above. Figure S9 shows that both free 1 and 1-loaded PEG-ASP CNAs were ineffective while both free 3 and 3-loaded PEG-ASP CNAs remained potent when cells were washed after an 8 hr exposure time. The effect of free 1 and 3 were consistent with past observations of poor cellular uptake of 1 in the dark and rapid cellular response to 3 (data not shown). More interesting was the effect of 1- and 3-loaded PEG-ASP CNAs. Assuming the CNAs were taken up by the cells, it appeared that 1 was rapidly released from the PEG-ASP CNAs as suggested in the preceding release studies detailed above (t1/2 = 0.8 ± 0.03 hrs with 98.7% maximum release) as the minimal cytotoxicity suggested little 1 was taken into cells. Conversely, 3-loaded PEG-ASP CNAs, which were shown to release entrapped complexes slowly (t1/2 = 10.7 ± 2.4 hrs with 37.0% maximum release) demonstrated a potency similar to free 3, suggesting that the majority of 3 remained entrapped inside the PEG-ASP CNAs, which were internalized within 8 hrs of exposure. Subsequently, 3 was released leading to potency similar to free 3.
The apparent disconnect between DNA binding and cell cytotoxicity for 3 and 3-loaded PEG-ASP CNAs may be due to numerous factors including differences in the mechanism of cellular uptake, dissimilarities in intracellular localization, or changes to the release rate of Ru complexes in cell media and intracellular environment compared to in a buffer solution. Generally, polypyridyl Ru complexes are thought to enter cells via non-endocytic processes such as passive diffusion.14b, 21 However, PEGylated nanoassemblies similar to PEG-ASP CNAs have been shown to enter cells through endocytic pathways, which could impact both the intracellular concentration and distribution of 3.18b, 22 In addition, the presence of proteins in the cell media and inside cells could have increased the release rate of hydrophobic 3 in comparison to previous release studies that were conducted in buffer solution. Furthermore, it has previously been shown that PEG-ASP CNAs cross-linked with fluorescent acridine yellow are internalized by cells within 3 hrs and translocate to the nucleus within 24 hrs.23 We have found that free 3 localizes in the cytosol and cell organelles, including the mitochondria, but does not enter the nucleus.1b Based on this, it is possible that 3-loaded PEG-ASP CNAs were able to enter the nucleus, altering cellular cytotoxicity, which was then balanced by potentially lower intracellular concentrations of 3 due to incomplete release from PEG-ASP CNAs, resulting in similar observed EC50 values. At this time it is unclear which of these possibilities accounts for the observed differences between DNA interactions and in vitro cell results. Additional experiments would be required to fully establish the cause or combination of causes that are responsible.
4 Conclusions
Light-reactive polypyridyl Ru complexes represent a promising new approach to cancer therapy; however, their small size and hydrophilicity will likely affect their ability to circulate in the bloodstream long enough to accumulate at the site of a tumor. Here, three polypyridyl Ru complexes were loaded into CNAs constructed from PEG-ASP block copolymers and the effect of complex properties and buffer conditions on complex loading, release, and in vitro anticancer potential analyzed. Both hydrophilic and hydrophobic Ru complexes were efficiently entrapped in PEG-ASP CNAs. The Ru complex-loaded PEG-ASP CNAs were stable in solution and entrapment in PEG-ASP CNAs did not inhibit photoactivation of 1 and 2. The release rate and the amount of complex released from PEG-ASP CNAs were found to depend strongly on complex hydrophobicity and on solution ionic strength, while variations in solution pH had almost no effect. This shows that PEG-ASP CNAs can serve as an effective carrier for polypyridyl Ru complexes and that by increasing the hydrophobicity of entrapped Ru complexes the release rate can be slowed and the amount of complex released can be tuned, thereby enhancing the circulation time of the Ru complex-loaded PEG-ASP CNA system. In vitro analysis of the activity of free Ru complexes and Ru complex-loaded PEG-ASP CNAs revealed that while PEG-ASP CNAs are capable of shielding hydrophobic Ru complexes from DNA interactions, after a sufficient period of time the Ru complexes are released and display potency indistinguishable from free Ru complexes. In summary, the use of PEG-ASP CNAs is a promising approach to improve the pharmacological properties of polypyridyl Ru complexes but further optimization is required to realize the full potential of this therapeutic approach.
Supplementary Material
Acknowledgments
This work was made possible through the support of the American Cancer Society (RSG-13-079-01-CDD). MD acknowledges the University of Kentucky Cancer Nanotechnology Training Center (UK-CNTC) postdoctoral traineeship, supported by the NCI/NIH and part of the National Cancer Institute Alliance for Nanotechnology in Cancer (5R25CA153954).
References
- 1.(a) Howerton BS, Heidary DK, Glazer EC. Strained ruthenium complexes are potent light-activated anticancer agents. J Am Chem Soc. 2012;134:8324–8327. doi: 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]; (b) Dickerson M, Sun Y, Howerton B, Glazer EC. Modifying charge and hydrophilicity of simple Ru(II) polypyridyl complexes radically alters biological activities: old complexes, surprising new tricks. Inorg Chem. 2014;53(19):10370–7. doi: 10.1021/ic5013796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Zhang CX, Lippard SJ. New metal complexes as potential therapeutics. Curr Opin Chem Biol. 2003;7:481–489. doi: 10.1016/s1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]; (b) Fricker SP. Metal based drugs: from serendipity to design. Dalton Trans. 2007;43:4903–4917. doi: 10.1039/b705551j. [DOI] [PubMed] [Google Scholar]; (c) Bruijnincx PCA, Sadler PJ. New trends for metal complexes with anticancer activity. Curr Opin Chem Biol. 2008;12:197–206. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen T, Liu Y, Zheng WJ, Liu J, Wong YS. Ruthenium polypyridyl complexes that induce mitochondria-mediated apoptosis in cancer cells. Inorg Chem. 2010;49:6366–6368. doi: 10.1021/ic100277w. [DOI] [PubMed] [Google Scholar]; (e) Groessl M, Zava O, Dyson PJ. Cellular uptake and subcellular distribution of ruthenium-based metallodrugs under clinical investigation versus cisplatin. Metallomics. 2011;3:591–599. doi: 10.1039/c0mt00101e. [DOI] [PubMed] [Google Scholar]
- 3.(a) Goldbach RE, Rodriguez-Garcia I, van Lenthe JH, Siegler MA, Bonnet S. N-acetylmethionine and biotin as photocleavable protective groups for ruthenium polypyridyl complexes. Chem Eur J. 2011;17:9924–9929. doi: 10.1002/chem.201101541. [DOI] [PubMed] [Google Scholar]; (b) Wachter E, Heidary DK, Howerton BS, Parkin S, Glazer EC. Light-activated ruthenium complexes photobind DNA and are cytotoxic in the phtodynamic therapy window. Chem Commun. 2012;48:9649–9651. doi: 10.1039/c2cc33359g. [DOI] [PubMed] [Google Scholar]; (c) Frasconi M, Liu Z, Lei J, Wu Y, Strekalova E, Malin D, Ambrogio MW, Chen X, Botros YY, Cryns VL, Sauvage JP, Stoddart JF. Photoexpulsion of surface-grafted ruthenium complexes and subsequent release of cytotoxic cargos to cancer cells from mesoporous silica nanoparticles. J Am Chem Soc. 2013;135:11603–11613. doi: 10.1021/ja405058y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hufziger KT, Thowfeik FS, Charboneau DJ, Nieto I, Dougherty WG, Kassel WS, Dudley TJ, Merino EJ, Papish ET, Paul JJ. Ruthenium dihydroxybipyridine complexes are tumor activated prodrugs due to low pH and blue light induced ligand release. J Inorg Biochem. 2014;130:103–111. doi: 10.1016/j.jinorgbio.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Novakova O, Kasparkova J, Vrana O, van Vliet PM, Reedijk J, Brabec V. Correlation between cytotoxicity and DNA binding of polypyridyl ruthenium complexes. Biochemistry. 1995;34:12369–12378. doi: 10.1021/bi00038a034. [DOI] [PubMed] [Google Scholar]; (b) Kladjner M, Hebraud P, Sirlin C, Gaiddon C, Harlepp S. DNA binding to an anticancer organo-ruthenium complex. J Phys Chem. 2010;114:14041–14047. doi: 10.1021/jp1044783. [DOI] [PubMed] [Google Scholar]; (c) Heidary DK, Glazer EC. A light-activated metal complex targets both DNA and RNA in a fluorescent in vitro transcription and translation assay. ChemBioChem. 2014;15:507–511. doi: 10.1002/cbic.201300681. [DOI] [PubMed] [Google Scholar]
- 5.(a) Grover N, Welch TW, Fairley TA, Cory M, Thorp HH. Covalent Binding of Aquaruthenium complexes to DNA. Inorg Chem. 1994;33:3544–3548. [Google Scholar]; (b) Allardyce CS, Dyson PJ. Ruthenium in medicine: Current clinical uses and future prospects. Platinum Metals Rev. 2001;45:62–69. [Google Scholar]; (c) Lentzen O, Moucheron C, Kirsch-De Mesmaeker A. Metallotherapeutic drugs and metal-based diagnostic agents. John Wiley & Sons, Ltd; 2005. 44Ru Perspectives of ruthenium complexes in cancer therapy; pp. 359–378. [Google Scholar]; (d) Boerner LJK, Zaleski JM. Metal complex-DNA interactions: from transcription inhibition to photoactivated cleavage. Curr Opin Chem Biol. 2005;9:135–144. doi: 10.1016/j.cbpa.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 6.(a) Chu G. Cellular responses to cisplatin the roles of DNA-binding proteins and DNA repair. J Biol Chem. 1994;269:787–790. [PubMed] [Google Scholar]; (b) Dronkert MLG, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]; (c) Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch JH, Rogers WP, Dwyer FP, Gyarfas EC. The metabolic fate of tris-1,10-phenanthroline 106ruthenium (II) perchlorate, a compound with anticholinesterase and curare-like activity. Aust J Biol Sci. 1957;10:342–350. [Google Scholar]
- 8.(a) Dwyer FP, Gyarfas EC. Biological activity of complex ions. Nature. 1952;170:190–191. doi: 10.1038/170190a0. [DOI] [PubMed] [Google Scholar]; (b) Brandt WW, Dwyer FP, Gyarfas EC. Chelate complexes of 1, 10-phenanthroline and related compounds. Chem Rev. 1954;54:959–1017. [Google Scholar]
- 9.Garcia-Fresnadillo D, Georgiadou Y, Orellana G, Braun AM, Oliveros E. 106. Singlet-Oxygen (1Δg) Production by Ruthenium(II) Complexes Containing Polyazaheterocyclic Ligands in Methanol and in Water. Helv Chim Acta. 1996;79:1222–1238. [Google Scholar]
- 10.Togano T, Nagao N, Tsuchida M, Kumakura H, Hisamatsu K, Howell FS, Mukaida M. One-pot and selective synthesis of a series of [RuCl6–2nLn] (L=bidentate ligand, n=0–3) types of complexes with polypyridyl ligands; another example of the synthetic utility of ‘ruthenium-blue’ solution. Inorg Chim Acta. 1992;195:221–225. [Google Scholar]
- 11.(a) Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Edit. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]; (b) Lee HJ, Bae Y. Cross-linked nanoassemblies from poly(ethylene glycol)-poly(aspartate) block copolymers as stable supramolecular templates for particulate drug delivery. Biomacromolecules. 2011;12:2686–2696. doi: 10.1021/bm200483t. [DOI] [PubMed] [Google Scholar]
- 12.Zava O, Zakeeruddin SM, Danelon C, Vogel H, Gratzel M, Dyson PJ. A Cytotoxic ruthenium tris(bipyridyl) complex that accumulates at plasma membranes. ChemBioChem. 2009;10:1796–1800. doi: 10.1002/cbic.200900013. [DOI] [PubMed] [Google Scholar]
- 13.Sangster J. Octanol-water partition coefficients of simple organic compounds. J Phys Chem Ref Data. 1989;18:1111–1227. [Google Scholar]
- 14.(a) Puckett CA, Barton JK. Methods to explore cellular uptake of ruthenium complexes. J Am Chem Soc. 2006;129:46–47. doi: 10.1021/ja0677564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Puckett CA, Barton JK. Mechanism of uptake of a ruthenium polypyridyl complex. Biochemistry. 2008;47:11711–11716. doi: 10.1021/bi800856t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pyle AM, Rehmann JP, Meshoyrer R, Kumar CV, Turro NJ, Barton JK. Mixed-ligand complexes of ruthenium(II): factors govering binding to DNA. J Am Chem Soc. 1989;111:3051–3058. [Google Scholar]
- 16.(a) Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57:171–185. doi: 10.1016/s0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]; (b) Mu L, Feng SS. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J Control Release. 2003;86:33–48. doi: 10.1016/s0168-3659(02)00320-6. [DOI] [PubMed] [Google Scholar]; (c) Hu Y, Jiang X, Ding Y, Zhang L, Yang C, Zhang J, Chen J, Yang Y. Preparation and drug release behaviors of nimodipine-loaded poly(caprolactone)-poly(ethylene oxide)-polylactide amphiphilic copolymer nanoparticles. Biomaterials. 2003;24:2395–2404. doi: 10.1016/s0142-9612(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 17.(a) Muller RH, Keck CM. Challenges and solutions for the delivery of biotech drugs - a review of drug nanocrystal technolgy and lipid nanoparticles. J Biotechnol. 2004;113:151–170. doi: 10.1016/j.jbiotec.2004.06.007. [DOI] [PubMed] [Google Scholar]; (b) Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 18.(a) Wang H, Zhao P, Liang X, Gong X, Song T, Niu R, Chang J. Folate-PEG coated cationic modified chitosan - cholesterol liposomes for tumor-targeted drug delivery. Biomaterials. 2010;31:4129–4138. doi: 10.1016/j.biomaterials.2010.01.089. [DOI] [PubMed] [Google Scholar]; (b) Zhang P, Ling G, Sun J, Zhang T, Yuan Y, Sun Y, Wang Z, He Z. Multifunctional nanoassemblies for vincristine sulfate delivery to overcome multidrug resistance by escaping P-glycoprotein mediated efflux. Biomaterials. 2011;32:5524–5533. doi: 10.1016/j.biomaterials.2011.04.022. [DOI] [PubMed] [Google Scholar]; (c) Wang G, Babadagli ME, Uludag H. Bisphosphonate-derivatized liposomes to control drug release from collagen/hydroxyapatite scaffolds. Mol Pharm. 2011;8:1025–1034. doi: 10.1021/mp200028w. [DOI] [PubMed] [Google Scholar]; (d) Su W, Wang H, Wang S, Liao Z, Kang S, Peng Y, Han L, Chang J. PEG/RGD-modified magnetic polymeric liposomes for controlled drug release and tumor cell targeting. Int J Pharm. 2012;426:170–181. doi: 10.1016/j.ijpharm.2012.01.013. [DOI] [PubMed] [Google Scholar]; (e) Al-Ahmady ZS, Al-Jamal WT, Bossche JV, Bui TT, Drake AF, Mason AJ, Kostarelos K. Lipid-peptide vesicle nanoscale hybrids for triggered drug release by mild hyperthermia in vitro and in vivo. ACS Nano. 2012;6:9335–9346. doi: 10.1021/nn302148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn U, Luelf H, Winkler HDF, Schalley CA, Vögtle F, De Cola L. Encapsulation of luminescent homoleptic [Ru(dpp)3]-type chromophores within an amphiphilic dendritic environment. Chem Eur J. 2012;18:15424–15432. doi: 10.1002/chem.201201126. [DOI] [PubMed] [Google Scholar]
- 20.Lee HJ, B Y. Pharmaceutical differences between block copolymer self-assembled and cross-linked nanoassemblies as carriers for tunable drug release. Pharm Res. 2013;30:478–488. doi: 10.1007/s11095-012-0893-3. [DOI] [PubMed] [Google Scholar]
- 21.(a) Gill MR, Garcia-Lara J, Foster SJ, Smythe C, Battaglia G, Thomas JA. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat Chem. 2009;1:662–667. doi: 10.1038/nchem.406. [DOI] [PubMed] [Google Scholar]; (b) Yadav A, Janaratne T, Krishnan A. Regression of lung cancer by hypoxia-sensitizing ruthenium polypyridyl complexes. Mol Cancer Ther. 2013;12:643–653. doi: 10.1158/1535-7163.MCT-12-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly (DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. The FASEB Journal. 2002;16:1217–1226. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]; (b) He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 23.Cao P, Ponta A, Kim J, Bae Y. Block copolymer crosslinked nanoassemblies co-entrapping acridine yellow and doxorubicin for cancer theranostics. Brit J Pharm Res. 2013;3:523–535. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.