

Abstract
Data-independent acquisition LC-MS/MS techniques complement supervised methods for peptide quantification. However, due to the wide precursor isolation windows, these techniques are prone to interference at the fragment ion level, which in turn is detrimental for accurate quantification. The “non-outlier fragment ion” (NOFI) ranking algorithm has been developed to assign low priority to fragment ions affected by interference. By using the optimal subset of high priority fragment ions these interfered fragment ions are effectively excluded from quantification. NOFI represents each fragment ion as a vector of four dimensions related to chromatographic and MS fragmentation attributes and applies multivariate outlier detection techniques. Benchmarking conducted on a well-defined quantitative dataset (i.e. the SWATH Gold Standard), indicates that NOFI on average is able to accurately quantify 11-25% more peptides than the commonly used Top-N library intensity ranking method. The sum of the area of the Top3-5 NOFIs produces similar coefficients of variation as compared to the library intensity method but with more accurate quantification results. On a biologically relevant human dendritic cell digest dataset, NOFI properly assigns low priority ranks to 85% of annotated interferences, resulting in sensitivity values between 0.92 and 0.80 against 0.76 for the Spectronaut interference detection algorithm.
Keywords: Data-Independent Acquisition, SWATH, Interference removal, Multivariate outlier detection, Mass Spectrometry, Quantification, Peptides
INTRODUCTION
In the life sciences, liquid chromatography coupled to mass spectrometry (LC-MS) is the preferred analytical platform for characterizing proteins and low molecular weight compounds in complex samples. Data-dependent acquisition (DDA)1 is traditionally used for discovery and qualitative workflows, whereas selected reaction monitoring (SRM, also referred to as MRM from multiple reaction monitoring) is the method of choice for quantitative studies.2,3 More recently, the combination of both qualitative and quantitative (QUAL/QUAN) aspects has been made possible by the emergence of data-independent acquisition (DIA) techniques and high-resolution mass spectrometers. Contrary to DDA which isolates precursor ions at unit mass resolution, DIA approaches use wider isolation windows and rely on mass analyzers of high resolving power at the fragment ion level to cope with the loss of selectivity from the first MS dimension. As a matter of fact, the application of combined QUAL/QUAN strategies using DIA methods and high-resolution MS has produced a paradigm shift in the drug metabolism and pharmacokinetics (DMPK) field.4-6 In proteomic studies, DIA is now established and provides the advantage of monitoring all detectable peptides with high sensitivity and reproducibility across large sample sets.7 In other words, DIA methods combine the high-throughput from DDA with the high reproducibility of SRM.8 Because DIA systematically parallelizes the fragmentation of all detectable ions within a wide m/z range regardless of intensity, it is particularly suitable to perform more consistent and accurate quantification by extracted ion chromatograms (XICs) at the fragment ion level.9 Indeed, Venable et al.10 observed that the selectivity afforded by XICs at the MS/MS level resulted in reduced noise with an average signal-to-noise improvement of 3.5-fold, as well as a larger dynamic range (by at least a factor of 2) compared to precursor ion XICs from MS spectra. Along the same line, Gillet et al.11 illustrated how SWATH, in combination with targeted data extraction using information from spectral libraries (generated experimentally by surveying the sample of interest by DDA or in silico from public repositories), allows consistent quantification of proteins/peptides and displays highly correlated values compared to SRM.
Despite the fact that more fragment ions provide better specificity for identification, once a peptide is confidently identified a subset of fragment ions can be used for quantification, in a similar way that proteins are quantified with a subset of their assigned peptides to favor quantification accuracy independently of the sequence coverage.12 From a quantitative perspective each peptide is typically quantified by the sum of its fragment ion XIC areas, but the number of fragment ions used for quantification varies and the “Top-N most intense” rule is often applied. Silva et al. have already described a similar concept in early applications of MSE based workflows for relative13,14 and absolute15 label-free quantification, where they proposed to quantify each protein by the average signal response of only its Top-3 peptides. Using a subset of fragment ions for quantification is particularly convenient for DIA, where the likelihood of interferences is much higher than DDA methods. Indeed, several precursor ions are isolated within the same MS window and are concurrently fragmented leading to composite MS/MS spectra. At the fragment ion level, interferences often arise due to m/z overlapping between fragment ions generated from different precursor ions which reduce quantification accuracy.11 The problem of fragment ion interferences has been addressed for decades in the bioanalysis of low molecular weight compounds using SRM-based quantitative methods. In early days, interferences due to cross-talk phenomena between SRM transitions were observed.16,17 More recently, new MS instrumentation prevents cross-talk phenomena and when an SRM transition is impaired by isobaric interferences an alternative SRM transition is selected for quantification.
Lately, several SRM data processing methods have attempted to exploit information of the fragmentation pattern to improve quantification results. Obviously, in absence of interferences, a peptide fragmentation pattern (viz. the relative fragment ion intensities or areas) is directly dependent to the sequence of that peptide and to the MS settings (e.g. collision energy, fragmentation technique) irrespective of its concentration. In this context, the algorithm named AuDIT18,19 detects fragment ions impacted by interferences by comparing the distribution of area ratios for the different fragment ions of the endogenous peptide to the analogous distribution from its isotopically-labeled version used as internal standard. The fragment ions showing discrepant distributions of area ratios are excluded from quantification. Bao et al.20 described another method that also utilizes the concept of intensity ratios but does not require isotopically-labeled internal standards. Nevertheless, several measurements of the analyte at different concentrations are necessary in order to detect the ratios that are not constant in all the concentrations and thus identify interferences. A more flexible algorithm named Anubis21 allows the user to input a reference list of expected intensity ratios. Instead of comparing distributions Anubis applies wavelet analysis to detect the peak group in the chromatogram that best correlates to the reference ratios and excludes interference-affected fragment ions in the quantification step.
Contrary to SRM methods where usually a limited number of transitions per peptide are analyzed, DIA and other targeted methods in which high resolution full MS/MS spectra are acquired for each target peptide such as Parallel Reaction Monitoring (PRM)22, generate data for all detectable fragment ions. These acquisition methods provide the possibility of computationally removing fragment ion interferences with no need for sample MS re-acquisition. Nonetheless, only a few software packages dedicated to DIA processing workflows have implemented interference correction tools, such as Spectronaut,23 DIANA,24 and more recently SWATHProphet25. Although initially developed for SRM, Anubis has been lately extended to DIA spectra within a set of tools called DIANA. In this implementation, the algorithm detects interferences by computing reference intensity ratios from the DDA spectral library. Instead of excluding interference-affected fragment ions, fragment areas are corrected based on the reference intensity ratios. Along the same lines, SWATHProphet includes two methods for correcting areas of fragment ions affected by interferences. The first method detects interferences between identified fragment ion groups corresponding to two different precursor ions contained in the spectral library. Because this method cannot detect interferences from precursor ions absent in the library, a second method that uses co-elution and peak shape scores in addition to the fragmentation pattern can pinpoint fragment ions with aberrant elution profiles and subsequently remove interferences. Scores are calculated with a set of rules imposing low score re-calculation in the absence of potentially impaired fragment ions to test for improvement, therefore this algorithm can be considered greedy and dependent on cutoff values.
These interference related algorithms applied to DIA spectra are typically integrated within software packages (i.e. DIANA, SWATHProphet and Spectronaut) that also perform several other processing steps such as peak picking, intensity normalization and scoring for identification, which limits a direct evaluation of the consequences of interference in terms of quantification only. Furthermore, the cited algorithms either attempt to correct the area of the affected fragment ion or to exclude it from quantification. Correcting the area is more error-prone, as including more fragment ions may increase the likelihood of including interferences unaccounted by the detection model. On the other hand, excluding only the impaired fragment ions produces an inconsistent number of selected fragment ions across peptides, which in turn may result in mixed statistical distributions. Alternatively, we addressed the problem by evaluating and finding a consistent number of fragment ions which are the most appropriate for quantification in order to reduce the impact of interferences and, at the same time, providing flexibility to the users regarding two aspects: 1) to use any identification/peak-integration software (where results can be exported as text format) and 2) to choose a consistent and optimal number of fragment ions for quantification according to the characteristics of the dataset. Here we describe the “non-outlier fragment ion” (NOFI) ranking algorithm which also uses information from peptide fragmentation and chromatographic features to feed an outlier detection technique ranking and prioritizing fragment ions for quantification. Instead of computing a weighted score to combine the different attributes, a four-dimensional vector is built to characterize each fragment ion thus enabling the detection of outliers in multiple dimensions. As a result, low priority ranks are assigned to fragment ions providing evidence of being affected by interferences due to co-elution and co-fragmentation and therefore effectively excluding them from quantification by using the optimal subset of high priority fragment ions. In particular, we have investigated: 1) how the number of fragment ions can impact quantification results by comparing a traditional intensity based ranking method against the proposed NOFI algorithm and 2) the sensitivity for interference detection provided by NOFI and compared to Spectronaut.
MATERIALS AND METHODS
Materials
Acetone, triethylammonium bicarbonate (TEAB) (1M, pH 8), sodium deoxycholate (Na-DOC), Tris-(2-carboxyethyl)phosphine (TCEP), methyl methanethiosulfonate (MMTS), isopropanol, trifluoroacetic acid (TFA), formic acid (FA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sequencing-grade modified trypsin was obtained from Promega (Madison, WI, USA). Acetonitrile (ACN, LC-MS grade) and water (LC-MS grade) were purchased from VWR (Radnor, PA, USA).
Proteomics sample preparation
Human dendritic cells (DC) were generated from peripheral blood mononuclear cells after isolation from buffy-coats derived from healthy donors. Buffy-coats were obtained from anonymous blood donors were provided by the Blood Transfusion Center of the Hematology Service of the University Hospital of Geneva by agreement with the service, after approval of our project by the Ethics Committee of the University Hospital of Geneva (Ref #0704). Acetone precipitated DC protein pellet was solubilized by dissolution buffer (0.5M TEAB, pH 8, and 1% Na-DOC), followed sequentially by reduction with 5 mM TCEP at 60°C for 1 hour, alkylation with 10mM MMTS at room temperature for 10 minutes and trypsin digestion at 37 °C for 16 hours with a 1:40 enzyme-to-protein ratio. Trypsin digestion was stopped by adding 2 μL of 50% formic acid and the concentration of the protein digest was adjusted to 0.2 μg/μL with 0.1% TFA in water. Before LC-MS/MS analysis, the mixture of iRT peptides (Biognosys AG, Switzerland) for DDA analysis and the HRM Calibration Kit (Biognosys AG, Switzerland) for SWATH analysis were added at a ratio of 1:10 v/v.
HPLC-MS/MS analysis
Protein tryptic digest was analyzed by reverse-phase HPLC-MS/MS using a NanoLC-Ultra 2D plus system (Eksigent, Dublin, CA, USA) coupled to a TripleTOF 5600 mass spectrometer (SCIEX, Concord, ON, Canada). 1μg of such digest was first desalted and concentrated on a C18 nano trap column (Acclaim PepMap100, 5 μm, 100 Å, 300 μm i.d. × 1 mm, Dionex, Sunnyvale, CA, USA), followed by LC separation on an nano LC column (Acclaim PepMap100, C18, 3 μm, 100 Å, 75 μm i.d. × 15 cm, Dionex, Sunnyvale, CA, USA) with mobile phase A (0.1% FA in water) and B (0.1% FA in ACN) using the following elution gradient setup: 0-2 min, 2% B; 2-5 min, 2-5% B; 5-65 min, 5-35% B; 65-72 min, 35-60% B; 72-73 min, 60-90% B; 73-80 min, 90% B; 80-82 min, 90-2% B; 82-85 min, 2% B at a flow rate of 300 nL/min. The mass spectrometer was operated in the positive ionization mode. For DDA, an MS survey scan with 100 ms accumulation time and 10 subsequent MS/MS experiments were performed with total cycle time of 1150 ms. The collision energy (CE) was automatically calculated by the “rolling CE” function of Analyst TF (v1.5.1, SCIEX) based on m/z and charge state of the precursor ion with a spread of 20. For SWATH acquisition, a set of 36 sequential Q1 variable isolation windows was used to cover the precursor m/z range of 350-1250 Da (Supporting Information - Table S1). The accumulation time for each SWATH experiment was 69 ms for a total cycle time of 2.5 sec. The CE of each Q1 window was determined by Analyst TF according to the calculation for a charge 2+ ion at the center of the window with a spread of 20.
Data processing
Targeted data extraction of SWATH files was performed by Skyline software (v2.5, Seattle, WA).26 SWATH raw files and assay built from DDA spectral library of the SWATH Gold Standard dataset (SGS)8 were downloaded from the PeptideAtlas repository (accession number PASS00289).
For the DC dataset, the MS raw files acquired by DDA (five technical replicates concatenated) were searched by ProteinPilot™ software (v4.2, SCIEX) using the Paragon algorithm with Thorough ID search effort and False Discovery Rate (FDR) analysis. The following sample parameters were used: trypsin digestion and cysteine alkylation with MMTS. Data files were searched against a reconstructed database (a combination of UniProt_Human and common laboratory contaminants, mostly from cRAP and MaxQuant databases, updated on 04-12-2014). ProteinPilot search result (.group file) were imported into Skyline to generate the spectral library using a cutoff of 5% local FDR. The same database used for ProteinPilot search was set as background proteome with the maximum missed cleavage of 1 for the tryptic digestion. The fragment ions (charge: +1 and +2, types: y- and b-ions, m/z range: 300-1500) from peptides with charge state from +2 to +4 were selected to perform the subsequent targeted data analysis.
For both datasets, XICs of the 10 most intense library fragment ions from each peptide that were possible to export from Skyline were extracted using MS/MS filtering function in Skyline with retention time (RT) window of 5 minutes centered in the predicted retention time (based on the iRT value calculated from the iRT peptides). The XIC group from each peptide was manually checked to ensure that Skyline peak picking was consistent across technical replicates. FDR analysis of XICs was performed by the mProphet algorithm included in Skyline using the second best peak option and peptides identified at 1% FDR (corresponding to Qvalue 0.01) in at least one technical replicate were kept.
Skyline reports with information at the fragment ion level including XIC intensity and the corresponding library intensity were exported in CSV format. R software (v3.1.1 x64) was used to perform the different calculations described hereafter. Visualization and comparison of results were done using the ggplot2 R package (v1.0.0).
Non-outlier fragment ion ranking algorithm
By means of Skyline, the fragment ion XICs were extracted from the SWATH spectra using as seeds the 10 most intense fragment ions per peptide contained in the DDA spectral library. Based on these XICs, four different parameters related to peptide chromatographic or fragmentation properties were defined in the non-outlier fragment ion ranking algorithm (Figure 1). Each peptide was analyzed individually considering its 10 most intense library fragment ions to compute the corresponding fragment ion scores for the following attributes: 1) Retention Time deviation (RTd): the difference between the retention time of each fragment ion and the mean retention time divided by the standard deviation within each technical replicate; 2) Full Width at Half Maximum deviation (FWHMd): the difference between the FWHM of the LC peak of each fragment ion and the mean FWHM divided by the standard deviation within each technical replicate; 3) Intensity Ratio deviation (IRd): the difference between the intensity ratio of each fragment ion and the median intensity ratio divided by the standard deviation within each technical replicate. It is important to note that instead of using intensity ratios between different fragment ions within the SWATH or the DDA spectra independently, ratios in the form SWATHj / DDAj were considered for each fragment ion. The intensity in the SWATH data is the height of the LC peak or XIC at the apex; 4) Intensity Ratio reproducibility (IRrep): for a given fragment ion, the difference between the intensity ratio and the mean intensity ratio across technical replicates divided by the standard deviation. As described for IRd, ratios in the form SWATHj / DDAj were considered for each fragment ion. However, the IRrep was computed across all technical replicates for each fragment ion independently, in contrast the IRd was computed using all the fragment ions of a peptide and within each technical replicate.
Figure 1.
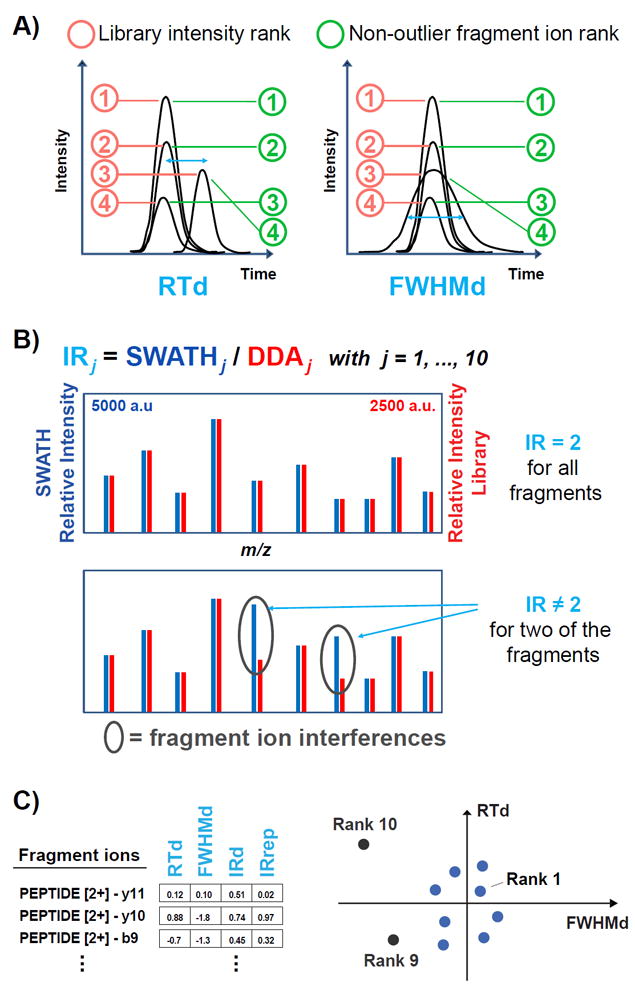
Illustration of the attributes considered in the non-outlier fragment ion (NOFI) ranking algorithm and visualization of the NOFI concept. A) The attributes related to chromatographic properties of the peptide are the retention time deviation (RTd) and the full width at half maximum deviation of the LC peak (FWHMd), as explained in Methods. The library intensity rank corresponds to the red circles, assuming that the SWATH intensities have the same fragment ion order as the DDA library intensities. The NOFI rank corresponds to the green circles, in which the fragment ion with deviated attribute values is pushed down to lower priority ranks. B) The attributes related to the fragmentation pattern of the peptide (i.e. IRd and IRrep) are computed for each fragment ion from the intensity ratio (IRj) between the SWATH and the DDA library spectra. Top panel: in the absence of interferences the relative intensities show the same pattern and the ratio of the absolute intensities is ideally constant (the absolute intensities in the SWATH spectrum have twice the values of the library spectrum, e.g. 5000 a.u. in SWATH compared to 2500 a.u. in DDA for the most intense fragment ion). Bottom panel: in the presence of interferences (grey ellipses), the intensity ratios of interference-affected fragment ions are deviated from the remaining fragment ions of the peptide. C) Left panel: vectors combining the four attributes represent the fragment ions of a peptide. Right panel: considering a projection on the first two dimensions (RTd and FWHMd attributes), high priority ranks are assigned to the fragment ions that are less deviated (closer to the center) and the deviated fragment ions are assigned lower priority ranks.
Then, these attributes were used in a vector representing each fragment ion in the four corresponding dimensions. The PCOut algorithm27 implemented in the R package mvoutlier (v.2.0.6) was used. This algorithm relies on simple properties of principal components to identify outliers. Based on the robustly sphered data, semi-robust principal components are computed for determining distances of each observation to the center and a final non-outlier weight inversely proportional to the distance. The PCOut algorithm using its default parameters was used to analyze each peptide independently. The final non-outlier weights returned by PCOut were used to generate the new “non-outlier fragment ion (NOFI)” rank taking the average weight of the technical replicates for each fragment ion. Since small values indicate potential multivariate outliers, fragment ions were sorted by decreasing weight and ranked.
The main aim of the NOFI approach consists in finding the best Top-N fragment ions per peptide which are the most suitable for quantification. The outline is as follows: 1) the input contains the list of SWATH fragment ion XICs from the identified and quantified peptides by software tools such as Skyline and OpenSWATH (typically using as seeds the 10 most intense fragment ions per peptide from a DDA spectral library); 2) the first step in NOFI is the computation of the 4 attributes (RTd, FWHMd, IRd and IRrep) used to represent each fragment ion as a vector; 3) multivariate outlier detection techniques are used to rank all the fragment ions from each peptide and to assign low priority NOFI ranks to fragment ions showing evidence of being affected by interferences; 4) several figures are generated to visualize the effect of the Top-N fragment ions over different indicators (coefficient of variation of the total area, cosine similarity, intensity and score distribution of the attributes; see the corresponding descriptions in the following sections) with N ranging from 1 to the maximum number of fragment ions per peptide; 5) finally, the user can choose the number of top fragment ions per peptide, thereby utilizing the optimal subset of high priority Top-N NOFIs for quantification while excluding the impaired fragment ions. In this way the rejection of outliers is performed based on the NOFI rank commonly set for all peptides.
The R script corresponding to the implementation of the NOFI ranking algorithm is available at http://nofiranking.sourceforge.net.
Performance assessment for quantification and interference detection
To assess the performance of our proposed “non-outlier fragment ion (NOFI)” ranking algorithm, two different datasets were used: the SWATH Gold Standard (SGS) dataset8 and the DC dataset. The original SGS dataset consists of synthesized stable isotope-labeled standard peptides added at known concentrations in three different backgrounds of different complexity (water and tryptic digests from yeast cell protein extract or from human HeLa cell lysate). From the complete SGS dataset, only peptides that have at least 10 fragment ions in the library were retained. Also, only the three experimental replicates of the five most concentrated dilution steps (viz. from 30.0 to 1.875 fmol/mL with a two-fold dilution between consecutive levels) were considered. After Skyline data processing, 182 peptides common to the three backgrounds were used to evaluate the quantification performance of the NOFI ranking algorithm. For that, the summed area of the Top-N fragment ions for each peptide at each dilution step was computed and then normalized by the most concentrated sample (viz. 30 fmol/mL) followed by a log2 transformation. Then several performance indicators were computed. The calibration curve slope and intercept were obtained from linear regression performed in log-log scale. The error calculated as the difference of the normalized summed area to its theoretical value from the 1:1 dilution curve in log-log scale. The difference was preferred over the distance or squared distance in order to visualize the distribution of both positive and negative values.
In addition, the following two indicators were computed for both the SGS and the DC datasets: 1) The coefficient of variation (CV) of the normalized summed areas across the experimental replicates; 2) The cosine similarity or cosine of the angle calculated between the SWATH spectrum and the DDA library spectrum considering the intensity vectors (square root transformation) from the Top-N fragment ions. This similarity is generally known as Dot Product because of the equivalence when the intensity vectors are normalized to have unit length.
The primary DC dataset consists of 2284 peptides with at least 10 library fragment ions common to the five experimental replicates. This initial dataset was manually checked to label interfered fragment ions and generate the annotated DC dataset according to the following criteria: 1) Retention time shift at the apex larger than half LC peak width (ca. 15 s) from the remaining members of the peak group; 2) Broad LC peak, the full width of the peak at half its maximum (FWHM) is approximately 50% larger than the remaining members of the peak group; 3) Neighbor peak, potential interference from another peak having the same m/z value and RT distance at the apex less than 1 minute; 4) Difference in the intensity rank larger than 5 positions, from a rank ≥ 8 in the library to a rank ≤ 2 in the SWATH data.
In total, 806 fragment ions from the 2284 peptides were considered as interferences by manual inspection. Prior to manual correction and annotation the whole primary DC dataset was analyzed by the NOFI ranking algorithm to compute the ranks of the 10 fragment ions from each peptide as well as by the Spectronaut™ software (v7.0, Biognosys AG).
RESULTS AND DISCUSSION
In the non-outlier fragment ion (NOFI) ranking algorithm, peptide chromatographic and fragmentation properties are encoded in four-dimensional vectors to represent fragment ions as opposed to providing values to compute a weighted score combining four attributes. The vectorial representation is used to detect outliers in multiple dimensions within the group of fragment ions from each peptide (applying the mvoutlier R package) thereby avoiding the need for optimization of individual weights when combining attributes in a final score. Besides the computational efficiency, this approach provides another advantage over traditional methods in terms of accuracy, since outliers spotted in a multidimensional space could be missed when looking at each dimension independently.
As shown in Figure 1A, two of the four dimensions, i.e. the retention time deviation (RTd) and the full width at half maximum deviation (FWHMd), are based on chromatographic properties favoring the discovery of fragment ions that have distorted elution profiles in comparison to the remaining ions of the XIC group of the peptide. The other two vectorial dimensions, i.e. the intensity ratio deviation (IRd) and the intensity ratio repeatability (IRrep), reflect information regarding the fragmentation pattern (Figure 1B). Ratios in the form SWATHj / DDAj for each fragment ion as opposed to intensity ratios across different fragment ions within the same MS/MS spectrum, are computed, considering that in absence of interferences SWATHj / DDAj ratios should be ideally constant within each peptide as illustrated in Figure 1B. The attributes related to the fragmentation pattern allow the detection of interferences in case of co-fragmented peptides with high degree of co-elution, viz. when the elution profile is not distorted (RT and FWHM are not affected) but the intensity of a single detected peak results from the contribution of two or more fragment ions with overlapping m/z values. This method has the additional advantage of being more robust against possible collision energy variations used to acquire either the DDA or the SWATH spectra.
For three of the considered attributes, i.e. RTd, FWHMd and IRrep, the deviation of each fragment ion is calculated to the mean of the group and divided by the standard deviation, similarly to the determination of Z-scores. However for IRd, the presence of interferences impairing the fragment ion intensity can dramatically change the intensity ratio. Therefore the deviation is calculated from the median of the intensity ratios assuming that only few of the fragment ions are affected by interferences and that the ratios of the affected fragment ions are sufficiently deviated from the median to be spotted as outliers. It is important to mention that in some cases the intensity ratio can also be negatively deviated (i.e. the SWATH intensity is lower than the DDA intensity and the corresponding IR is lower than the median IR of the peptide), for example in the case of low intensity fragment ions, which typically are noisy and have more variability.
Performance assessment for quantification
To assess NOFI and in particular to investigate how interferences can impact quantification performance, a subset of the SWATH Gold Standard (SGS) dataset8 was used. The subset consisted of 182 peptides, which were spiked in and identified from three sample mixtures with increasing complexity (i.e. water, yeast cell lysate and HeLa cell lysate) at five concentrations and analyzed in triplicates. The performance of the commonly used Top-N most intense fragment ions selection method was compared against NOFI. In either case, N ranged from 1 to 10 and indicated the number of fragment ions that were added together to build the calibration curve and to perform linear regression analysis for each peptide.
Figure 2 shows results pertaining to the first technical replicate (the complete set of technical replicates is shown in Supporting Information - Figure S1) from the two different ion ranking approaches for two representative peptides spiked in water vs. in human HeLa cell lysate digest using either the 3 or the 10 most intense fragment ions selected from the library MS/MS spectrum. The peptide total area (viz. the summed areas of the selected N fragment ions) was plotted vs. the dilution factor on a scatterplot. Since the five consecutive concentrations were derived from a series of 2-fold dilution steps, a calibration curve similar to the theoretical 1:1 dilution curve with a slope of 1 and an intercept of 0 is expected when data are plotted in log2-log2 scale (assuming a linear relationship between peak area and concentration without interferences impairing the fragment ion XICs). When considering only the Top-3 ions in the context of the water background the two scoring methods are equivalent to each other for peptide VDPGQVISVR (Figure 2A). However, the NOFI algorithm significantly outperformed the library ranking method when peptides were spiked at diluted concentrations (ca. 1.875 to 7.5 fmol/mL) in the complex background where the presence of interference is expected (viz. human HeLa cell lysate digest – Figure 2C). Additionally, the interference effect was not apparently true for all peptides, indicating that in some cases the Top-3 most intense library fragment ions are unaffected by interferences even with complex backgrounds. For example, in the case of peptide LFIGGLNTETNEK (Figure 2, B and D), the two approaches gave similar results regardless of the background condition.
Figure 2.
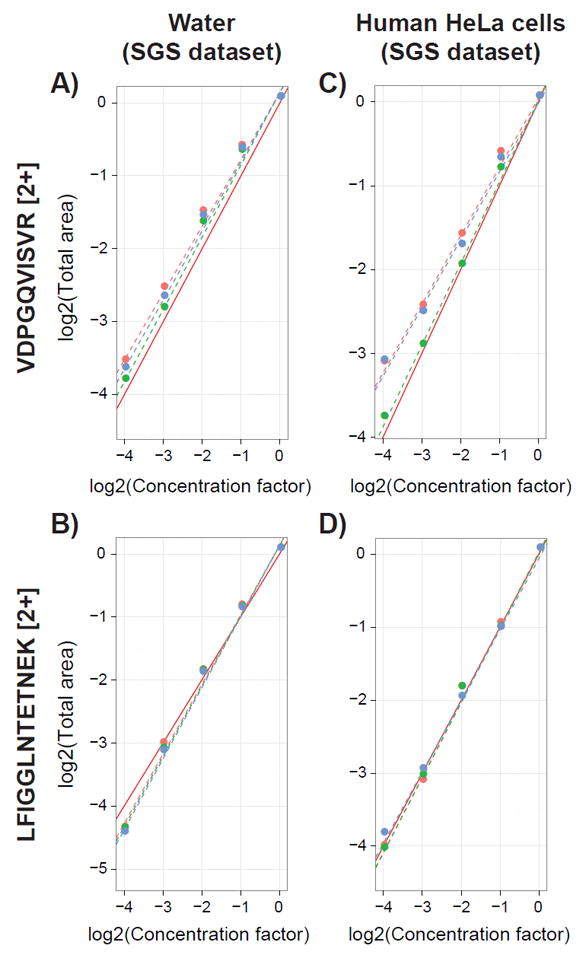
Calibration curves for selected examples illustrating the background effect and impact of interferences for quantification of peptides from the SGS dataset. Top panels (A and C) show peptide VDPGQVISVR [2+] and bottom panels (B and D) peptide LFIGGLNTETNEK [2+] spiked respectively in water (left panels A and B) or in human HeLa cell lysate digests (right panels C and D). Linear regressions were computed using the Top-3 most intense fragment ions obtained from the library (red dashed line), from the NOFI ranking algorithm (green dashed line), as well as for all ten-fragment ions from the library (blue dashed line). Dots show the corresponding total areas computed as the sum of the fragment areas normalized by the most concentrated sample. The solid red line shows the theoretical 1:1 dilution curve.
Considering the divergent behavior observed for the two peptides initially investigated, all 182 peptides from the SGS dataset were analyzed with N ranging from 1 to 10 to compare the two ranking methods for the three different background complexities. The resulting distributions of calibration curve slopes are shown as box plots in Figure 3 A-C (intercepts are shown in Supporting Information - Figure S2), whereas Figure 3 D-F illustrates the distribution of the error corresponding to the difference of the observed experimental value (viz. the summed area of the Top-N fragments) from the theoretical value obtained from the 1:1 dilution curve.
Figure 3.
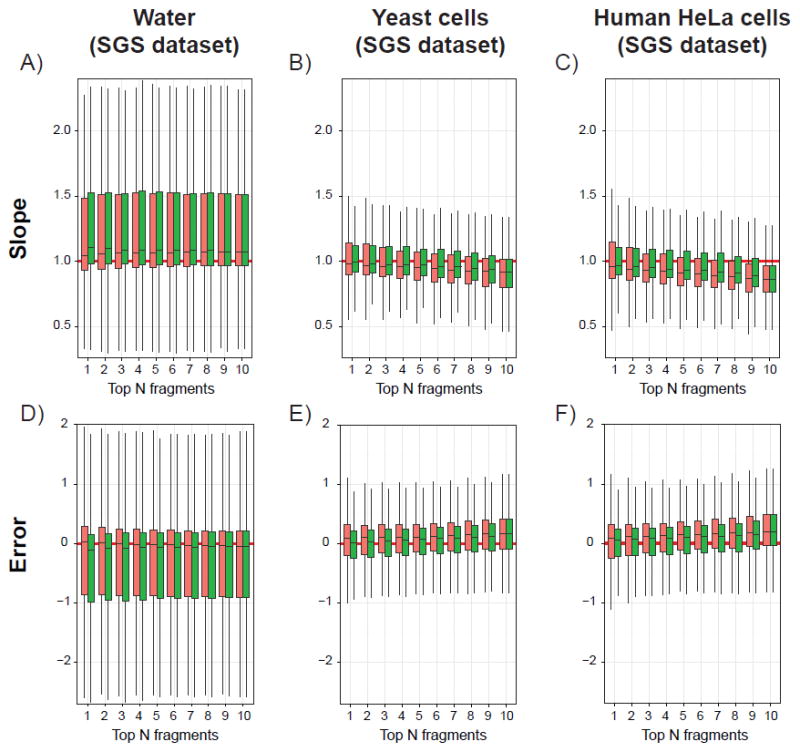
Box-plots illustrating the distributions of slopes (top panels A-C) and errors on the summed areas (bottom panels D-F) calculated with the library intensity (red) or the NOFI ranking methods (green). Results are obtained from 182 peptides selected from the SGS dataset spiked in water (A and D), yeast cells (B and E) or human HeLa cells (C and F) lysate digests. Solid horizontal red lines indicate expected values according to the theoretical 1:1 dilution curve.
The distributions of slopes and errors are clearly wider and more skewed when peptides are spiked in a background without matrix (i.e. water, Figure 2A) than in tryptic digests. In addition, the medians of the slope distributions are higher than the expected theoretical value (i.e. slope of 1) and correlate well with the negative error values (Figure 2D), which corroborates the hypothesis from Röst et al. that the spiked-in peptides are prone to surface absorption or solubility issues during sample preparation preventing an accurate quantification of the diluted samples.8 When peptides are spiked in more complex backgrounds (i.e. yeast and human cell digests), the performance of either ranking method decreases as the number of fragment ions increases, suggesting that adding more fragment ions potentially increases the quantification error due to increased likelihood of additional interfering ions.
Although the difference is minimal, a trend is observed in complex backgrounds suggesting that, when summing areas of the Top-N fragment ions, the NOFI algorithm is more effective in prioritizing interference-free fragment ions than the library intensity method, where interference issue is not considered. This is shown by the slope and intercept distributions where the median values are closer to the theoretical values of 1 and 0 respectively. In addition, when averaging the absolute error observed for each peptide over all concentration levels, the NOFI ranking algorithm invariably produced an increased fraction of quantifiable peptide identifications within a given error tolerance, as compared to the library intensity method. An example is given when summing the Top-3 fragment ions for the different backgrounds (Supporting Information - Figure S3 A-C). In cell lysate digests (Supporting Information - Figure S3 bottom tables) the NOFI ranking algorithm allows on average to accurately quantify 11-25% more peptides than the library intensity method, depending on the number of fragment ions considered for the total area.
In addition to the accuracy, the precision (i.e. reproducibility) of a quantitative assay needs to be assessed across the different experimental replicates. Therefore, coefficients of variation (CV) were calculated on the basis of the summed areas for each peptide with both ranking methods, for both the SGS dataset described above (182 peptides, three replicates) and the DC dataset, which is an experimental dataset obtained by analyzing a lysate digest of primary human dendritic cells (2284 peptides, five replicates). As observed in Figure 4 A and B, the CV distributions tend to become narrower regardless of the ranking method with more selected fragment ions. Their median values reaches a plateau when N ≥ 3-4 for the SGS dataset and when N ≥ 5-6 for the DC dataset. Interestingly the median values obtained when using the NOFI ranking algorithm are relatively higher than those obtained using the library intensity rank. This increased variability could be explained by the fact that with NOFI the top ranked fragment ions (viz. with the highest priority for quantification) are generally less intense than those selected with the library intensity method (Supporting Information - Figure S4). In practice, lower CVs are preferred for quantification. However, quantifying using a peak area with low CV but impaired by interferences (i.e. the peak area is arising from the contribution of the areas of several fragment ions from different peptides) produces incorrect quantification results, which are particularly detrimental for low-intensity peptides. Thus, our results suggest that despite the fact that high-intensity fragment ions ranked first by the library intensity method are less variable (presumably due to higher signal-to-noise ratio and better peak integration), they are not necessarily the most suitable for quantification since they might be impaired by DIA interferences. Therefore, a balance between sensitivity and reproducibility is a key factor for quantification in DIA, which in our hands appears to be achieved by summing enough top-ranked fragment ion areas to obtain a lower CV value. It should also be noted that, the Top-N fragment ion areas of the NOFI rank could reach comparable CV values to the library intensity rank while providing more accurate quantification results.
Figure 4.
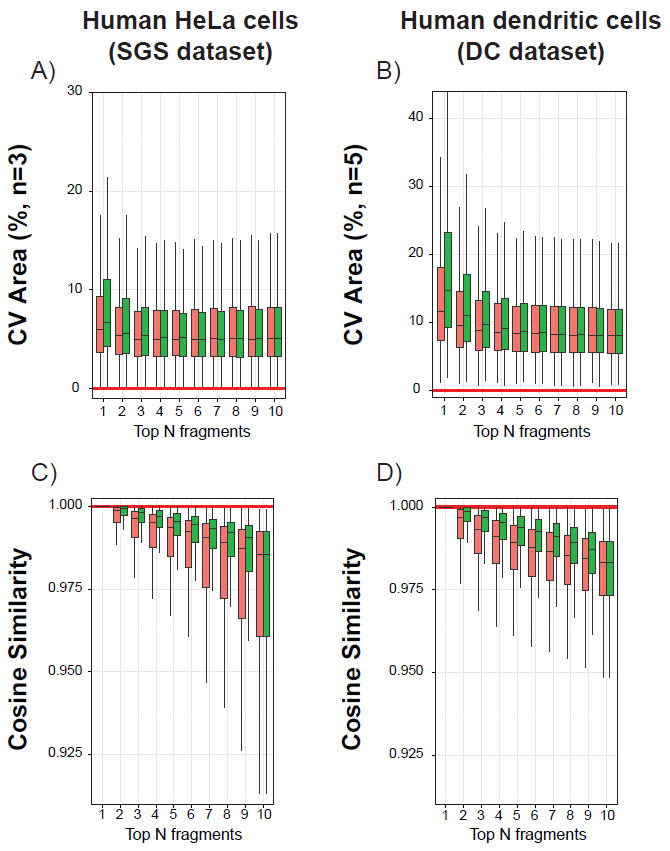
Box-plots illustrating the distributions of the coefficient of variation (CV) of the normalized summed areas among experimental replicates (top panels A and B), as well as the distributions of cosine similarity between SWATH and DDA library intensity vectors (bottom panels C and D). Results were calculated with the library intensity (red) or the NOFI ranking method (green) from the SGS dataset 182 selected peptides spiked in human HeLa cell lysate digests (A and C) and from the 2284 DC dataset peptides (B and D). Solid horizontal red lines indicate best case values. Similar trends were observed for the SGS dataset results corresponding to water and yeast cells (data not shown).
Another way to compare the two ranking methods is to compute the cosine similarity between vectors containing the experimental SWATH spectrum intensities from the Top-N fragment ions and intensities of the same fragments from the DDA library MS/MS spectrum (Figure 4). A value of 1 indicates a perfect correlation between the two vectors, which reflects an absence of interference and a perfect linear relationship between the respective fragment intensities of the experimental and library spectra. It is clear from Figure 4 C and D that the proposed NOFI ranking algorithm outperforms the library intensity ranking method. This was expected due to the IRd attribute used in the NOFI ranking algorithm. The median values are indeed always closer to 1 and the cosine similarity distributions have a much-reduced spread, reflecting the higher similarity between the SWATH and the DDA fragment ions when considering the Top-N NOFI rank.
Performance assessment for interference detection
The NOFI ranking algorithm relies on four attributes reflecting chromatographic and MS fragmentation related parameters. For each peptide four individual scores are computed for every of the 10 most intense library fragment ions and gathered into vectors. The NOFI ranking algorithm uses these vectors to detect outliers that are not suitable for quantification. When looking at each attribute, the score distribution according to the fragment rank can be plotted for each background and dataset. For three of the attributes (i.e. RTd, FWHMd and IRd), a clear trend towards a larger fraction of high scores is observed for low priority fragment ions, thus confirming the relevance of these attributes for outlier detection (Supporting Information - Figure S5). As expected for the last attribute (i.e. IRrep), the score distribution is nearly constant across the ranks due to the reproducible nature of collision-induced dissociation used in peptide fragmentation. However, the DC dataset shows higher scores compared to the SGS dataset. This difference arises from the DC dataset containing fragment ions with overall lower signal-to-noise ratios and this reduces the intensity ratio repeatability among technical replicates.
Another way to visualize the results after assigning the NOFI rank is categorizing the fragment ions to reflect the attribute that is the most affected or deviated from the expected value. This was done by finding the attribute with the maximum absolute score per fragment as illustrated in Figure 5. The fragment ions ranked with lower priority (viz. rank 10) show higher values (dark blues) and the intensity ratio deviation (IRd) contains a large fraction of fragment ions. In other words, the IRd tends to be the most influential attribute for detection of fragment ion outliers, followed by the RTd and FWHMd attributes. On the contrary, fragment ions at high priority ranks (viz. ranks 1-4) have mostly low attribute values (light blues) and the largest fraction of fragment ions fall in the IRrep category, which is the attribute with overall less deviation. In addition, the IRrep attribute allows the consideration of the fragment ion intensity in an implicit manner. As discussed previously, MS signal intensities are usually proportional to the reproducibility (Coefficient of variation in Figure 4 as well as Intensity in Supporting Information - Figure S4). Therefore in the absence of interferences when the remaining attributes are not affected, the fragment ions are ranked according to the repeatability which is modeled by the IRrep attribute.
Figure 5.
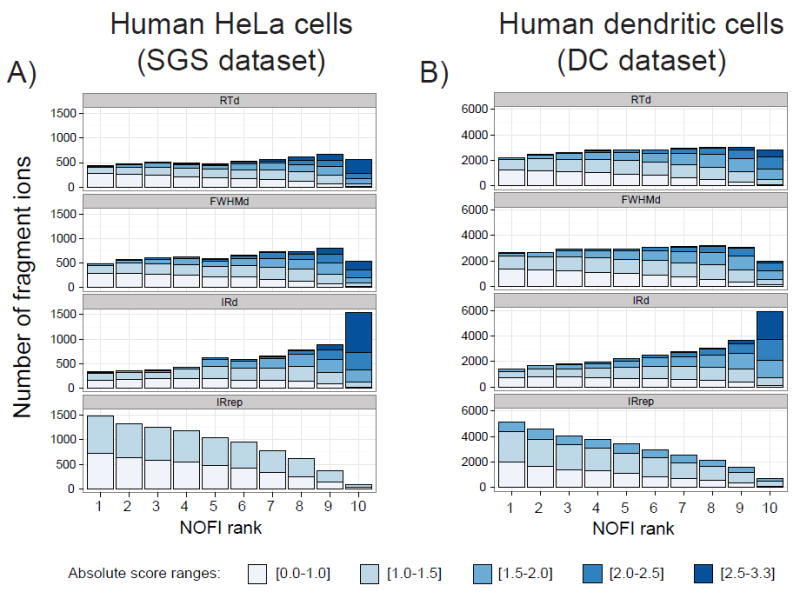
Bar charts illustrating the impact of each attribute used in the NOFI ranking algorithm when selecting only the attribute with the highest score for each fragment ion. The different datasets are: 182 SGS peptides spiked in human HeLa cell lysate digests (A) and 2284 DC native peptides from human dendritic cell lysate digests (B). The color intensity scale corresponds to five absolute score ranges, darker blue being the range with a higher likelihood of outlier detection. Similar trends were observed for the SGS dataset results corresponding to water and yeast cells (data not shown).
The outlier detection performance of NOFI was evaluated under experimentally relevant conditions by using the DC dataset which contains 2284 peptides with at least 10 fragment ions per peptide. Despite the fact that the SWATH acquisition method used was enhanced to minimize interference by employing precursor isolation windows of optimized variable widths,28 806 fragment ions identified from the DC dataset were judged as being affected by interferences during a manual annotation process. When processing the full DC dataset of 2284 peptides, these 806 ions were ranked by using either the library intensity ranking method or the NOFI ranking algorithm (Figure 6). As observed, the likelihood for a fragment ion to be affected by interference was almost equal for all fragment ions regardless of intensity (viz. library intensity rank from 1 to 10). However, in the case of NOFI, 85% (686 ions) of the same fragment ions were assigned low priority (i.e. ranks higher or equal to 6) and 50% of them (404 ions) were ranked to the lowest possible priority, effectively excluding them from quantification. Additionally, it is worth mentioning that only 2% and 3% of the ions annotated as affected by interferences were found in the first and second ranked-categories respectively, which is a significant reduction with respect to the 9% and 8% that were found in the same positions when using the library intensity approach. Furthermore, when investigating the number of impaired fragment ions per peptide based on the 806 annotated interferences, 486 peptides corresponded to a single annotated interference, 112 peptides contained 2 annotated interferences, 25 peptides contained 3 annotated interferences, 4 peptides contained 4 annotated interferences and the maximum number of annotated interferences was 5 for only one of the peptides. In the latter case, all 5 annotated interferences were correctly assigned to the low priority positions (NOFI rank > 5). This suggests that 5 non-outlier fragment ions (equivalent to 50%) per peptide are sufficient for NOFI to successfully assign low priority positions to the remaining 5 fragment ions affected by interferences.
Figure 6.
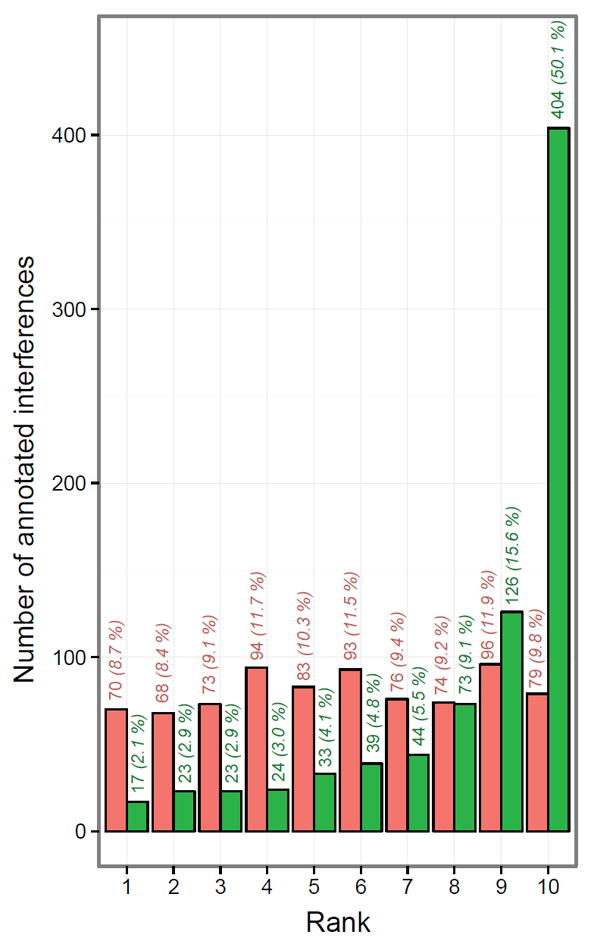
Comparison of the rank distribution of the 806 fragment ions manually annotated as being affected by interferences in the DC dataset showing the performance of the NOFI ranking algorithm to assign lower priority ranks. Ranks are attributed using either the library intensity ranking method (red bars) or the NOFI ranking algorithm (green bars). Clearly, NOFI outperforms the traditional method in assigning a lower priority rank to a higher frequency of interference-impacted ions. Indicated above each bar are the absolute and relative numbers of ions assigned to each category by the utilized ranking algorithm.
In order to further evaluate the interference detection performance of NOFI, the full DC dataset of 2284 peptides was also processed by the Spectronaut software. It is important to mention that the approaches implemented in other software tools such as DIANA and SWATHProphet are conceptually different from the NOFI approach because these tools attempt to correct the fragment ion area instead of excluding it from quantification. In contrast, Spectronaut contains an algorithm for interference detection and an option to exclude the impaired fragment ion or not from all replicates in the quantification step. However, Spectronaut requires the use of a commercial set of calibrating peptides (i.e. HRM Calibration Kit), thus the SGS dataset could not be processed since this kit was not used by the authors when the dataset was generated.
Therefore, the NOFI algorithm was compared against Spectronaut in terms of sensitivity by using the 806 fragment ions from the DC dataset that had been manually recognized as being affected by interferences. The complete DC dataset of 2284 peptides was processed by both algorithms independently and then the results were compared. Spectronaut was able to detect 613 out of the 806 annotated interferences. In order to carry out the comparison, the Top-N fragment ions were selected for NOFI, and the annotated interferences within the Top-N NOFIs were considered false negatives whereas the remaining ones were considered true positives, since these impaired fragment ions are successfully excluded from quantification. Table 1 illustrates that the NOFI algorithm outperformed Spectronaut at detecting interferences when using the Top 3-6 NOFIs, resulting in the detection of annotated interferences with sensitivity values between 0.92 and 0.80 against 0.76 for Spectronaut. However, when investigating the Spectronaut detection rate and each of the NOFI ranks for the annotated interferences (Supporting Information - Figure S6) it can be seen that Spectronaut was able to detect some of the annotated interferences that NOFI failed to rank at low priority positions (viz. the green fraction of the Top 1-6 bars where NOFI assigned high priority ranks), possibly due to the low signal-to-noise ratios that were observed in most of the cases. At the same time, NOFI successfully assigned low priority ranks to some of the annotated interferences that Spectronaut failed to detect (viz. the red fraction of the bars corresponding to the 7-10 NOFIs that were assigned low priority ranks), presumably due to the consideration of the IRd attribute, which is the most influential one for outlier detection, as discussed previously.
Table 1.
NOFI compared to Spectronaut in terms of sensitivity for interference detection. The comparison is based on the 806 fragment ions annotated as affected by interferences in the DC dataset. Once the Top-N number of fragment ions is selected for NOFI, the annotated interferences within the Top-N NOFIs are considered false negatives (FN) and the remaining ones are considered true positives (TP), since these interferences are effectively excluded for quantification. The “Top-N fragments” column is not applicable for Spectronaut.
| Algorithm | Top-N fragments | True positives | False negatives | Sensitivity |
|---|---|---|---|---|
| NOFI | 3 | 743 | 63 | 0.92 |
| 4 | 719 | 87 | 0.89 | |
| 5 | 686 | 120 | 0.85 | |
| 6 | 647 | 159 | 0.80 | |
| Spectronaut | NA | 613 | 193 | 0.76 |
Sensitivity = TP / (TP + FN).
Additionally, among the few interference-containing annotated fragment ions that were not considered as outliers by the NOFI algorithm and ranked in the first 3 positions, the presence of a closely eluting peak (hereafter called “neighboring peak”) or of a shoulder peak was often observed in the fragment ion XIC trace. Under these circumstances, accurate peak integration can be challenging. Therefore to avoid quantification inaccuracies another fragment ion without interference is generally preferred. Both of these situations are illustrated in Figure S7 (Supporting Information) for LSLEGDHSTPPSAYGSVK [2+]. The y11 fragment ion with a shoulder peak is detected as an outlier by the NOFI ranking algorithm and is given the lowest priority rank. In contrast, the b9 fragment with a neighboring peak is ranked first because this case is beyond the detection power of the four attributes used. Indeed when looking more closely at the chromatographic attribute scores, the RTd is low and the FWHMd is not sufficient to spot it as an outlier since the peak width deviation (compared to the other fragments) is calculated at 50% of its height. One solution to detect fragment ions with closely eluting peaks would be to add one or several attributes to better describe the chromatographic peak shape, such as the asymmetry first proposed by Foley and Dorsey29 or the width measured at the baseline. Moreover, as mentioned above significantly low signal-to-noise values were observed for the annotated interferences that were detected by Spectronaut and failed by NOFI, mostly due to a lack of an adequate signal-to-noise measure of the LC peak in Skyline reports. In fact, the signal-to-noise is an important measure of the quality and provides information often uncaptured by the LC peak width. For instance, setting the integration boundaries is not a trivial task and software tools usually set a single integration boundary common to the entire group of fragment ion XICs of the peptide (as shown in Supporting Information - Figure S7). In such cases where the attributes are determined at the integration boundaries, the peak width at any height is relatively unaffected but the signal-to-noise is able to discriminate the impaired fragment ion. Indeed, depending on the peak detection software and its available metrics (e.g. peak asymmetry, baseline determination, signal-to-noise, etc.), additional attributes can be implemented into each vector representing the fragment ions to expand the outlier detection power of the algorithm and reduce the priority of impacted fragment ions for quantification.
CONCLUSIONS
When performing label-free peptide quantification using data-independent acquisition LC-MS/MS, the number of fragment ions used to express the total area representing the peptide amount is often difficult to select. Most of the time a selection of the Top-N most intense ions (typically from 3 up to 10) based on a spectral library is made. Our results suggest that the use of a subset of fragment ions instead of the entire identification set provides higher accuracy for DIA quantification results. More importantly, this subset does not necessarily include the most intense fragment ions since the likelihood of resulting interfering ions from precursor ions isolated within the same precursor window increases during DIA. In fact when selecting the Top-N most intense fragment ions as by the library intensity ranking method, some of the corresponding fragment ion traces from DIA spectra may be impaired by interferences and therefore are not the most appropriate for accurate and precise quantification results. In contrast, our algorithm based on outlier detection ranks these fragment ions with lower priority regardless of their intensity. Consequently, the use of more than 3 fragment ions for identification paired with the NOFI ranking allows the selection of an optimal subset in the subsequent quantification step. Additional morphological chromatographic attributes, such as asymmetry, baseline peak width or signal-to-noise, could be incorporated to increase the dimensionality of the vector space. Such improvements are expected to reduce potential pitfalls arising from the assumption that the peak group is correctly picked during integration. For example, in the current implementation the selection of a wrong peak, as in the case of closely eluting peaks that are not baseline resolved from the fragment ion XIC, could conceivably decrease outlier detection performance.
Despite these limitations, we have shown that outlier detection techniques applied to a multivariate space based on chromatographic and MS fragmentation related attributes, can lead to an optimal subset of fragment ions and yield more reliable quantification results, which in turn are essential for producing biologically significant MS results of complex proteomes.
Supplementary Material
Acknowledgments
The authors would like to thanks Dario Bottinelli (University of Geneva) for providing the human dendritic cell samples, as well as Markus Mueller (SIB) for fruitful discussions regarding features of the algorithm.
This work was supported by the Swiss National Science Foundation Sinergia grant CRSII3_136282 7, the SystemsX.ch IPhD grant 2014/241 and by National Institutes of Health (USA) grants RO1111809 and DP1DA034990.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interest.
References
- 1.Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopfgartner G, Varesio E. New approaches for quantitative analysis in biological fluids using mass spectrometric detection. TrAC Trends in Anal Chem. 2005;24:583–589. [Google Scholar]
- 3.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 4.Hopfgartner G, Tonoli D, Varesio E. High-resolution mass spectrometry for integrated qualitative and quantitative analysis of pharmaceuticals in biological matrices. Anal Bioanal Chem. 2012;402:2587–2596. doi: 10.1007/s00216-011-5641-8. [DOI] [PubMed] [Google Scholar]
- 5.Ramanathan DM. Looking beyond the SRM to high-resolution MS paradigm shift for DMPK studies. Bioanalysis. 2013;5:1141–1143. doi: 10.4155/bio.13.95. [DOI] [PubMed] [Google Scholar]
- 6.Tonoli D, Varesio E, Hopfgartner G. Quantification of acetaminophen and two of its metabolites in human plasma by ultra-high performance liquid chromatography low and high resolution tandem mass spectrometry. J Chromatogr B. 2012;904:42–50. doi: 10.1016/j.jchromb.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Hüttenhain R, Collins B, Aebersold R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert review of molecular diagnostics. 2013;13:811–825. doi: 10.1586/14737159.2013.845089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinovi SM, Schubert OT, Wolski W, Collins BC, Malmström J, Malmström L, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 9.Bilbao A, Varesio E, Luban J, Strambio-De-Castillia C, Hopfgartner G, Müller M, Lisacek F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics. 2015;15:964–980. doi: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- 10.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 11.Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cappadona S, Baker PR, Cutillas PR, Heck AJ, Breukelen B van. Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino acids. 2012;43:1087–1108. doi: 10.1007/s00726-012-1289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ, McKenna T, Nold MJ, Richardson K, Young P, et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187–2200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 14.Silva JC, Denny R, Dorschel C, Gorenstein MV, Li GZ, Richardson K, Wall D, Geromanos SJ. Simultaneous Qualitative and Quantitative Analysis of the Escherichia coli Proteome A Sweet Tale. Mol Cell Proteomics. 2006;5:589–607. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Mansoori BA, Dyer EW, Lock CM, Bateman K, Boyd RK, Thomson BA. Analytical performance of a high-pressure radio frequency-only quadrupole collision cell with an axial field applied by using conical rods. J Am Soc Mass Spectrom. 1998;9:775–788. [Google Scholar]
- 17.Morris M, Pierre T, K BR. Characterization of a high-pressure quadrupole collision cell for low-energy collision-induced dissociation. J Am Soc Mass Spectrom. 1994;5:1042–1063. doi: 10.1016/1044-0305(94)85066-6. [DOI] [PubMed] [Google Scholar]
- 18.Abbatiello SE, Mani D, Keshishian H, Carr SA. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem. 2010;56:291–305. doi: 10.1373/clinchem.2009.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mani D, Abbatiello SE, Carr SA. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinformatics. 2012;13:S9. doi: 10.1186/1471-2105-13-S16-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao Y, Waldemarson S, Zhang G, Wahlander A, Ueberheide B, Myung S, Reed B, Molloy K, Padovan J, Eriksson J, et al. Detection and correction of interference in SRM analysis. Methods. 2013;61:299–303. doi: 10.1016/j.ymeth.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teleman J, Karlsson C, Waldemarson S, Hansson K, James P, Malmström J, Levander F. Automated selected reaction monitoring software for accurate label-free protein quantification. J Proteome Res. 2012;11:3766–3773. doi: 10.1021/pr300256x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol Cell Proteomics. 2012;11:1475–1478. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernhardt OM, Selevsek N, Gillet LC, Rinner O, Picotti P, Aebersold R, Reiter L. Spectronaut: A fast and efficient algorithm for MRM-like processing of data independent acquisition (SWATH-MS) data. 60th ASMS Conference on Mass Spectrometry and Allied Topics; Vancouver, Canada. 2012. pp. 20–24. [Google Scholar]
- 24.Teleman J, Röst H, Rosenberger G, Schmitt U, Malmström L, Malmström J, Levander F. DIANA-algorithmic improvements for analysis of data-independent acquisition MS data. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu686. btu686. [DOI] [PubMed] [Google Scholar]
- 25.Keller A, Bader SL, Shteynberg D, Hood L, Moritz RL. Automated Validation of Results and Removal of Fragment Ion Interferences in Targeted Analysis of Data Independent Acquisition MS using SWATHProphet. Mol Cell Proteomics. 2015 doi: 10.1074/mcp.O114.044917. mcp–O114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filzmoser P, Maronna R, Werner M. Outlier identification in high dimensions. Computational Statistics & Data Analysis. 2008;52:1694–1711. [Google Scholar]
- 28.Zhang Y, Bilbao A, Bruderer T, Luban J, Strambio-De-Castillia C, Lisacek F, Hopfgartner G, Varesio E. The use of variable Q1 isolation windows improves selectivity in LC-SWATH-MS acquisition. J Proteome Res. 2015 doi: 10.1021/acs.jproteome.5b00543. [DOI] [PubMed] [Google Scholar]
- 29.Foley JP, Dorsey JG. Equations for calculation of chromatographic figures of merit for ideal and skewed peaks. Anal Chem. 1983;55:730–737. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.