

Abstract
Rapid plasma membrane repair is essential to restore cellular homeostasis and improve cell survival after injury. Several mechanisms for plasma membrane repair have been proposed, including formation of an intracellular vesicle patch, reduction of plasma membrane tension, lesion removal by endocytosis, and/or shedding of the wounded membrane. Under all conditions studied to date, plasma membrane repair is strictly dependent on the entry of calcium into cells, from the extracellular medium. Calcium-dependent exocytosis of lysosomes is an important early step in the plasma membrane repair process, and defects in plasma membrane repair have been observed in cells carrying mutations responsible for serious lysosomal diseases, such as Chediak–Higashi (Huynh, Roth, Ward, Kaplan, & Andrews, 2004) and Niemann–Pick Disease type A (Tam et al., 2010). A functional role for release of the lysosomal enzyme acid sphingomyelinase, which generates ceramide on the cell surface and triggers endocytosis, has been described (Corrotte et al., 2013; Tam et al., 2010). Therefore, procedures for measuring the extent of lysosomal fusion with the plasma membrane of wounded cells are important indicators of the cellular repair response. The importance of carefully selecting the methodology for experimental plasma membrane injury, in order not to adversely impact the membrane repair machinery, is becoming increasingly apparent. Here, we describe physiologically relevant methods to induce different types of cellular wounds, and sensitive assays to measure the ability of cells to secrete lysosomes and reseal their plasma membrane.
1. OVERVIEW OF WOUNDING METHODS AND PLASMA MEMBRANE REPAIR MECHANISMS
Plasma membrane repair is an important cellular function that allows maintenance and restoration of cellular integrity after wounding events. Such events are frequent under physiological conditions, and include tears in the sarcolemma of muscle fibers exposed to mechanical stress or attack by pathogen or immune system proteins that have membrane-damaging activity (Gonzalez, Bischofberger, Pernot, van der Goot, & Frêche, 2008; Keefe et al., 2005). In all cases, plasma membrane resealing occurs within a few seconds (Idone et al., 2008; McNeil, Vogel, Miyaki, & Terasaki, 2000; Steinhardt, Bi, & Alderton, 1994) and requires the influx of extracellular calcium to induce the first step of the process, exocytosis of intracellular vesicles. Vesicle secretion, a process observed within seconds of lesion formation and calcium influx, was originally proposed to promote repair by generating a patch to fill the wound or by releasing membrane tension to allow the lipid bilayer to reseal (McNeil & Steinhardt, 2003). Subsequently, lysosomes were identified as the calcium-regulated secretory vesicles that mediate plasma membrane resealing (Chakrabarti et al., 2003; McNeil, 2002; Reddy, Caler, & Andrews, 2001). While lysosomes were initially thought to provide membrane for patching wounds, new evidence indicates that lysosomes promote resealing by secreting acid sphingomyelinase (ASM), an enzyme that generates ceramide by cleaving the abundant membrane lipid sphingo-myelin, triggering endocytosis and removal or closure of different types of wounds (Corrotte et al., 2013; Idone et al., 2008), from large mechanical wounds to stable transmembrane pores formed by bacterial toxins. Additional mechanisms for plasma membrane repair that involve extracellular shedding of membrane buds have been proposed (Babiychuk, Maonastyrskaya, & Draeger, 2008; Jimenez et al., 2014), and the role of ceramide platforms proposed in one of these studies (Babiychuk, Maonastyrskaya, & Draeger, 2008) is also consistent with a possible involvement of sphingomyelinase.
Regardless of the mechanism used by cells to repair their plasma membrane, the ability to induce proper physiological membrane wounding is important for the study of this process. Mechanical wounding can be achieved by inducing cellular contraction, scraping attached cells from the substrate, or by exposing cell monolayers to abrasive agents such as microscopic glass beads. These methods mimic the forms of mechanical wounding that are predicted to occur as cells move and contract in vivo, and are likely to generate large lesions in the plasma membrane (>100 nm in diameter) that lead to rapid and massive elevations in the intracellular calcium concentration. On the other hand, the use of bacterial pore-forming toxins allows a more tightly controlled generation of smaller membrane wounds (<100 nm). These toxins can be prebound to cells and then activated to cause cell permeabilization, and titrated to achieve different levels of injury. The ability to perform dose-dependent and synchronized wounding greatly facilitates studies of the kinetics of plasma membrane repair and the importance of cellular factors in the process. Plasma membrane wounding with lasers has been widely used and offers the advantage of allowing the generation of much localized lesions and real-time imaging of the repair response (Defour, Sreetama, & Jaiswal, 2014). However, laser wounding is very different from more physiological forms of injury because it involves very high increases in local temperature, which can cause denaturation of proteins and lipids and thus interfere with the correct interpretation of results. The size of wounds generated with lasers varies greatly and cells have been reported to remain permeabilized for several minutes before resealing (Jimenez et al., 2014), a response that differs significantly from the known kinetics of plasma membrane repair (Idone et al., 2008; McNeil et al., 2000; Steinhardt et al., 1994). Thus, here we will focus our discussion on plasma membrane wounding techniques that mimic more physiological conditions.
Once the plasma membrane has been wounded, it is important to have sensitive and fast assays that allow precise measurement of the efficiency of repair, if one is to understand the various steps and molecular factors involved in the process. Here we will focus on the use of Propidium Iodide (PI), a fluorescent, membrane-impermeant dye that binds to nucleic acids, and on the lipophilic dye FM1-43, which increases two orders of magnitude in fluorescence once it is intercalated in phospholipid bi-layers and only reaches intracellular organelles when there is a breach in plasma membrane integrity (Betz, Mao, & Smith, 1996). To assess and quantify cellular responses required for plasma membrane repair, we also describe several assays for detecting lysosomal exocytosis and the secretion of lysosomal enzymes.
2 . PROCEDURES FOR PLASMA MEMBRANE WOUNDING
To study plasma membrane repair, various methods have been developed that allow controlled wounding under conditions that resemble the forms of stress or pathogen attack that mammalian cells often encounter in vivo. These methods range from mechanical forms of wounding that include cellular contraction, scraping from the substrate, passage through thin needles, or exposure to abrasive objects such as glass beads, to exposure of cells to bacteria expressing the type III secretion system or to purified pore-forming toxins.
2.1 MECHANICAL WOUNDING BY THREE-DIMENSIONAL CELLULAR CONTRACTION
This method is based on the ability of fibroblasts to form abundant polymerized actin stress fibers and strong focal attachments when embedded in a collagen matrix, which is kept under isometric tension by forces exerted by the cells. Release of the collagen matrix from the substrate in one single, fast step results in simultaneous contraction of the fibroblasts, compaction of the collagen matrix, and tearing of focal adhesion sites on the plasma membrane of the cells (Grinnell, 2000).
Materials
Neutralized Vitrogen 100 collagen (Cohesion Corp.)
Dulbecco’s modified Eagle medium (DMEM) (Life Technologies)
Fetal Bovine Serum (FBS) (Life Technologies)
Human foreskin fibroblasts (Celleng Tech)
24-well tissue culture-treated dishes (Falcon)
Ascorbic acid (Sigma) Texas red dextran 10 kDa (Life Technologies)
Thin metal spatula
Paraformaldehyde (PFA)
Triton X-100
Alexa Fluor 488-phalloidin (Life Technologies)
ProLong® Gold mounting solution (Life Technologies)
Methods
Prepare a 1.5 mg/mL collagen solution in serum-free DMEM and sterilize by filtration.
Trypsinize a culture of human foreskin fibroblasts and resuspend the cells at 5 × 106/mL in the collagen medium.
Warm 0.2 mL aliquots of the cell–collagen mixture at 37 °C for 5 min and add them to the bottom of a well in a 24-well plate.
Incubate at 37°C/5% CO2 for 60 min to allow for collagen polymerization.
Add 1 mL of DMEM 10% FBS containing 50 μg/mL ascorbic acid to each well.
Incubate at 37°C/5% CO2 for 48 h.
To trigger fibroblast contraction and wounding, release the anchored collagen matrices from the wells by inserting a thin flat spatula between the matrix and the dish.
After various periods of incubation at 37 °C (0, 30 s, 1 min, and 5 min) add 1 mg/mL 10 kDa Texas red dextran to the medium. When added at the time of matrix detachment, intracellular dextran will reveal all the initially wounded cells; after various periods of incubation at 37 °C to allow repair, staining will reflect cells that failed to reseal.
At each time point, wash the released matrices several times in cold phosphate-buffered saline (PBS) with calcium and fix in 4% PFA in PBS for 1 h at room temperature.
Permeabilize the fixed matrices in 0.5% Triton X-100 for 10 min and incubate with 1 U/mL Alexa Fluor 488-phalloidin, followed by three washes in PBS.
Mount the matrix in a glass slide and image the red (dextran, indicating wounded cells) and green (phalloidin) fluorescence on a confocal microscope. Identification and quantification of wounded cells is done through detection of red cytosolic staining and loss of phalloidin-positive actin stress fibers.
2.1.1 Aspects to consider
The separation of the collagen matrix from the dish with a spatula should be done very quickly, in a single movement, to ensure a strong gel contraction and synchronized cell wounding. The collagen matrix, initially transparent when attached to the dish, should undergo significant volume reduction and become opaque. The collagen gel is highly porous, so the attached matrices can be incubated with various drugs and inhibitors prior to wounding and quantification of plasma membrane repair. This method, although more labor-intensive than other procedures described below, is particularly adequate when analysis of a highly physiological form of plasma membrane injury is desired. Calcium can be removed from the medium during the contraction and resealing period at 37 °C as a negative control for resealing.
2.2 MECHANICAL WOUNDING BY SCRAPING CELLS FROM THE SUBSTRATE
Scraping monolayers of attached cells leads to tearing of the plasma membrane at points of contact with the substrate, a form of wounding that is likely to occur in vivo in tissues under mechanical stress. This methodology was originally described as a useful method to load macromolecules into cells (McNeil, Vogel, Miyaki, & Terasaki, 1989). Below we describe a modification of this procedure that uses a membrane-impermeant fluorescent dye and flow cytometry to quantify the extent of wounding and resealing in a population of adherent cells.
Materials
NRK rat cells or another cell line with good substrate attachment
Flat-edge plastic cell scrapers
DMEM with and without calcium (Life Technologies)
PI (Sigma)
Flow cytometry plastic tubes
Methods
Grow cells to ~70% confluency in 35 mm dishes (6-well plates).
Add 1 mL DMEM prewarmed at 37 °C with or without calcium, and rapidly scrape cells from the dish. For reproducibility of the extent of wounding, all dishes should be scraped using the same pattern, as follows: The flat edge of the scraper is placed on the bottom of the well near the wall and rapidly moved in a full circle, followed by diagonal scraping of the central area of the dish.
Pipette the medium containing the resuspended cells up and down three times using a 5 mL plastic pipette and transfer to a FACS tube maintained at 37 °C in a water bath for 5 min.
Place the tubes in an ice water bath for 30 s to stop any further membrane repair reaction and add 50 μg/mL PI. In the absence of calcium, PI-stained cells will reflect the extent of plasma membrane wounding; with calcium, PI-stained cells will reflect the cell population that did not repair their plasma membrane.
After 5 min, analyze the PI fluorescence in at least 10,000 cells per sample using a flow cytometer, as previously described (Idone et al., 2008; Tam et al., 2010) (Figure 1(A)).
FIGURE 1.

Flow cytometry measurement of plasma membrane repair in mammalian cell populations wounded by different methods. (A) NRK cells were wounded by scraping from the dish, stained with PI for 5 min, and analyzed by flow cytometry. (B) C2C12 myoblasts/myotubes were wounded by four passages through a 30 gauge needle, stained with PI for 5 min, and analyzed by flow cytometry. (C) NRK cells were wounded with the pore-forming toxin SLO, stained with PI for 5 min, and analyzed by flow cytometry. In all conditions, cells were wounded in medium with or without calcium, and after flow cytometry the percentage of wounded (PI-positive) or resealed (PI-negative) cells was determined by applying a gate to the data based on the calcium-free condition, which does not allow plasma membrane repair. (See color plate)
2.2.1 Aspects to consider
All samples should be analyzed by flow cytometry as soon as possible, 5 min after addition of PI—shorter or longer staining periods can cause variations in the amount of staining. This method allows a very quantitative assessment of plasma membrane repair in an entire cell population after a defined period of time, and can also be used to analyze calcium-dependent exocytosis of lysosomal enzymes after removing the cells and assaying the supernatant. Cell scraping coupled to PI staining of the whole population is not well suited for kinetic analysis of the plasma membrane repair process, because cell resealing occurs in seconds but adequate PI staining for flow cytometry quantification requires 4–5 min.
2.3 MECHANICAL WOUNDING USING A NEEDLE/SYRINGE
Forcing cells through a very narrow orifice such as a 30 gauge hypodermic needle produces a shear stress that wounds the plasma membrane. This technique, termed “syringe loading,” was originally adopted to load macromolecules into cells (Clarke & McNeil, 1992). This method of membrane wounding is well suited for cells that do not adhere well to the substrate.
Materials
Mammalian cells, adherent or in suspension
1 mL plastic syringe
30 gauge hypodermic needle
DMEM with and without calcium
PI
Flow cytometry (FACS) plastic tubes
Methods
Resuspend the cells at 3 × 105/mL in DMEM with or without calcium.
Using a 10 mL plastic pipette, load 1 mL of the medium containing the resuspended cells into a 1 mL syringe. Gently and firmly push the plunger down, forcing the cell suspension through a 30 gauge hypodermic needle into a flow cytometry tube.
Load the cell suspension back into the same syringe and pass the cells again through the needle, ensuring that the distance from the tip of the needle to the bottom of the tube and the pressure applied are constant each time. Passage the cell suspension for a total of 2 or 4 times through the syringe (this will have to be adjusted, depending on the size and type of cell).
Incubate the cells at 37 °C in a water bath for 5 min, to allow plasma membrane repair.
Place the tubes in an ice water bath for 30 s and add 50 μg/mL PI.
After 5 min, analyze the PI fluorescence in at least 10,000 cells per sample using a flow cytometer, as previously described (Idone et al., 2008; Tam et al., 2010), gating the cell population based on fluorescence intensity to calculate the fractions of resealed and non-resealed cells (Figure 1(B)).
2.3.1 Aspects to consider
Samples should be analyzed by flow cytometry shortly after adding PI to avoid variations arising from differences in staining. To ensure reproducibility, the distance from the bottom of tube, the pressure applied, and the time taken to pass the cells through the syringe should be kept constant. This experiment should be performed in triplicate, to average out variations. Depending on the type and size of cells, the number of passages through the needle can be either increased or decreased to optimize the extent of membrane wounding. The ideal condition is a level of wounding (determined based on the level of PI staining in the absence of calcium) that affects most of the cell population, but that still allows plasma membrane repair (determined based on the level of PI staining in the presence of calcium) (Figure 1(B)).
2.4 MECHANICAL WOUNDING USING GLASS BEADS
Glass bead wounding is a form of mechanical wounding that results in tearing of the plasma membrane by abrasion, and is generally thought to generate relatively large lesions. With this approach, most of cell monolayer remains attached to the substrate, allowing the use of microscopy after fixation to detect wounded cells and analyze changes in protein localization. This procedure can also be coupled to electron microscopy and protein–protein interaction assays (such as co-immunoprecipitation) (Corrotte et al., 2013), but is not suited for live imaging due to interference of the glass beads with the image acquisition process. This method is also not adequate for whole cell population analysis by flow cytometry, since only a small fraction of the cells become wounded.
Materials
NRK rat cells or another cell line with good substrate attachment
Acid-washed glass beads ≤106 μm (Sigma)
DMEM with and without calcium (Life Technologies)
Methods
Grow cells to 70% confluency on coverslips, 35 mm MatTek glass-bottom dishes, or 10 cm dishes.
Replace the culture medium by DMEM with (repair condition) or without calcium (nonrepair condition) at 37 °C.
Sprinkle cells with 0.05 g (coverslips or MatTek dishes) or 0.1 g (10 cm dishes) of acid-washed glass beads from a small plastic conical tube held at a distance of about 1 cm above the dish.
Rock the dish gently three times as described in (Reddy et al., 2001) and process for various assays to assess plasma membrane wounding and repair, such as PI staining (Figure 2(A)).
FIGURE 2.
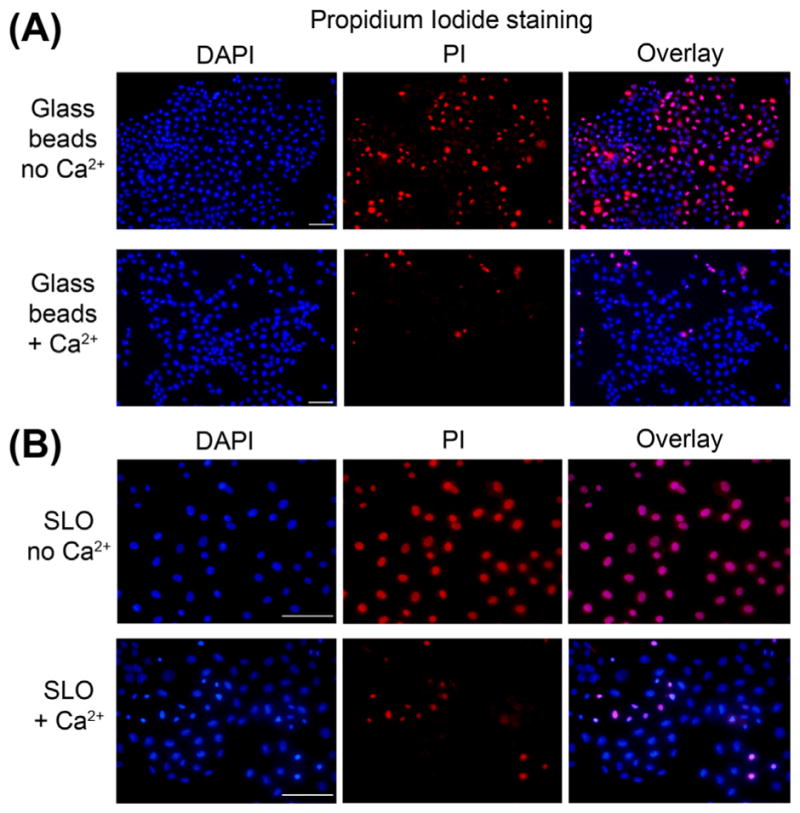
Use of PI to detect wounded cells by microscopy. NRK cells were wounded by contact with glass beads (A) or by permeabilization with the pore-forming toxin SLO (B). Glass beads lead to wounding of a smaller fraction of the cell population when compared to SLO, as indicated by the number of PI-positive cells in the calcium-free condition. In both cases, the presence of calcium in the medium leads to extensive cell resealing and exclusion of PI. Bars = 50 μm. (See color plate)
2.4.1 Aspects to consider
For reproducibility, it is important to maintain the distance from which the glass beads are added to the cell monolayer and the time of rocking as constant as possible. The extent of wounding is likely to vary with cell type, so different amounts of glass beads should be tested to avoid excessive wounding and cell loss, or too little wounding.
2.5 WOUNDING USING PORE-FORMING PROTEINS
The major advantages of this assay are: (1) the cell-wounding step can be tightly synchronized; (2) the lesions formed are highly homogenous in size, and the number of lesions can be titrated by using different concentrations of the pore-forming protein. Various bacterial pore-forming toxins have been used for membrane permeabilization (Gonzalez, Bischofberger, Pernot, van der Goot, & Freche, 2008), as well as proteins derived from mammalian immune cells such as perforin (Rosado et al., 2008; Thiery et al., 2010). The best characterized bacterial pore-forming toxins are the cholesterol-dependent cytolysins, which include Streptolysin O (SLO) (Tweten, 2005), the toxin used for the procedure described below. SLO monomers bind to cholesterol on the plasma membrane, followed by oligomerization, insertion into the lipid bilayer, and formation of a protein-lined pore of about 30 nm in diameter. Prebinding of the toxin at 4 °C allows for very good synchronization of the pore-forming process, which is triggered when the temperature is shifted to 37 °C.
Materials
Mammalian cells, attached or in suspension
DMEM without calcium + 5 mM EGTA (washing solution)
DMEM with calcium (Life Technologies)
PBS without calcium
Purified SLO pore-forming toxin (100 μg/mL stock solution in PBS).
Methods
Grow the cells to about 70% confluency, and either keep them attached to the dish or trypsinize the monolayer to obtain cells in suspension.
Wash cells in ice-cold calcium-free DMEM + EGTA and place dishes on a metal plate covered by a wet paper towel kept over ice.
Wash cells again with ice-cold calcium-free DMEM and add purified SLO at various concentrations (the 100–500 ng/mL is usually adequate for most cell types) in cold DMEM with or without calcium, followed by 5 min on ice.
Remove cells from the ice and add DMEM with or without calcium prewarmed at 37 °C
Incubate cells at 37 °C for various periods of time to induce pore formation. In the absence of calcium, the cells will be permeabilized but will not reseal; in the presence of calcium, the plasma membrane repair response is triggered.
Place the cells back on ice, remove the medium, and wash with calcium-free PBS to stop the plasma membrane repair process.
For assays with cells in suspension, after trypsinization resuspend cells in DMEM 10% FBS to stop the trypsin, centrifuge at 200 g for 10 min, resuspend in cold DMEM with or without calcium, add SLO, and then transfer to a 37 °C water bath (without centrifugation, to avoid damaging the cells) for the desired periods of plasma membrane repair.
Process samples for various assays to detect permeabilized cells, such as PI staining followed by imaging (Figure 2(B)) or flow cytometry (Figure 1(C)), or cell lysis and western blot analysis (Corrotte et al., 2013).
2.5.1 Aspects to consider
Good-quality, purified SLO is essential for good results in this assay. Instead of using commercial sources, it is preferable to purify 6 × histidine-tagged SLO carrying a cysteine deletion that eliminates the need for thiol activation (cloned into Invitrogen pTrcHisA, provided by R. Tweeten, University of Oklahoma, Norman, OK) after expression in BL21 Escherichia coli. The purified toxin should be aliquoted and stored at −80 °C, and kept on ice when thawed. Pore-forming activity decreases with freeze and thaw cycles, so it is advisable to titrate toxin concentrations in each assay to achieve the desired levels of cell permeabilization. The extent of plasma membrane repair decreases with the increasing concentrations of SLO used (Idone et al., 2008; Tam et al., 2010). For robust synchronization of plasma membrane wounding, cells must not be allowed to warm above 4 °C during the toxin prebinding step.
3 . PROCEDURES FOR MEASURING THE EXTENT OF PLASMA MEMBRANE REPAIR
Plasma membrane repair is strictly calcium dependent, so high extracellular concentrations of calcium (1 mM or above) are essential for triggering resealing. The calcium-containing media and buffers described in this section and all others in this chapter contain 1.8 mM calcium.
3.1 PI INFLUX (MICROSCOPY AND FLOW CYTOMETRY)
PI is a membrane-impermeant 668 Da fluorescent molecule that binds to DNA and RNA by intercalating between bases, at a ratio of one PI molecule per 4–5 base pairs. When bound to nucleic acids, it has a fluorescence excitation maximum of 535 nm, and an emission maximum of 617 nm, so excitation energy can be supplied by a xenon or mercury-arc lamp or a 488 line of an argon laser. PI can only enter the cytosol of cells and stain nuclear DNA by going through plasma membrane lesions, and for this reason it is a sensitive tool for the detection of wounded cells in a population. In plasma membrane repair assays, PI is very well suited to determine the fraction of cells that were not capable of resealing their plasma membrane.
Materials
PI stock solution 5 mg/mL (Sigma)
4% PFA fixative solution
4′,6-diamidino-2-phenylindole (DAPI) 10 mM stock solution(Sigma)
Glass slides and Prolong® Gold mounting solution (Life Technologies)
Methods
Microscopy
Cell monolayers on glass coverslips, wounded by the methods described above, are incubated for 5 min with 50 μg/mL PI (Sigma) in ice-cold calcium-free DMEM. Cells are then washed in ice-cold calcium-free PBS and fixed in 4% PFA for 10 min at room temperature. The cell-permeable nucleic acid dye DAPI (fluorescent stain that binds strongly to A–T rich regions in DNA with an excitation maximum at 358 nm (ultraviolet range) and emission maximum at 461 nm) is then added at 10 μM to stain all nuclei in the cell population, followed by washes in PBS, mounting of the coverslips on glass slides using Prolong® Gold antifade, and immediate microscopic imaging using a fluorescence microscope equipped with a digital camera and the adequate filters to detect PI and DAPI (Figure 2(A) and (B)). Quantification of the number of PI-positive and negative cells is done by counting nuclei stained with DAPI and/or PI (a total of ~200 per sample) in random microscopic fields, in triplicate assays in images taken with a 10× or 32× objective).
Flow cytometry
Cells injured as described above are resuspended (by trypsinization, if the assay was performed with attached cells), centrifuged at 200 g for 5 min, and resuspended at 10–12 × 105 cells/mL in ice-cold DMEM with or without calcium, depending on assay conditions. Samples of 0.5 mL of the cell suspensions are placed on ice and about 104 cells are analyzed immediately by flow cytometry, using an instrument capable of detecting PI (such as the FACSCanto, Becton Dickinson). Data is then analyzed using Flow-Jo software (Three Star, Inc.), and fluorescence level gating for assigning cells to a PI-negative or positive population are determined based on the background fluorescence of noninjured cells, and on the fluorescence of cells injured under nonrepair conditions (absence of calcium) (Figure 1(A)–(C)).
3.1.1 Issues to consider
PI is not fixable and can rapidly diffuse out of nuclei, so microscopic imaging should be done as soon as possible after completion of the assay. Cells should be kept on ice until imaging, both microscopically and by flow cytometry. With some cell types, flow cytometry may detect cells with intermediate levels of fluorescence intensity after plasma membrane repair, which may represent more heavily wounded cells. The gates used to quantify the populations of resealed and non-resealed cells should be carefully determined according to the experimental conditions.
3.2 LIVE IMAGING OF FM1-43 DYE INFLUX
FM1-43 is a styryl lipophilic dye that reversibly partitions into the outer leaflet of cell membranes without permeating, due to two cationic charges located in its head group. It is virtually nonfluorescent in aqueous medium, but when bound to phospholipid bilayer membranes it fluoresces with an excitation maximum of 479 nm, and an emission maximum of 598 nm (Betz et al., 1996). When there is a breach in the membrane, FM1-43 rapidly flows into cells and intercalates into intracellular membranes. Thus, microscopic live imaging of FM1-43 influx coupled to image analysis allows a precise determination of the kinetics of cell injury and resealing in real-time (Tam, Flannery, & Andrews, 2013), complementing end-point assays based on PI detection by flow cytometry. Live imaging of membrane repair is therefore an important complementary assay to the study of membrane repair that can help identify differences between essential actors and regulators of membrane repair.
Materials
FM1-43 dye (Life Technologies)
Glass-bottom dishes (MatTek)
SLO stock solution at 100 μg/mL
DMEM with and without calcium
Confocal imaging system equipped with an environmental chamber (temperature, CO2, and humidity control)
Methods
Grow cells to 70% confluency on MatTek 35 mm glass-bottom dishes and wash the monolayers twice in ice-cold calcium-free DMEM ± 5 mM EGTA.
Preincubate cells with various concentrations of SLO (100–500 ng/mL) for 5 min at 4 °C in DMEM with or without calcium and 4 μM of FM1-43 dye.
Remove the medium from the dishes leaving a small volume over the glass coverslip containing the cells, and transfer the cells to an environmental chamber at 37 °C with 5% CO2 mounted on an inverted spinning disk confocal microscope (such as the LiveCell System, Pathology Devices, Westminster, MD). Keep the additional dishes on ice until imaged.
Rapidly select an appropriate field of cells and set the focus to visualize plasma membrane-associated FM1-43. Add 2 mL of prewarmed DMEM with or without calcium and 4 μM of FM1-43. Acquire images using a 40× NA 1.3 objective at the rate of one frame every 10 s, for a period of 8 min.
Using the appropriate imaging software, determine the fluorescence intensity in a defined area inside all cells in the microscopic field, without overlapping with the plasma membrane. Export the data as an excel table and calculate the fluorescence intensity at each time point as fold change over time = 0. Plot the results as average and standard error of the mean (SEM) of the fold increase in fluorescence in the Y axis and time on the X axis (Figure 3(B)). Representative images for each condition may be selected at various time points to assemble a gallery showing the progression of FM1-43 influx, under conditions that allow repair (calcium) or not (no calcium) (Figure 3(A)).
FIGURE 3.
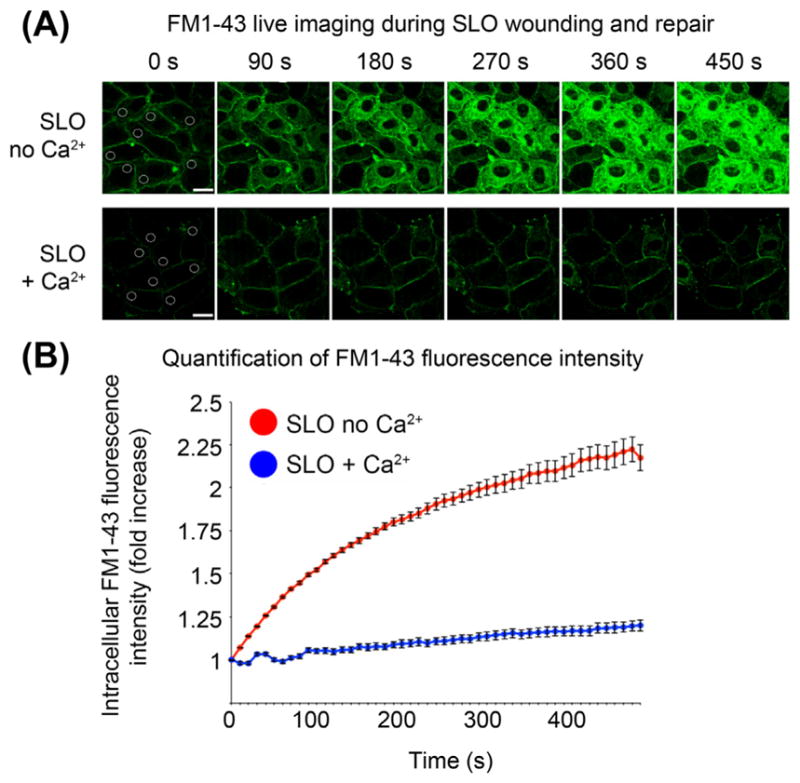
Live imaging of FM1-43 influx to determine the kinetics and extent of plasma membrane injury and repair. (A) Images of NRK cells that were preincubated with SLO at 4 °C in medium with or without calcium, transferred to a 37°C/5% CO2 environmental chamber mounted on the stage of a spinning disk confocal microscope, and imaged every 10 s for 8 min. (B) FM1-43 fluorescence intensity in a spot placed within the cells (as shown in the 0 s images in A) was determined using the image analysis software Volocity. Bars = 20 μm. (See color plate)
3.2.1 Aspects to consider
The FM1-43 dye can also be added at the time of imaging, but prestaining of the cells at 4 °C preferable because it allows a faster focusing and selection of the microscopic fields to be imaged. Since plasma membrane repair occurs within 30 s of injury, delays may prevent the acquisition of images before the cells fully reseal and thus not allow a determination of the kinetics of resealing. Before image analysis, the movies acquired should be played until the end to verify whether there were changes in cell shape or cell movement that resulted in overlap of the region of interest with FM1-43-stained plasma membrane. If segments showing these events are detected, the corresponding frames should be edited out of the movie before analysis of the intracellular fluorescence intensity.
4 . PROCEDURES TO MEASURE EXOCYTOSIS OF LYSOSOMES
Lysosomes behave as secretory vesicles following calcium influx (Rodriguez, Webster, Ortego, & Andrews, 1997) and are necessary for plasma membrane repair (McNeil, 2002; Reddy et al., 2001). However, while originally exocytosis was thought to originate a patch to repair wounds, recent evidence shows that secretion of the lysosomal content plays a significant role in the resealing mechanism. One of the lysosomal enzymes found to be secreted by wounded cells, ASM, was recently shown to be required for plasma membrane repair (Corrotte et al., 2013; Tam et al., 2010). Thus, detection of lysosomal enzymes secreted into the culture medium or lysosomal membrane proteins on the cell surface are useful tools to study the calcium-dependent, lysosome-mediated plasma membrane repair process.
4.1 SURFACE EXPOSURE OF LAMP1 LUMINAL EPITOPES
Lamp1 is a highly abundant glycoprotein present on the membrane of mammalian lysosomes. Highly specific monoclonal antibodies that react with luminal epitopes of Lamp1 from several species (Developmental Studies Hybridoma Bank) are available, allowing sensitive immunofluorescence detection of lysosomal exocytosis by the procedure below.
Materials
Adherent cell line
Culture dishes with glass coverslips
PBS 4.5% dextrose with and without calcium
Cell wounding agent (glass beads, pore-forming toxin, etc.)
Anti-Lamp1 monoclonal antibodies to the appropriate species, such as 1D4B for mouse cells, or H4A3 for human cells (Developmental Studies Hybridoma Bank)
PBS 10% bovine serum albumin (BSA)
PFA 4% fixative (in PBS)
Appropriate secondary antibodies coupled to a fluorophore, such as Alexa 488 (Life Technologies)
Glass slides and ProLong® Gold mounting solution (Life Technologies)
Methods
Grow cells to 70% confluency on dishes containing glass coverslips.
Prepare a solution of the anti-Lamp1 antibodies at the appropriate final concentration (this can be predetermined by performing immunofluorescence assays in saponin-permeabilized cells) in PBS 10% BSA and keep on ice.
Place a metal plate over ice and cover with a wet paper towel.
Wash cells in ice-cold PBS/dextrose with or without calcium and wound cells by adding glass beads or a pore-forming toxin such as SLO.
Immediately after wounding, add to the coverslips 50 μL of the anti-Lamp1 antibody solution, and place the dish on the wet paper towel on ice for 30 min.
Wash the cells three times with ice-cold PBS/dextrose with or without calcium and fix for 1 h with 4% PFA.
Wash the cells with PBS three times and incubate at room temperature for 30 min with the appropriate fluorescent-labeled secondary antibodies diluted in PBS 10% BSA.
Wash the coverslips in PBS, mount on glass slides with the antifade mounting medium, and image in a fluorescence microscope. When calcium-dependent lysosomal exocytosis is triggered by wounding, punctate Lamp1 staining is detected on the cell surface.
4.1.1 Aspects to consider
This assay is based on the fact that antibodies cannot cross the plasma membrane of live, intact cells. Fixed cells, even without detergent permeabilization, can become permeabilized, so it is critical to perform the first antibody binding step, using anti-Lamp1, in live cells kept at 4 °C. Caution must be taken for the temperature not to increase, or the primary anti-Lamp1 antibody may be endocytosed, leading to misleading results. After the first step at 4 °C in live cells, the cells can be fixed and handled as in conventional immunofluorescence protocols.
4.2 SECRETION OF LYSOSOMAL ENZYMES
Permeabilization with the pore-forming toxin SLO is a very effective method to trigger synchronous, fast, and reproducible calcium-triggered lysosomal exocytosis, facilitating assays to detect released hydrolases. To verify enzymatic activity, several chromogenic and fluorogenic commercial substrates are available, and specific antibodies can also be used to detect specific lysosomal enzymes by western blot.
Materials
Mammalian cell line to be analyzed
35 mm plastic dishes
SLO stock solution (100 μg/mL)
PBS containing 4.5% dextrose (PBS/dextrose) with and without calcium
10% Triton X-100
Cathepsin B activity assay kit (Abcam, ab65300)
Amplex Red Sphingomyelinase assay kit (Invitrogen, A12220)
Rabbit anti-ASM antibodies (Abcam, ab83354)
Methods
Seed 2 × 105 cells in individual 35 mm dishes and culture for 48 h, or until the culture reaches 80–90% confluency.
Place dishes on a wet metal plate over ice and wash cells in ice-cold calcium-free PBS/dextrose.
Add SLO to cells at various final concentrations (100–500 ng/mL, depending on cell type) in 2 mL of cold calcium-free PBS/dextrose and keep on ice for 5 min.
Prewarm PBS/dextrose with or without calcium at 37 °C.
Wash cells with ice-cold calcium-free PBS/dextrose, aspirate all the supernatant, and add 250 μL of prewarmed PBS/dextrose with calcium. Place dishes floating in a water bath at 37 °C and harvest 50 μL samples at increasing periods of time, starting at 5 s. Since the calcium-triggered lysosomal exocytosis response is rapid, being completed between 20 and 30 s of cell wounding and calcium influx, there is no need to take measures to prevent evaporation of the buffer added to the dishes. But if longer time points are used, the dishes should be moved to a humidified incubator.
Place the supernatant buffer taken from the dishes on ice and spin for 15 s on maximum speed on a refrigerated microfuge to remove any cellular debris.
Assay the supernatant samples for the presence of lysosomal hydrolases, making sure to analyze in parallel buffer alone, that was not incubated with cells, for determination of the assay background. Figure 4(A) and (B) shows results of enzymatic activity assays using fluorimetric kits for the detection of the lysosomal cysteine protease cathepsin B (Abcam) and ASM (Invitrogen), released from NRK cells permeabilized with SLO in the presence of calcium.
Lyse the remaining cells attached to each dish in 250 μL lysis buffer (0.5% Triton X-100, 20 mM Tris, 100 mM NaCl, 1 mM EDTA containing a cocktail of protease inhibitors, 20 μL per 106 cells) for 15 min on ice, followed by 10 min centrifugation at maximum speed in a refrigerated microfuge, and assay of a diluted sample of the supernatant for enzyme activity. This will allow expression of the results in percent activity released over the total enzyme content in the cells. For most cell types, maximum levels of exocytosis range from 3% to 10% of the total cellular content (Rodriguez et al., 1997; Tam et al., 2010). If adding protease inhibitors to the total cell lysate is not feasible (such as when proteases are the enzymes to be detected), total lysates should still be prepared to allow normalization of the activity data by the protein content of each dish (this is a more precise measurement than cell counting).
For detection of secreted proteases by western blot, place 250 μL of the cell supernatant containing the exocytosed enzyme (obtained as described above) on ice immediately after collection and add a broad protease-inhibitor cocktail to avoid protein degradation. Concentrate the sample about 10 times (such as by centrifugation at 4 °C for 3 h at 4500 g through 10 kDa Centricon filter, Milipore), add reduced SDS sample buffer, and boil the sample, prior to SDS-PAGE and western blot with specific antibodies. Figure 4(C) shows lysosomal ASM detected in the supernatant by western blot.
FIGURE 4.

Detection of lysosomal enzymes secreted after SLO permeabilization. (A) Kinetics of degradation of a substrate specific for cathepsin B by the supernatant of NRK cells collected from untreated control cells, or 5 s after permeabilization with SLO. (B) Degradation of a substrate specific for ASM by the supernatant of NRK cells collected from untreated control cells, or 5 s after permeabilization with SLO. (C) Western blot with rabbit anti-ASM antibodies of the supernatant of NRK cells collected from untreated control cells, or 5 s after permeabilization with SLO. (See color plate)
4.2.1 Aspects to consider
Lysosomal exocytosis assays can also be adapted to cells in suspension, but the need for centrifugation to harvest the cells can cause heating of the sample, interfering with the quality of the results. Best results are obtained with adherent cells cultured for 48 h, which ensures better attachment to the dish and good supernatant medium recovery without contamination with detached cells. Values obtained from non-wounded cells (no SLO controls, if the wounding agent used is SLO) should be very low, serving as a good control spontaneous cell lysis or detachment during the assay. If the sensitivity to SLO of the cell type used is unknown, it is important to perform SLO dose-response assays using PI as an indicator of cell permeability, to find the optimum SLO concentration that triggers robust calcium-dependent lysosomal secretion. In the case of NRK cells, the maximum amounts of lysosomal enzyme exocytosis are usually achieved with 50–100 ng/mL of SLO. Ideally, the enzyme activity assays should be done kinetically, to allow selection of data points within the linear phase of the substrate cleavage reaction. Total cell lysates should be diluted 10-fold or more to allow detection levels comparable to the exocytosed fraction.
Acknowledgments
Work in the Andrews laboratory is funded by the National Institutes of Health (grants R37 AI34867 and R01 GM064625). Thiago Castro-Gomes is supported by a fellowship from the Brazilian National Research Council for Scientific and Technological Development (CNPq).
References
- Babiychuk EB, Monastyrskaya K, Draeger A. Fluorescent annexin A1 reveals dynamics of ceramide platforms in living cells. Traffic. 2008;9:1757–1775. doi: 10.1111/j.1600-0854.2008.00800.x. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Current Opinion in Neurobiology. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, et al. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. Journal of Cell Biology. 2003;162:543–549. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MS, McNeil PL. Syringe loading introduces macromolecules into living mammalian cell cytosol. Journal of Cell Science. 1992;102:533–541. doi: 10.1242/jcs.102.3.533. [DOI] [PubMed] [Google Scholar]
- Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife. 2013;2:e00926. doi: 10.7554/eLife.00926. http://dx.doi.org/10.7554/eLife.00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defour A, Sreetama SC, Jaiswal JK. Imaging cell membrane injury and sub-cellular processes involved in repair. Journal of Visualized Experiments. 2014;85:e51106. doi: 10.3791/51106. http://dx.doi.org/10.3791/51106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MR, Bischofberger M, Pernot L, van der Goot FG, Frêche B. Bacterial pore-forming toxins: the (w)hole story? Cellular and Molecular Life Sciences. 2008;65:493–507. doi: 10.1007/s00018-007-7434-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F. Fibroblast-collagen-matrix contraction: growth factor signalling and mechanical loading. Trends in Cell Biology. 2000;10:362–365. doi: 10.1016/s0962-8924(00)01802-x. [DOI] [PubMed] [Google Scholar]
- Huynh C, Roth D, Ward DM, Kaplan J, Andrews NW. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proceedings of the National Academy of Sciences of the USA. 2004;101:16795–16800. doi: 10.1073/pnas.0405905101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. Journal of Cell Biology. 2008;180:905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343:1247136. doi: 10.1126/science.1247136. [DOI] [PubMed] [Google Scholar]
- Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–262. doi: 10.1016/j.immuni.2005.08.001. [DOI] [PubMed] [Google Scholar]
- McNeil PL. Repairing a torn cell surface: make way, lysosomes to the rescue. Journal of Cell Science. 2002;115:873–879. doi: 10.1242/jcs.115.5.873. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Vogel SS, Miyaki K, Terasaki M. Patching plasma membrane disruptions with cytoplasmic membrane. Journal of Cell Science. 2000;113:1891–1902. doi: 10.1242/jcs.113.11.1891. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annual Reviews of Cellular and Developmental Biology. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Vogel SS, Miyaki K, Terasaki M. Incorporation of macromolecules into living cells. Methods in Cell Biology. 1989;29:153–173. doi: 10.1016/s0091-679x(08)60193-4. [DOI] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as conventional Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. Journal of Cell Biology. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, et al. The MACPF/CDC family of pore-forming toxins. Cellular Microbiology. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Tam C, Flannery AR, Andrews NW. Live imaging assay for assessing the roles of Ca2+ and sphingomyelinase in the repair of pore-forming toxin wounds. Journal of Visualized Experiments. 2013;78:e50531. doi: 10.3791/50531. http://dx.doi.org/10.3791/50531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. Journal of Cell Biology. 2010;189:1027–1038. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J, Keefe D, Saffarian S, Martinvalet D, Walch M, Boucrot E, et al. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood. 2010;115:1582–1593. doi: 10.1182/blood-2009-10-246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming proteins. Infection and Immunity. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]