

Abstract
In vitro -in vivo correlation (IVIVC) is a predictive mathematical model describing the relationship between an in vitro property and a relevant in vivo response of drug products. Since the U.S. Food and Drug Administration (FDA) published a regulatory guidance on the development, evaluation, and applications of IVIVC for extended release (ER) oral dosage forms in 1997, IVIVC has been one of the most important issues in the field of pharmaceutics. However, even with the aid of the FDA IVIVC Guidance, only very limited Abbreviated New Drug Application (ANDA) submission for ER oral drug products included adequate IVIVC data to enable the completion of bioequivalence (BE) review within first review cycle. Establishing an IVIVC for non-oral dosage forms has remained extremely challenging due to their complex nature and the lack of in vitro release methods that are capable of mimicking in vivo drug release conditions. This review presents a general overview of recent advances in the development of IVIVC for complex non-oral dosage forms (such as parenteral polymeric microspheres/implants, and transdermal formulations), and briefly summarizes the knowledge gained over the past two decades. Lastly this review discusses possible directions for future development of IVIVC for complex non-oral dosage forms.
Keywords: In vitro-in vivo correlation (IVIVC), non-oral, parenteral, prediction, in vitro release testing, modeling
Graphical abstract
1. Introduction
In vitro-in vivo correlation (IVIVC) is defined by the U.S. Food and Drug Administration (FDA) as “a predictive mathematical model describing the relationship between an in vitro property of a dosage form and a relevant in vivo response” [1]. Generally the in vitro property is the rate or extent of drug dissolution or release, while the in vivo response is the plasma drug concentration or amount absorbed. In the case of non-oral drug products (e.g. transdermal and ophthalmic dosage forms), an in vitro property could be in vitro drug permeation across the membrane of interest, while an in vivo property could be in vivo drug permeation. The history of IVIVC can be traced back to as early as 1950s, when pharmaceutical scientists attempted to correlate in vitro drug dissolution profiles of oral formulations with their respective in vivo pharmacokinetic profiles by means of mathematical modeling [2, 3]. In 1997, the U.S. FDA published a regulatory guidance related to the development, evaluation, and applications of IVIVC for extended release oral dosage forms. Since then, the establishment and application of IVIVC has increasingly gained more significance in the field of pharmaceutics. Generally, IVIVC can be categorized into five different levels: Levels A, B, C, D, and multiple Level C (Figure 1).
Figure 1.

Levels of IVIVC.
Level A represents a point-to-point relationship between in vitro and in vivo profiles. Generally the correlations are linear. However, non-linear correlations are also acceptable [4]. A Level A correlation is considered the most informative and is recommended by the U.S. FDA. It is also the only level of IVIVC that can be used to obtain biowaiver.
Level B correlation utilizes the principles of statistical moment analysis. A mean in vitro dissolution time (MDTin vitro) is compared to either a mean in vivo residence (MRTin vivo) or dissolution time (MDTin vivo). Similar to a Level A IVIVC, a Level B correlation compares all in vitro and in vivo data available. However, since various in vivo release profiles may result in the same MRTin vivo or MDTin vivo, a Level B correlation is not considered to be a point-to-point correlation, and does not necessarily reflect the actual in vivo plasma profile and hence may lack sufficient predictability.
Level C correlation establishes a single point relationship between a dissolution parameter (e.g. the time required for 50% dissolution, T50%) and a pharmacokinetic parameter such as Cmax, Tmax or AUC. Since it is based on a single point analysis, it is does not reflect the complete shape of the plasma concentration time curve, which is critical to define in vivo performance of a drug product. Accordingly, a Level C IVIVC is limited in predicting in vivo drug performance. Nevertheless, Level C correlations may be useful in the early stages of formulation development when pilot formulations are being selected.
Multiple Level C correlation relates multiple dissolution time points to one or more pharmacokinetic parameter(s) (e.g. Cmax, Tmax or AUC). A multiple Level C correlation should be based on at least three dissolution time points covering the early, middle, and late stages of the dissolution profile. A multiple Level C correlation can be as useful as a Level A correlation. However, if a multiple Level C correlation is obtainable, then the development of a Level A correlation should also be feasible and is more preferable.
Level D correlation is a rank order correlation comparing in vitro and in vivo release profiles. A level D correlation is only qualitative and is not adopted in the U.S. FDA IVIVC Guidance.
A meaningful IVIVC can be used to guide formulation and/or process development changes in the various stages of drug product development. In addition, an IVIVC can be used to support and/or validate the use of an in vitro dissolution method and can help set clinically relevant dissolution specifications to ensure product quality [5]. Most importantly, when a Level A IVIVC is established and validated, the in vitro release method can be used as a surrogate for bioequivalence studies when pre-approval and post-approval changes are required (e.g. formulation composition, as well as manufacturing process, equipment and site) [6–8]. Through the successful development and application of a meaningful IVIVC, the in vivo performance may be accurately predicted from the in vitro performance of drug products and therefore, human or animal studies can be minimized and the regulatory burden can be reduced [9, 10].
Despite the publication of the FDA IVIVC guidance on ER oral dosage forms nearly two decades ago, only 14 ANDA submissions had IVIVC data, most of which were deficient and thereby, not acceptable [11]. Compared to the ER oral dosage forms, the establishment of an IVIVC for non-oral drug products (e.g. parenteral microspheres and implants, as well as transdermal and ophthalmic products) has been even more challenging due to their complex characteristics as well as the lack of standardized, compendial in vitro release testing methods [10]. In recent years, there has been significant interest within the pharmaceutical industry, academia, and regulatory agencies in developing suitable in vitro release testing methods as well as establishing IVIVCs for complex non-oral drug products. Notably, the U.S. FDA has funded over 20 research grants to advance in vitro equivalence methods for complex non-oral drug products and drug-device combinations in the past two years. Through collective and collaborative efforts in the field of pharmaceutics and drug delivery, some “ground-breaking” progress has been achieved. This review highlights recent advances in the development of IVIVC for complex non-oral dosage forms and briefly summarizes the knowledge gained over the past two decades. Lastly this review discusses possible directions for future development of IVIVC for these complex dosage forms.
2. Current State-of-the-Art
To date, there is no regulatory IVIVC guidance available for complex non-oral drug products. The same principles of developing IVIVC for ER oral dosage forms as detailed in the FDA IVIVC Guidance have been applied to develop IVIVC for various complex non-oral dosage forms such as parenteral polymeric microspheres and implants [12–17], transdermal patches/gels [18, 19], as well as ocular inserts [20].
2.1. Approaches to develop IVIVCs
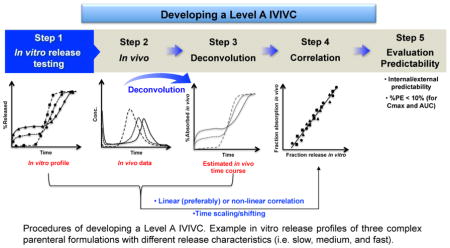
A Level A IVIVC is generally considered the highest level of correlation and is desirable from a regulatory point of view. Typically, developing a Level A IVIVC involves the following procedures (Figure 2): 1) obtaining formulations (preferably, three or more) with different release rates (e.g. slow, medium, and fast) or using one formulation if its in vitro dissolution is independent of dissolution testing conditions (e.g. pH, media, and agitation); 2) obtaining in vivo plasma concentration profiles or in vivo dissolution profiles of the selected formulations; 3) estimating in vivo absorption or dissolution time course of each formulation using an appropriate deconvolution technique (e.g. model-dependent, and model-independent numerical) (Table 1); 4) establishing a correlation/relationship between the estimated fraction in vivo release/absorption and the faction in vitro release, using a linear (preferably) or non-linear model (e.g. Sigmoid, Hixon-Crowell, Weibull, Higuchi, and Logistic) [21]; and 5) evaluating the predictability of the developed IVIVC internally and/or externally. Based on the FDA IVIVC Guidance, an average percentage prediction error (%, PE) of 10% or less for pharmacokinetic parameters of interest (e.g. Cmax or AUC) establishes the predictability of a developed IVIVC. When developing a Level A IVIVC, there may be disparity between deconvoluted in vivo and in vitro dissolution profiles due to the intrinsic difference between in vitro and in vivo dissolution conditions. Accordingly, time shifting/scaling may be utilized to allow the deconvoluted in vivo data to be on the same time scale as the in vitro dissolution data, which in turn makes it possible to establish a correlation/relationship between in vitro and in vivo release data.
Figure 2.

Procedures of developing a Level A IVIVC. Example in vitro release profiles of three complex parenteral formulations with different release characteristics (i.e. slow, medium, and fast). The figure in Step 4 shows a one-to-one linear correlation.
Table 1.
Some deconvolution techniques.
| Deconvolution Technique | Function | ||
|---|---|---|---|
| Model-Dependent | Wagner-Nelson (one-compartment model) |
|
|
| Loo-Riegelman (two-compartment model) |
|
||
| Mechanistic, Physiologically-based | |||
| Model-Independent | Numerical |
|
|
Ct: the plasma concentration at time t; Ke: the elimination rate constant; (Xp)/t: the amount of drug in the peripheral compartment as a function of time; Vc: the apparent volume of the central compartment; K10: the apparent first order elimination rate constant of the drug from the central compartment estimated from an intravenous study of the same subject; rabs: the absorption rate time course; Cδ: the concentration time profile.
Although a Level A IVIVC is most informative and recommended by the U.S. FDA, other levels of IVIVC (e.g. multiple Level C, and Level B) can be helpful to assure product quality, and to assist in formulation development. When developing a Level B IVIVC, at least three formulations are required. Based on the principles of statistical moment analysis, a mean residence time (MRTin vivo), mean absorption time (MATin vivo), or mean in vivo dissolution time (MDTin vivo) is calculated and related to a mean in vitro dissolution time (MDTin vitro) (Figure 3A). All parameters determined are model-independent. In the case of developing a multiple Level C correlation, one or more pharmacokinetic parameters (e.g. Cmax, Tmax or AUC) are correlated with at least three dissolution time points covering the early, middle, and late stages of the dissolution profile. Based on the U.S. FDA IVIVC Guidance, the recommendations for assessing the predictability of Level C correlations depend on the type of application for which the correlation is to be used. The methods and criteria for assessing the predictability are the same as that for Level A correlations described above.
Figure 3.

Example Level B IVIVC (A) and Levy plot obtained from times for 0–90% drug released in vitro and absorbed in vivo (B).
The development of IVIVCs for non-oral drug products is a complicated process, due to not only their complex characteristics (e.g. multi-phasic release) but also the lack of suitable in vitro release testing methods. Despite that extensive efforts have been devoted in this area, there are only a few literature reports on the establishment of IVIVCs for these drug products based on multiple formulations, albeit with different in vitro release testing methods such as USP apparatus 4 methods [15, 16], dialysis membrane methods [14, 22], and Franz diffusion cells [18–20] (Table 2).
Table 2.
Overview on IVIVCs for complex non-oral dosage forms.
| Dosage Forms | IVIVCs | Correlation | In Vitro Release Testing |
|---|---|---|---|
| Method | |||
| Parenteral Microspheres/Implants | |||
| Microspheres | 1:1 linear correlation | In vitro release vs in vivo absorption | Flow through [15, 16]; dialysis membrane [14, 22]; sample-and-separate [17, 23–25] |
| In vitro release vs fractional AUC | Dialysis membrane [14, 22]; sample-and-separate [23] | ||
| Level B | MDTin vitro vs MRTin vivo | Sample-and-Separate [26] | |
| Implants | 1:1 linear correlation | In vitro release vs in vivo release; Levy plot | Sample-and-Separate [17, 27] |
| Level B | MDTin vitro vs MRTin vivo | Sample-and-Separate [17, 28] | |
| Implantable devices | 1:1 linear correlation | In vitro release vs in vivo absorption/r AUC | In-House flow-cell apparatus [29] |
| Intra-Articular Injectable depots | 1:1 linear correlation | Levy plot (the time for in vitro disappearance vs the time for in vivo joint disappearance; the time for in vitro release vs the time for in vivo absorption | Rotating dialysis [30] |
| Transdermal (TDDS) | |||
| Patches1 | Level A1 | In vitro permeability vs in vivo bioavailability | Franz diffusion cells [18] |
| Gel | 1:1 linear correlation | In vitro permeability vs in vivo permeability | Franz diffusion cells [19, 31] |
| Iontophoretic patches | 1:1 linear correlation | Cumulative amounts delivered in vitro vs in vivo absorption | Franz diffusion cells [32] |
| Ophthalmic | |||
| Ocular insert | 1:1 linear correlation | in vitro release vs in vivo release | Franz diffusion cells [20] |
The only developed Level A IVIVC was validated internally and externally (PE%<15%).
2.2. IVIVCs for parenteral polymeric microspheres/implants
Parenteral polymeric microspheres/implants, particularly poly(lactic-co-glycolic acid) (PLGA)/poly(lactic acid) (PLA)-based microsphere/implant drug products have been one of the most successful complex non-oral polymeric drug products on the market. The PLGA/PLGA-based microsphere/implant drug products are biodegradable, biocompatible, and possess the capability of delivering a variety of therapeutics (e.g. small molecules and biologics) in a controlled manner over periods of days to several months [33–36]. These ER parenteral drug products normally contain substantial amounts of potent therapeutics. Therefore, it is critical to assure consistent product performance and safety through in vitro quality control tools such as discriminatory in vitro release testing methods, as well as reliable IVIVCs or in vitro-in vivo relationship (IVIVR) in the event that an IVIVC is not feasible. Over the past two decades, the development of IVIVCs for polymeric microspheres/implants has received the most attention, as a result of which considerable progress has been achieved (Table 2). However, most reported literature are “proof-of-concept” research that only demonstrated the possibility of developing point-to-point linear correlations or Level B correlations based on one formulation. Encouragingly, Level A IVIVCs established using two or more microsphere formulations with different release characteristics have recently be presented [14, 16, 22, 23]. It should be noted that multiple formulations with different release characteristics are essential to develop a reliable IVIVC.
One of the most challenging aspects of developing IVIVCs for complex microsphere/implant drug products is to design in vitro release studies in such a way that the in vivo behavior of these products is reflected as much as possible. PLGA/PLA-based polymeric microspheres/implants are normally administrated into subcutaneous or muscular tissues or directly injected into local areas (e.g. knee joints). Following injection/implantation, therapeutics are slowly released from microspheres/implants into the tissue fluids via complex release mechanisms (e.g. diffusion, polymer erosion or a combination thereof) [37, 38], and are subsequently transported into the systemic blood circulation system via diffusion and/or convective processes [39–41]. Due to the lack of compendial in vitro release methods, various in vitro release methods (e.g. sample-and-separate [23, 26, 27], membrane dialysis [14, 22], and flow through [15, 16]) have been utilized to determine in vitro drug release characteristics and to develop IVIVCs. Although it is feasible to develop IVIVCs for parenteral microspheres/implants based on a simple sample-and-separate method [17, 23–25], there are limitations associated with this method such as poor hydrodynamic conditions, loss of product (e.g. microspheres) during sampling as well as inability to mimic different in vivo drug release conditions. For example, the presence of the in vivo boundary layers as well as the small interstitial fluid volume available for drug release at the administration sites. It has been reported that the correlation/relationship between the in vitro and in vivo data of huperzine microspheres was sensitive to the route of administration. Additionally, the sample-and-separate method appeared to better reflect drug release from PLGA microspheres in muscular tissues compared to that in subcutaneous tissues, thus a better correlation was obtained for the intramuscular route [23]. Compared to the sample-and-separate method, membrane dialysis and flow though (USP apparatus 4) methods have more complex apparatus setup. However, they appear to be more capable of mimicking in vivo drug release conditions of parenteral microspheres/implants (e.g. microspheres/implants are exposed to a limited volume of release media at a time). Accordingly, these two release methods may be more suitable to develop a Level A IVIVC for parenteral microspheres/implants. A Level A IVIVC based on three olanzapine microsphere formulations with different release rates (i.e. fast, medium, and slow) has been recently reported using a membrane dialysis method [13]. In our ongoing research, we have successfully developed a Level A IVIVC using different combinations of three risperidone microsphere formulations that are compositionally equivalent but prepared using different manufacturing processes. Notably, both external predictability and robustness (IVIVCs not affected by the formulation combinations) of the developed IVIVC have been validated. Compared to the sample-and-separate method, the USP apparatus 4 method demonstrated not only better discrimination against compositionally equivalent microsphere formulations with manufacturing differences, but also better predictability of the in vivo PK profiles obtained from the animal study using a rabbit model. More importantly, the USP apparatus 4 method demonstrated better predictability of the in vivo initial burst release phase, whereas the sample-and-separate method was not able to discriminate the initial burst release phase. Reliable detection of the in vitro initial burst can be important, especially in regulatory applications when two formulations are being evaluated for equivalence.
To help understand drug release mechanisms and guide the establishment of IVIVCs for polymeric microspheres/implants, different mathematical models (e.g. Higuchi, and Weibull) have often been used (Table 3). These mathematical models assume zero dissolution at time zero and complete dissolution at sufficient time t. By introducing a parameter that represents the degree of dissolution, these mathematical models can easily be modified to account for incomplete dissolution (if any). For example, a Level A IVIVC was established for buserelin implants, from which drug release can be described using the Higuchi model [17]. Interestingly, when buserelin release from implants was governed by a combination of diffusion, dissolution as well as erosion, a Level B IVIVC appeared more suitable [17].
Table 3.
Some of the functions that have been used to describe in vitro drug dissolution.
| Description | Function | |
|---|---|---|
| Exponential | F1 (θ1, t) = 1 − e (−θ11t) | |
| Weibull | F1 (θ1, t) = 1 − e (−θ11tθ12) | |
| Higuchi | F1 (θ1, t) = kH (t)0.5 | |
| Logistic |
|
|
| Korsmeyer-Peppas |
|
θ1i (i = 1 and 2): parameter θ1 at observation times t1 and t2; kH: the Higuchi dissolution constant; Mt: the amount of drug released at time t; M∞: the amount of drug released at infinite time; M∞: the kinetic constant; n: the release exponent.
Another challenging aspect of developing IVIVCs for complex microspheres/implants is how to deconvolute in vivo data and correlate that with in vitro release data. Due to their complex release characteristics (e.g. bi- or tri-phasic release profiles), it may be very difficult to correlate deconvoluted in vivo data with multi-phasic in vitro release data using a simple mathematical model. In some cases, it may not be possible to predict the initial in vivo drug release based on the in vitro release data from the burst release phase, since the rate-limiting step for the initial in vivo drug availability may actually be drug permeation across the tissue barriers. Although an IVIVC may still be developed using post-burst in vivo and in vitro release data, the “post-burst” IVIVC may have limited prediction capability [26]. Furthermore, pharmacokinetic parameters of testing dosage forms are normally needed when using the deconvolution approach. For drugs that fit into a two-compartment model, an extra in vivo study (intravenous administration) is required in order to perform deconvolution. Accordingly, in order to simplify the development of IVIVCs and avoid the use of pharmacokinetic parameters, other approaches (e.g. fractional AUC) have been reported [13, 14, 23, 29]. The factional AUC is determined by dividing cumulative AUC at time “t” with cumulative AUC0-t and plotting along with the percent drug release in vitro. In a manner similar to the deconvolution approach, the in vitro and in vivo drug release data are then compared. It has been demonstrated that in vivo profiles of PLGA microspheres obtained using both the Wagner-Nelson and fractional AUC methods were nearly superimposable, suggesting that fractional AUC could be used as an alternative to the commonly used Wagner-Nelson method [14, 22]. However, it should be noted that these methods are not adopted in the FDA IVIVC Guidance and therefore, may not be useful for regulatory purposes. In addition to comparing the drug absorption percentage with the amount of drug released at different time points, the time for 0% to 90% absorbed in vivo with the time for releasing the same amounts of the drug in vitro (Levy plot, Figure 3B) has been compared and good linear correlation was shown [17, 30].
Considering that real-time in vitro release testing of parenteral microspheres/implants normally requires extended periods of time, it may be necessary to develop IVIVCs based on discriminatory accelerated in vitro release tests. To this end, a relationship between accelerated and real-time in vitro release data should be established such that the accelerated in vitro test could maintain “bio-relevance”. Ideally, drug release from real-time and accelerated tests should follow the same release mechanism with a one-to-one correlation between the release profiles [42]. However, it is possible that the drug mechanism(s) may change since accelerated release tests are typically performed under extreme conditions (e.g. high temperatures, extreme pH conditions) [43]. Nevertheless, developing an IVIVC based on accelerated testing may still be feasible as long as all microsphere/implant formulations experience similar changes and their release characteristics can be differentiated. The possibility of developing an IVIVC based on accelerated testing for PLGA microspheres has recently been demonstrated using commercial Risperdal® Consta® [15]. Despite that the accelerated in vitro release profiles obtained at elevated temperatures (50°C and 54.5°C) did not show a good linear correlation with the real-time in vitro release profile, a one-to-one linear correlation between the accelerated in vitro data and the time scaled in vivo data of Risperdal® Consta® was shown [15].
Physiologic responses (e.g. foreign body response) to biomaterials is another important issue that must be considered when developing a reliable IVIVC as they may result in polymer degradation mechanism changes in vivo. For instance, acidic PLGA degradation products may accumulate at the local sites and hence lower the pH in the interstitial space immediately surrounding the microspheres. This may result in a change in the degradation mechanism of PLGA microspheres from bulk erosion to surface erosion, thus accelerating polymer degradation with subsequent increased drug release in vivo [16, 44]. On the other hand, chronic inflammation in response to the presence of microspheres in the interstitial site, and fibrosis may form and isolate microspheres, thus slowing down drug absorption/drug release in vivo [45]. The formation of fibrosis was speculated to be responsible for the slower in vivo release/absorption of Risperdal® Consta® 30 days following administration to humans [15]. Interestingly, it was noted in our recent research that Risperdal® Consta® demonstrated faster drug release in rabbits compared to in vitro real time release. This suggests that differences in drug absorption and drug release between animals and humans must be taken into consideration, and an IVIVC study based on animal data may not be fully extrapolated to humans.
2.3. IVIVCs for transdermal dosage forms
Transdermal drug delivery systems (TDDS) are one of the first generation controlled release drug products that appeared on the market [47, 48]. Since the first transdermal drug product (Transdermal Scop®) was approved by the U.S. FDA in 1979, there have been different generations of transdermal drugs products (e.g. passive transdermal patches, and iontophoretic transdermal devices) developed and commercialized [48, 49]. Unfortunately, unlike parenteral microspheres/implants, the development of IVIVCs for TDDS has not yet been given substantial attention. To date, there are only very few literature reports on IVIVCs for TDDS (Table 2).
Transdermal drug delivery involves a few consecutive steps: i) drug release from the formulation; ii) drug penetration/diffusion into/through skin; and iii) drug arriving at the site of action to trigger a pharmacological response. In most cases, drug penetration/diffusion through the skin is the rate-limiting step, which can be described using different mathematical models (Table 4). The development of IVIVC for TDDS is somewhat different compared to that of parenteral microspheres/implants. One of challenges to develop an IVIVC for TDDS is to mimic the process of drug permeation across human skin as much as possible. Various in vitro dissolution methods (e.g. apparatus 5 (paddle over disk method), apparatus 6 (rotating cylinder method), and apparatus 7 (reciprocating holder method)) have been recommended as quality control tools for transdermal/topical drug products (such as transdermal patches and films) [50]. However, these dissolution methods may not reflect complex mechanism(s) of drug permeation/diffusion across skin. Accordingly, instead of in vitro dissolution, in vitro skin permeation is more often used for the development of IVIVCs for TDDS [18, 19].
Table 4.
Some mathematical models describing drug release mechanisms of TDDS [53].
| TDDS | Release Mechanism | Function | |
|---|---|---|---|
| Passive transdermal patches | Fick s law of diffusion |
|
|
| Iontophoretic systems | Iontophoretic transport |
|
J: the flux of a drug through the skin; D: the diffusion coefficient of the drug through the skin; K: the partition coefficient; h: the thickness of the skin; Cv: the drug solubility in the vehicle; Cb: the plasma concentration; ψ: the electric potential in the membrane; x: the position in the membrane; ε: the combined porosity and tortuosity factor of the membrane; v: the average velocity of the convective flow.
Franz diffusion cells are the most widely used apparatus for determining in vitro drug permeation of TDDS and for the development of IVIVCs. Franz diffusion cells consist of a donor compartment and receptor compartment with the membrane of interest (e.g. excised human or animal skin) mounted as a barrier between the two compartments. Due to the inherent variability in absorption between individuals and between anatomical sites (e.g. abdominal vs forearm skin), it is important to control for skin source and viability, as well as to evaluate in vitro permeability across skin from several donors in cases where in vitro skin permeation data is correlated [51]. Excised human skin has been demonstrated to be the most appropriate in vitro skin model that may potentially be used as a surrogate for more costly and time-consuming in vivo bioequivalence studies [52]. Other skin models (such as excised porcine skin [19] and rat skin [32]) have also been used for in vitro drug permeation testing, and for the development of IVIVCs.
Generally IVIVCs for transdermal drug products could be categorized into the same levels as those described in the FDA IVIVC Guidance for extended release oral dosage forms (i.e. Levels A, B, C, and multiple C). However, since in vitro dissolution is not commonly used for the development of IVIVCs, Level B (which uses mean dissolution time) and Level C IVIVCs may not be applicable for TDDS [51]. A Level A IVIVC that correlates in vitro drug permeation across skin with in vivo drug permeation or absorption is more desirable. Similar to parenteral microspheres/implants, different deconvolution techniques (Table 1) can be utilized to deconvolute in vivo data and correlate with in vitro drug permeation data. In some cases, the steady state flux in vitro is extrapolated to determine the in vivo plasma concentration using pharmacokinetic modeling [53]. Most recently, a Level A IVIVC (a second order polynomial correlation) between the in vitro permeation percent and the deconvoluted in vivo absorption percent was established using three estradiol TDDS [18]. In this study, the Wagner-Nelson method was used to deconvolute human pharmacokinetic data obtained from the literature. The developed Level A IVIVC was validated internally and externally and showed less than 15% PE for both Cmax and AUC.
In order to facilitate the development of a good in vitro predictive model for TDDS, some improvements on current in vitro testing apparatus and in vivo analytical techniques have been reported. An inherent problem with Franz diffusion cells is the lack of microvasculature, which is present in the in vivo environment and helps in rapid clearance of the drug. To overcome this, flow-through diffusion cells have been designed in such a way that the receptor buffer is continuously removed to help maintain the sink conditions in vitro [54], which may be more relevant to the actual in vivo conditions and hence, could potentially benefit the development of IVIVCs for TDDS. In addition, new technologies (e.g. microdialysis and Confocal Raman Spectroscopy (CRS)) have been implemented to obtain in vivo drug adsorption data in a continuous fashion [31, 32]. For example, the real-time drug disposition in skin was monitored using CRS and correlated with in vitro human skin permeation data obtained using Franz diffusion cells. In this “proof-of-concept” study, a good correlation was obtained between the in vitro flux of niacinamide and signal intensity of niacinamide permeated into the mid ventral forearm at 4 μm in vivo after a 30 min application [31].
3. Future perspectives
Despite the encouraging progress that has been made over the past two decades, the development and application of IVIVCs for complex non-oral dosage forms still remains at an infant stage. At present, there is sparse or no literature reports on IVIVC for most complex non-oral dosage forms (e.g. parenteral nanoparticulate systems, and ophthalmic dosage forms). With the ongoing commercialization of novel and generic complex non-oral drug products, it is essential to initiate the development of IVIVCs to help assure the product performance and safety, as well as to assist in product development in a timely and cost-effective fashion.
One of the biggest hurdles yet to conquer in developing IVIVCs for complex non-oral drug products is the dearth of bio-relevant in vitro dissolution methods that are capable of reflecting the complex and dynamic in vivo environment these dosage forms are encountered. This is particularly the case for nanoparticulate systems such as liposomal products, as well as ophthalmic drug products. Liposomal formulations have often showed a poor correlation between their in vitro and in vivo performance, due to their possible multiple fates in the blood circulation [55]. For example, “stealth” liposomal drug products are designed to be stable with no (or very little) drug release prior to reaching their target organs and cells (e.g. Doxil®), while other liposomal drug products are designed to provide sustained release of therapeutics (e.g. DepoDur®). In addition, a vast lipid membrane pool in the systemic circulation, the presence of macrophages, as well as the physiological conditions at tumor sites, affect the disposition of liposomes and further complicates the in vivo release kinetics. Over the past few years, some breakthrough research in mimicking the complex in vivo performance of liposomes has been reported [56, 57]. A novel drug release assay that utilizes excess multilamellar vesicles to simulate the physiological presence of the lipid membrane “sink” has been developed [56]. Compared to the commonly used dialysis membrane method, this method demonstrated better correlations between in vitro and in vivo release data for different liposomal formulations (e.g. doxorubicin, verapamil and ceramide liposomes) [56]. In another study, a novel “two-stage” in vitro dissolution method was developed to mimic the projected fate of passively targeted liposomes in the body (including the first phase when the liposomes are circulating in the body prior to uptake at the target site; and the second phase when the liposomes breakdown at the target site to release the encapsulated drug) [57]. In vivo prediction of this simple “two-stage” in vitro dissolution method has yet to be demonstrated.
In the case of ophthalmic drug products, the development of IVIVCs is even more challenging due to the unique anatomy and physiology of the eye. For example, it may not be possible to use current in vitro methods to reflect the complex pre-corneal constraints of the eye (such as continuous clearance of dosage forms and the released drug from the cul-de-sac area through tear drainage, tear dilution, lacrimation as well as tear turnover), which may be critical to develop a reliable in vitro predictive model. In addition, different ocular diseases often require different administration routes, which complicates the development of IVIVCs. For example, topical instillation of ophthalmic dosage forms can be used to treat ocular diseases that manifest on the ocular surface or in the anterior segment, while treating other ocular diseases may require direct injection/implantation of dosage forms/therapeutics into different eye regions or tissues (e.g. anterior chamber (AC), subconjunctival space, vitreous cavity, or the back of the eye) (Figure 4). Accordingly, it may be necessary to develop in vitro dissolution methods and IVIVCs for different ophthalmic dosage forms based on their administration routes or the regions that they are intended to be delivered to. Recently, extensive efforts have been devoted to developing suitable in vitro dissolution methods for various ophthalmic dosage forms such as intravitreal implants, topical ophthalmic emulsions, suspensions, as well as semi-solid dosage forms. A previous study showed a good correlation between in vitro release of ocular inserts and their in vivo drug release (calculated based on the remaining drug in the inserts taken from the lower conjunctiva cul-de-sac of rabbits) [20]. However, this developed IVIVC may not be able to predict in vivo performance of these ocular inserts since the drug clearance at the cul-de-sac area and the distribution of the drug into different eye tissues were not taken into consideration.
Figure 4.
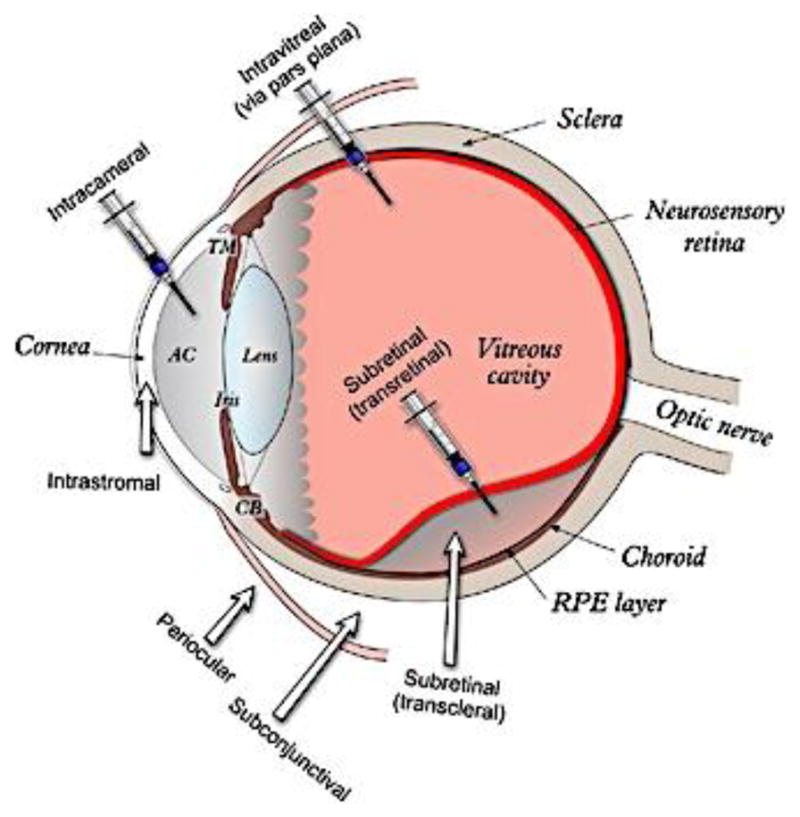
A schematic representation of the human eye and the different ophthalmic administration routes. From reference [58] with permission.
Another difficulty that hinders the establishment of IVIVCs for complex non-oral dosage forms is the lack of reliable analytical techniques to monitor in vivo drug performance. Typically, in vivo drug release can be determined through analyzing drug content in the plasma or blood fluid (e.g. aqueous humour), which may require frequent sampling, complicated procedures (to extract the drug from biological samples), high sensitive analytical instruments, as well as extensive animal/human experiments. Recent advances in biomedical engineering/instrumentation have enabled relatively simple and more accurate monitoring of in vivo drug performance. For example, microdialysis has been utilized to help continuously monitor drug concentrations in ocular tissues and fluids [59], which in turn can facilitate the development of IVIVCs for ophthalmic dosage forms. Surface Plasmon Resonance (SPR) was utilized to evaluate interactions between blood proteins and liposomes and demonstrated the potential for predicting blood clearance of liposomes [60]. In another study, non-invasive Micro-Positron Emission Tomography (PET) imaging was used to monitor in vivo drug release from liposomes following the intraperitoneal injection. Convoluted in vitro release data showed good prediction of in vivo profiles compared with that obtained using Micro-PET [61], suggesting that PET-imaging may be very useful to determine in vivo drug release and facilitate the development of IVIVCs for complex non-oral dosage forms with reduced animal testing. Despite these exciting research efforts, new analytical techniques and/or creative implementation of existing analytical/imaging instruments may help fully characterize the in vivo performance of complex non-oral dosage forms.
Last but not least, better mathematical methods and simulation techniques are needed to correlate in vitro performance with complicated in vivo performance of non-oral drug products, which may include drug distribution at the site of action and plasma drug distribution, as well as ultimately to relate in vitro drug performance to clinical performance.
4. Summary and Outlook
Development of in vitro models that allow accurate prediction of in vivo performance will revolutionize the development of both innovative and generic complex non-oral drug products. This area is still young, and it is not surprising that more time is needed to surmount all obstacles to obtain predictive and reliable IVIVCs for these complex drug products. What is important is not only innovative in vitro testing methods or complicated mathematical methods/simulation techniques, but also standardized in vitro testing methods to facilitate inter-laboratory comparisons and advance the regulatory review process. Furthermore, to propel the development of generic non-oral drug products, it may be essential to develop IVIVCs using dosage forms that are equivalent in formulation composition and components but with manufacturing differences.
Acknowledgments
Support is acknowledged from the FDA 1U01FD004931-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.FDA Guidance for Industry Extended Release Oral dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. U.S. Department of Human Health and Human Services, Food and Drug Administration; 1997. [Google Scholar]
- 2.Nelson E. Solution rate of theophylline salts and effects from oral administration. J Am Pharm Assoc Am Pharm Assoc (Baltim) 1957;46:607–614. doi: 10.1002/jps.3030461012. [DOI] [PubMed] [Google Scholar]
- 3.Edwards LJ. The dissolution and diffusion of aspirin in aqueous media. Transactions of the Faraday Society. 1951;47:1191–1210. [Google Scholar]
- 4.Uppoor VR. Regulatory perspectives on in vitro (dissolution)/in vivo (bioavailability) correlations. J Control Release. 2001;72:127–132. doi: 10.1016/s0168-3659(01)00268-1. [DOI] [PubMed] [Google Scholar]
- 5.Roudier B, Davit BM, Beyssac E, Cardot JM. In vitro-in vivo correlation's dissolution limits setting. Pharm Res. 2014;31:2529–2538. doi: 10.1007/s11095-014-1349-8. [DOI] [PubMed] [Google Scholar]
- 6.Burgess DJ, Crommelin DJ, Hussain AS, Chen ML. Assuring quality and performance of sustained and controlled released parenterals. Eur J Pharm Sci. 2004;21:679–690. doi: 10.1016/j.ejps.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Siewert M, Dressman J, Brown C, Shah V. FIP/AAPS guidelines for dissolution/in vitro release testing of novel/special dosage forms. Dissol Technol. 2003;10:6–15. [Google Scholar]
- 8.Anand O, Yu LX, Conner DP, Davit BM. Dissolution testing for generic drugs: an FDA perspective. AAPS J. 2011;13:328–335. doi: 10.1208/s12248-011-9272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emami J. In vitro - in vivo correlation: from theory to applications. J Pharm Pharm Sci. 2006;9:169–189. [PubMed] [Google Scholar]
- 10.Young D. In vitro-in vivo correlation for modified release parenteral drug delivery systems. In: Chilukuri DM, Sunkura G, Young D, editors. Pharmaceutical Product Development: In Vitro-In Vivo Correlation. Informa Healthecare; New York: 2007. pp. 141–151. [Google Scholar]
- 11.Kaur P, Jiang X, Duan J, Stier E. Applications of In Vitro-In Vivo Correlations in Generic Drug Development: Case Studies. AAPS J. 2015;17:1035–1039. doi: 10.1208/s12248-015-9765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlugt-Wensink KD, de Vrueh R, Gresnigt MG, Hoogerbrugge CM, van Buul-Offers SC, de Leede LG, Sterkman LG, Crommelin DJ, Hennink WE, Verrijk R. Preclinical and clinical in vitro in vivo correlation of an hGH dextran microsphere formulation. Pharm Res. 2007;24:2239–2248. doi: 10.1007/s11095-007-9433-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Souza S, Faraj JA, Giovagnoli S, Deluca PP. IVIVC from Long Acting Olanzapine Microspheres. Int J Biomater. 2014;2014:407065. doi: 10.1155/2014/407065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Souza S, Faraj JA, Giovagnoli S, Deluca PP. In vitro-in vivo correlation from lactide-co-glycolide polymeric dosage forms. Prog Biomater. 2014;3:131–142. doi: 10.1007/s40204-014-0029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rawat A, Bhardwaj U, Burgess DJ. Comparison of in vitro-in vivo release of Risperdal((R)) Consta((R)) microspheres. Int J Pharm. 2012;434:115–121. doi: 10.1016/j.ijpharm.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Zolnik BS, Burgess DJ. Evaluation of in vivo-in vitro release of dexamethasone from PLGA microspheres. J Control Release. 2008;127:137–145. doi: 10.1016/j.jconrel.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Schliecker G, Schmidt C, Fuchs S, Ehinger A, Sandow J, Kissel T. In vitro and in vivo correlation of buserelin release from biodegradable implants using statistical moment analysis. J Control Release. 2004;94:25–37. doi: 10.1016/j.jconrel.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Manda P, Pavurala N, Khan MA, Krishnaiah YS. Development and validation of in vitro-in vivo correlation (IVIVC) for estradiol transdermal drug delivery systems. J Control Release. 2015;210:58–66. doi: 10.1016/j.jconrel.2015.05.263. [DOI] [PubMed] [Google Scholar]
- 19.Mateus R, Moore DJ, Hadgraft J, Lane ME. Percutaneous absorption of salicylic acid--in vitro and in vivo studies. Int J Pharm. 2014;475:471–474. doi: 10.1016/j.ijpharm.2014.08.061. [DOI] [PubMed] [Google Scholar]
- 20.Gorle AP, Gattani SG. Design and evaluation of polymeric ocular drug delivery system. Chem Pharm Bull (Tokyo) 2009;57:914–919. doi: 10.1248/cpb.57.914. [DOI] [PubMed] [Google Scholar]
- 21.Dunne A. Approaches to developing in vitro-in vivo correlation models. In: Chilukuri DM, Sunkura G, Young D, editors. Pharmaceutical Product Development: In Vitro-In Vivo Correlation. Informa Healthecare; New York: 2007. pp. 47–70. [Google Scholar]
- 22.D'Souza S, Faraj JA, Giovagnoli S, Deluca PP. IVIVC from Long Acting Olanzapine Microspheres. Int J Biomater. 2014;2014:407065. doi: 10.1155/2014/407065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu DF, Fu XQ, Liu WH, Liu K, Li YX. Pharmacokinetics and in vitro and in vivo correlation of huperzine A loaded poly(lactic-co-glycolic acid) microspheres in dogs. Int J Pharm. 2006;325:116–123. doi: 10.1016/j.ijpharm.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Zhao Z, Li L, Zhou T, Lu W. Pharmacokinetics, in vitro and in vivo correlation, and efficacy of exenatide microspheres in diabetic rats. Drug delivery. 2015;22:86–93. doi: 10.3109/10717544.2013.871760. [DOI] [PubMed] [Google Scholar]
- 25.Su Z, Sun F, Shi Y, Jiang C, Meng Q, Teng L, Li Y. Effects of Formulation Parameters on Encapsulation Efficiency and Release Behavior of Risperidone Poly(<small>D</small>,<small>L</small>-lactide-co-glycolide) Microsphere. Chem Pharm Bull (Tokyo) 2009;57:1251–1256. doi: 10.1248/cpb.57.1251. [DOI] [PubMed] [Google Scholar]
- 26.Blanco-Prieto MJ, Campanero MA, Besseghir K, Heimgatner F, Gander B. Importance of single or blended polymer types for controlled in vitro release and plasma levels of a somatostatin analogue entrapped in PLA/PLGA microspheres. J Control Release. 2004;96:437–448. doi: 10.1016/j.jconrel.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Negrin CM, Delgado A, Llabres M, Evora C. In vivo-in vitro study of biodegradable methadone delivery systems. Biomaterials. 2001;22:563–570. doi: 10.1016/s0142-9612(00)00214-3. [DOI] [PubMed] [Google Scholar]
- 28.Amann LC, Gandal MJ, Lin R, Liang Y, Siegel SJ. In vitro-in vivo correlations of scalable PLGA-risperidone implants for the treatment of schizophrenia. Pharm Res. 2010;27:1730–1737. doi: 10.1007/s11095-010-0152-4. [DOI] [PubMed] [Google Scholar]
- 29.Prescott JH, Krieger TJ, Lipka S, Staples MA. Dosage form development, in vitro release kinetics, and in vitro-in vivo correlation for leuprolide released from an implantable multi-reservoir array. Pharm Res. 2007;24:1252–1261. doi: 10.1007/s11095-007-9243-2. [DOI] [PubMed] [Google Scholar]
- 30.Frost AB, Larsen F, Ostergaard J, Larsen SW, Lindegaard C, Hansen HR, Larsen C. On the search for in vitro in vivo correlations in the field of intra-articular drug delivery: administration of sodium diatrizoate to the horse. Eur J Pharm Sci. 2010;41:10–15. doi: 10.1016/j.ejps.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Mohammed D, Matts PJ, Hadgraft J, Lane ME. In vitro-in vivo correlation in skin permeation. Pharmaceutical research. 2014;31:394–400. doi: 10.1007/s11095-013-1169-2. [DOI] [PubMed] [Google Scholar]
- 32.Chaturvedula A, Joshi DP, Anderson C, Morris R, Sembrowich WL, Banga AK. Dermal, subdermal, and systemic concentrations of granisetron by iontophoretic delivery. Pharm Res. 2005;22:1313–1319. doi: 10.1007/s11095-005-5335-z. [DOI] [PubMed] [Google Scholar]
- 33.Hoffman AS. The origins and evolution of “controlled” drug delivery systems. J Control Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Burgess DJ. Microsphere Technologies. In: Wright JC, Burgess DJ, editors. Long Acting Injections and Implants. Springer US; 2012. pp. 167–194. [Google Scholar]
- 35.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655–672. doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao S, Guo C, Shi Y, Li LC. Recent advances in polymeric microspheres for parenteral drug delivery--part 1. Expert Opin Drug Deliv. 2012;9:1161–1176. doi: 10.1517/17425247.2012.709844. [DOI] [PubMed] [Google Scholar]
- 37.Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur J Pharm Sci. 2002;15:355–366. doi: 10.1016/s0928-0987(02)00023-4. [DOI] [PubMed] [Google Scholar]
- 38.Alexis F. Factors affecting the degradation and drug-release mechanism of poly(lactic acid) and poly[(lactic acid)-co-(glycolic acid)] Polym Int. 2005;54:36–46. [Google Scholar]
- 39.Larsen C, Larsen SW, Jensen H, Yaghmur A, Ostergaard J. Role of in vitro release models in formulation development and quality control of parenteral depots. Expert Opin Drug Deliv. 2009;6:1283–1295. doi: 10.1517/17425240903307431. [DOI] [PubMed] [Google Scholar]
- 40.Iyer SS, Barr WH, Karnes HT. Profiling in vitro drug release from subcutaneous implants: a review of current status and potential implications on drug product development. Biopharm Drug Dispos. 2006;27:157–170. doi: 10.1002/bdd.493. [DOI] [PubMed] [Google Scholar]
- 41.Medlicott NJ, Waldron NA, Foster TP. Sustained release veterinary parenteral products. Adv Drug Deliv Rev. 2004;56:1345–1365. doi: 10.1016/j.addr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Zolnik BS, Leary PE, Burgess DJ. Elevated temperature accelerated release testing of PLGA microspheres. J Control Release. 2006;112:293–300. doi: 10.1016/j.jconrel.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 43.Shen J, Burgess DJ. Accelerated in-vitro release testing methods for extended-release parenteral dosage forms. J Pharm Pharmacol. 2012;64:986–996. doi: 10.1111/j.2042-7158.2012.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang G, Woo BH, Kang F, Singh J, DeLuca PP. Assessment of protein release kinetics, stability and protein polymer interaction of lysozyme encapsulated poly(D,L-lactide-co-glycolide) microspheres. J Control Release. 2002;79:137–145. doi: 10.1016/s0168-3659(01)00533-8. [DOI] [PubMed] [Google Scholar]
- 45.Anderson FD, Archer DF, Harman SM, Leonard RJ, Wilborn WH. Tissue response to bioerodible, subcutaneous drug implants: a possible determinant of drug absorption kinetics. Pharmaceutical research. 1993;10:369–380. doi: 10.1023/a:1018932104577. [DOI] [PubMed] [Google Scholar]
- 46.Clark BC, Dickinson PA, Pyrah IT. Case study: in vitro/in vivo release from injectable microspheres. In: Burgess DJ, editor. Injtecable Dispersed Systems: Formulation, Processing and Performance. Taylor & Francis; Boca Raton: 2005. pp. 543–570. [Google Scholar]
- 47.Park K. Controlled drug delivery systems: past forward and future back. J Control Release. 2014;190:3–8. doi: 10.1016/j.jconrel.2014.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiedersberg S, Guy RH. Transdermal drug delivery: 30+ years of war and still fighting! J Control Release. 2014;190:150–156. doi: 10.1016/j.jconrel.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Prausnitz MR, Mitragotri S, Langer R. Current status and future potential of transdermal drug delivery. Nat Rev Drug Discov. 2004;3:115–124. doi: 10.1038/nrd1304. [DOI] [PubMed] [Google Scholar]
- 50.FDA-Recommned Dissolution Methods. http://www.accessdata.fda.gov/scripts/cder/dissolution/
- 51.Van Buskirk GA, Arsulowicz D, Basu P, Block L, Cai B, Cleary GW, Ghosh T, Gonzalez MA, Kanios D, Marques M, Noonan PK, Ocheltree T, Schwarz P, Shah V, Spencer TS, Tavares L, Ulman K, Uppoor R, Yeoh T. Passive transdermal systems whitepaper incorporating current chemistry, manufacturing and controls (CMC) development principles. AAPS PharmSciTech. 2012;13:218–230. doi: 10.1208/s12249-011-9740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franz TJ, Lehman PA, Raney SG. Use of Excised Human Skin to Assess the Bioequivalence of Topical Products. Skin Pharmacol Physiol. 2009;22:276–286. doi: 10.1159/000235828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaturvedula A, Banga AK. In vitro-in vivo correlation: transermal drug delivery systems. In: Chilukuri DM, Sunkura G, Young D, editors. Pharmaceutical Product Development: In Vitro-In Vivo Correlation. Informa Healthecare; New York: 2007. pp. 153–176. [Google Scholar]
- 54.Cordoba-Diaz M, Nova M, Elorza B, Cordoba-Diaz D, Chantres JR, Cordoba-Borrego M. Validation protocol of an automated in-line flow-through diffusion equipment for in vitro permeation studies. J Control Release. 2000;69:357–367. doi: 10.1016/s0168-3659(00)00306-0. [DOI] [PubMed] [Google Scholar]
- 55.Florence AT. “Targeting” nanoparticles: the constraints of physical laws and physical barriers. J Control Release. 2012;164:115–124. doi: 10.1016/j.jconrel.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 56.Shabbits JA, Chiu GN, Mayer LD. Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems. J Control Release. 2002;84:161–170. doi: 10.1016/s0168-3659(02)00294-8. [DOI] [PubMed] [Google Scholar]
- 57.Xu X, Khan MA, Burgess DJ. A two-stage reverse dialysis in vitro dissolution testing method for passive targeted liposomes. Int J Pharm. 2012;426:211–218. doi: 10.1016/j.ijpharm.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 58.Balaggan KS, Ali RR. Ocular gene delivery using lentiviral vectors. Gene Ther. 2012;19:145–153. doi: 10.1038/gt.2011.153. [DOI] [PubMed] [Google Scholar]
- 59.Boddu SH, Gunda S, Earla R, Mitra AK. Ocular microdialysis: a continuous sampling technique to study pharmacokinetics and pharmacodynamics in the eye. Bioanalysis. 2010;2:487–507. doi: 10.4155/bio.10.2. [DOI] [PubMed] [Google Scholar]
- 60.Crielaard BJ, Yousefi A, Schillemans JP, Vermehren C, Buyens K, Braeckmans K, Lammers T, Storm G. An in vitro assay based on surface plasmon resonance to predict the in vivo circulation kinetics of liposomes. J Control Release. 2011;156:307–314. doi: 10.1016/j.jconrel.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 61.Huhn E, Buchholz HG, Shazly G, Maus S, Thews O, Bausbacher N, Rosch F, Schreckenberger M, Langguth P. Predicting the in vivo release from a liposomal formulation by IVIVC and non-invasive positron emission tomography imaging. Eur J Pharm Sci. 2010;41:71–77. doi: 10.1016/j.ejps.2010.05.020. [DOI] [PubMed] [Google Scholar]