

Abstract
Progression of chronic obstructive pulmonary disease (COPD) is linked to episodes of exacerbations caused by bacterial infections due to Streptococcus pneumoniae. Our objective was to identify during COPD, factors of susceptibility to bacterial infections among cytokine network and their role in COPD exacerbations. S. pneumoniae was used to sub-lethally challenge mice chronically exposed to air or cigarette smoke (CS) and to stimulate peripheral blood mononuclear cells (PBMC) from non-smokers, smokers and COPD patients. The immune response and the cytokine production were evaluated. Delayed clearance of the bacteria and stronger lung inflammation observed in infected CS-exposed mice were associated with an altered production of IL-17 and IL-22 by innate immune cells. This defect was related to a reduced production of IL-1β and IL-23 by antigen presenting cells. Importantly, supplementation with recombinant IL-22 restored bacterial clearance in CS-exposed mice and limited lung alteration. In contrast with non-smokers, blood NK and NKT cells from COPD patients failed to increase IL-17 and IL-22 levels in response to S. pneumoniae, in association with a defect in IL-1β and IL-23 secretion. This study identified IL-17 and IL-22 as susceptibility factors in COPD exacerbation. Therefore targeting such cytokines could represent a potent strategy to control COPD exacerbation.
Abbreviations: COPD, chronic obstructive pulmonary disease; CS, cigarette smoke; Sp, Streptococcus pneumoniae; CFU, colony forming unit; BAL, broncho-alveolar lavage; APC, antigen presenting cells; AM, alveolar macrophages; DC, dendritic cells; PBMC, peripheral blood mononuclear cells; NK, natural killer cells; NKT, natural killer T cells
Keywords: Chronic obstructive pulmonary disease, Innate immunity, Bacterial infection, IL-22
Highlights
-
•
Increased bacterial susceptibility during COPD is related to a defect in Th17 cytokines.
-
•
Cigarette smoke alters the production of immunoregulatory cytokines by lung APC.
-
•
Immunotherapy restoring the defective IL-22 response could represent an ideal therapy to prevent exacerbation in COPD patients.
The progression of chronic obstructive pulmonary disease (COPD) is linked to episodes of exacerbations mostly due to bacterial infections. It is not well understood why COPD patients are more susceptible to infections. In our experimental model of COPD as well as in COPD patients, we identified a defect in the IL-17/IL-22 response to S. pneumoniae, leading to the bacterial outgrowth. This was mainly due to the alteration of lung antigen-presenting cells by cigarette smoke. Restoring the defective IL-22 response represents a promising therapeutic approach for the treatment and/or the prevention of COPD exacerbations.
1. Introduction
Chronic obstructive pulmonary disease (COPD) remains a major cause of morbidity and mortality worldwide. This will be the third cause of deaths worldwide in 2020 according to the WHO. COPD is a lung disorder characterized by progressive and irreversible airflow limitation. Cigarette smoking is a primary risk factor for the development of COPD, although other factors, including pollution and genetic determinants, have been described. Cigarette smoke (CS) chronically triggers inflammatory processes which ultimately alter pulmonary barrier functions and reduce immune defense mechanisms, thus leading to increased susceptibility to respiratory infections (Agusti et al., 2003, Barnes and Stockley, 2005, Fletcher and Peto, 1977, Soler et al., 1999, Soler-Cataluna et al., 2005).
Such infections further alter the clinical status of COPD patients thereby indirectly causing extensive morbidity and mortality (Soler et al., 1999). Acute exacerbation of COPD patients is associated with a greater decline in lung function, enhanced oedema as well as airway and systemic inflammation (Agusti et al., 2003). Among major bacterial species causing COPD exacerbation are Streptococcus pneumoniae, non-typeable Haemophilus influenzae and Moraxella catarrhalis (Sethi and Murphy, 2008). Cigarette smoking is associated with diminished antibacterial immune responses and delayed clearance of microbial agents (Drannik et al., 2004). However, it is not well understood how these alterations are controlled during COPD and why COPD patients are more susceptible to infections (Soler-Cataluna et al., 2005). Considering the increasing prevalence of COPD, there is an urgent need to better understand mechanisms leading to exacerbation in COPD patients in order to propose novel therapeutics (Barnes and Stockley, 2005).
Among the factors orchestrating the anti-bacterial response, Th17 cytokines, including interleukin (IL)-17 and IL-22, play a major role (Eidenschenk et al., 2014, Ivanov et al., 2013). These cytokines are produced by various cells of the adaptive and innate immune system. These include conventional T lymphocytes, natural killer (NK) cells, non-conventional T cells (such as γδ T cells, NKT cells and invariant mucosal-associated T (MAIT) cells) and type 3 innate lymphoid cells (ILC3). Production of Th17 cytokines is strongly dependent on IL-1β, IL-23 and IL-6 secretion by antigen presenting cells (APC) (Doisne et al., 2011, Ivanov et al., 2014). Anti-bacterial effects of Th17 cytokines comprise the induction of antimicrobial peptides and neutrophil chemoattractants by airway epithelial cells (Aujla et al., 2008, Wolk et al., 2004). Both IL-17 and IL-22 amplify the granulopoiesis by increasing the expression of G-CSF. In addition, IL-22 plays a central role in the maintenance of the epithelium integrity by limiting cellular apoptosis and by favoring repair/regeneration processes (Sonnenberg et al., 2011).
Since Th17 cytokines play major functions in the control of bacterial, including pneumococcal, outgrowth, we hypothesized that their production upon respiratory bacterial challenge could be altered in the context of COPD. Indeed, our data indicate a default in Th17 cytokine production, especially IL-22, in response to S. pneumoniae in a mouse model of COPD induced by chronic CS exposure (Pichavant et al., 2014) and ex vivo in COPD patients. This reduced response was associated with diminished production of Th17 cytokine inducing factors by pulmonary APC. Remarkably, administration of recombinant IL-22 in CS-exposed mice just before the bacterial challenge resulted in accelerated pneumococcal clearance and lowered pulmonary inflammation. Thus, targeting Th17 cytokines might be valuable to limit COPD exacerbation due to bacterial infections.
2. Material and Methods
2.1. Mice
Six- to eight-week-old male wild-type (WT) C57BL/6 (H-2Db) mice were purchased from Janvier (Le Genest-St.-Isle, France). All animal work conformed to the guidelines of Animal Care and Use Committee from Nord Pas-de-Calais (agreement no. AF 16/20,090). Mice were exposed to CS (5 cig/day, 5 days/week) during 12 weeks as previously described in order to generate a COPD-like disease (Wolk et al., 2004), or ambient air as control. Six to ten mice were used per group and per experiment. Experiments were repeated at least 3 times.
2.2. Patients with COPD
Peripheral blood was collected in stable COPD patients (n = 12), in smokers (without COPD, n = 13)) and in non-smoker healthy controls (n = 14) (CPP 2008-A00690-55) (see Table 1). Written informed consent was received from participants prior to inclusion in the study, according ethics committee on human experimentations. COPD patients at steady state included subjects with a GOLD score between 2 and 4 and did not received oral corticosteroids.
COPD patients included subjects with a gold score between 2 and 4 and are matched for age and sex with controls. FEV1%, predicted amount as a percentage of the forced expiratory lung volume in one second; PO2, partial pressure of oxygen; BODE: Bode index combining Body mass index, airflow obstruction (VEMS), dyspnea (MRC score) and exercise (6 minutes walk test [6 MWT]). Results were expressed as mean ± SEM. ND: not determined.
Table 1.
Characteristics of COPD patients, smokers and non-smoker subjects.
| Group | COPD | Smokers | Non smokers |
|---|---|---|---|
| Nb | 12 | 14 | 14 |
| Sexes (M/F) | 11/1 | 11/3 | 10/4 |
| Age | 59.2 ± 17.1 | 43.9 ± 4.7 | 45.5 ± 5.7 |
| Smoking (pack/year) | 57 ± 5.9 | 37.6 ± 5.2 | 0 |
| FEV1% | 51.3 ± 4.1 | 94.7 ± 1.3 | 95.3 ± 3.5 |
| PO2 | 77.1 ± 6.6 | ND | ND |
| BODE | 3. 1 ± 0.5 | ND | ND |
| Body mass index | 25.6 ± 1.2 | ND | ND |
| VEMS% | 56.9 ± 4.6 | ND | ND |
| MRC score | 1.5 ± 0.31 | ND | ND |
| 6 MWT | 410 ± 34.8 | ND | ND |
| Inhaled corticosteroid | 4 | 0 | 0 |
Peripheral blood mononuclear cells (PBMC) were purified on Ficoll Paque gradient (GE healthcare). Cells (3 × 106 in 1 ml) were cultured in RPMI1640 (GIBCO, Invitrogen Corporation) supplemented with 10% FCS, 200 U/ml penicillin/streptomycin (PS) and then exposed to S. pneumoniae (Sp, MOI = 2) or to phytohemagglutinin (1 μg/ml) (PHA, Difco) as a positive control. After 90 min, antibiotics were added to stop bacteria growth and supernatants were collected 24 h later. Cell viability was not affected. Some cells were incubated with brefeldin A (10 μg/ml, Sigma) for 4 h and used for intracellular staining of cytokines.
2.3. Reagents and Antibodies
Monoclonal antibodies (mAbs) against mouse CD3 (APC-conjugated), CD5 (FITC-conjugated), NK1.1 (PerCp-Cy5.5–conjugated), TCR-β (V450-conjugated), CD25 (APC-conjugated), CD69 (Alexa700-conjugated), CD11b (V450–conjugated), Ly-6G (APC-Cy7-conjugated), CD8 (V500-conjugated), CD4 (APC-conjugated), CD103 (PE-conjugated), CD11c (APC-conjugated), CD45 (Q-dot605-conjugated), F4/80 (PerCP-Cy5.5-conjugated), Siglec F (PE-conjugated), CD64 (APC-conjugated), CD86 (PE-conjugated), CD40 (PE-conjugated), I-Ab (FITC-conjugated), IFN-γ (PE-conjugated), IL-17 (APC-conjugated), CD11c (PE-Cy7-conjugated), F4/80 (PerCP-Cy5.5-conjugated), CD11b (V450-conjugated) and CD103 (PE-conjugated) and isotype controls were purchased from Biolegend (Le Pont de Claix, France). mAbs against human CD were also used including anti-CD11c, CD14, CD19, CD20 (PE-CF594-conjugated), CD117, TCRγδ (V450-conjugated), CD4, CD3 (Alexa-700 conjugated), CD8, CD127 (V500 conjugated), CD196, CD3 (BV605 conjugated), CD25, CD86 (APC-conjugated), CD56, Vα7.2 (PerCP-Cy5.5 conjugated), TCR Vα24Jα18, CD161 (PE-Cy7 conjugated) and CD45 (APC-H7 conjugated) (BD Biosciences, Biolegend and Myltenyi Biotech) as well as the Alexa488 anti-IFN-γ, Alexa647 anti-IL-17 (BD Biosciences) and PE anti-IL-22 antibodies (e-Biosciences) and the isotype controls. 3R4F research cigarettes were purchased from University of Kentucky (USA) and used to induce COPD like symptoms (Pichavant et al., 2014). Gating strategy for flow cytometry analysis of Th17 producing cells is depicted in Supplemental Fig. 5.
Supplemental Fig. 5.

PBMC were stimulated for 24 h with S. pneumoniae. IL-17 and IL-22 production was analyzed by intracellular staining and flow cytometry analysis, (a) Gating strategy used to identify Lin− cells, NK, NKT, Tγδ, MAIT cells as well as CD4+ and CD8+ T cells (bold lines), (b) Expression of IL-22 and IFN-y in NK and Lin′ cells after stimulation with S. pneumoniae or with PBS (Mock) in a non-smoker control, a smoker donor and a COPD patient. This is a representative experiment out of at least 10.
2.4. Primers
Quantitative RT-PCR was performed to quantify mRNA of interest (Table 2). Results were expressed as mean ± SEM of the relative gene expression calculated for each experiment in folds (2− ΔΔCt) using GAPH as a reference, and compared to controls.
Table 2.
Primer sequences for qRT-PCR in mice.
| GAPDH | F | TGCCCAGAACATCATCCCTG |
| R | TCAGATCCACGACGGACACA | |
| DefB2 | F | AAAGTATTGGATACGAAGCAGAACTTG |
| R | GGAGGACAAATGGCTCTGACA | |
| DefB3 | F | TGAGGAAAGGAGGCAGATGCT |
| R | GGAACTCCACAACTGCCAATC | |
| Cathelicidin | F | CAGAGCGGCAGCTACCTGAG |
| R | TCACCACCCCCTGTTCCTT | |
| s100a9 | F | CACCCTGAGCAAGAAGGAAT |
| R | TGTCATTTATGAGGGCTTCATTT | |
| Il-1b | F | TCCCCAACTGGTACATCAGCA |
| R | ACACGGATTCCATGGTGAAGTC | |
| Il-6 | F | AGCCTCCGACTTGTGAAGTG |
| R | CTGATGCTGGTGACAACCAC | |
| Il-12p40 | F | GACCCTGCCCATTGAACTGGC |
| R | CAACGTTGCATCCTAGGATCG | |
| Il-23p19 | F | CACCAGCGGGACATATGAA |
| R | CCTTGTGGGTCACAACCAT |
2.5. Infection by S. pneumoniae and bacterial counts
Mice were inoculated by the intranasal route with a clinical isolate of Sp serotype 1 described elsewhere (Marques et al., 2012). Mice were anesthetized and administered intranasally with 5 × 104 or 5 × 105 colony-forming units (CFU) in 50 μl. Mice were daily monitored for illness and mortality up to 7 days. Bacterial burden in the broncho-alveolar lavages (BAL), lungs and blood was measured by plating samples onto chocolate plates. CFU were enumerated 24 h later. In some experiments, CS-exposed and air mice received recombinant murine IL-22 (1 μg/mouse; Myltenyi Biotech) by intranasal route 24 h before Sp challenge.
2.6. Assessment of airway inflammation
Mice were sacrificed for sampling BAL, lungs, spleen and blood. Total cell numbers per BAL was determined. For histopathology, lungs were fixed by inflation and immersion in paraformaldehyde (PFA; 4%) and embedded in paraffin. To evaluate airway inflammation, lung sections (4-μm thick) were stained by hematoxylin & eosin.
Pulmonary cells from air or CS-exposed mice were prepared as previously described (van der Poll and Opal, 2009) and were analyzed by flow cytometry. To analyze cytokine profiles, pulmonary cell suspensions were incubated with phorbol 12-myristate 13-acetate (PMA; 20 ng/ml) and ionomycin (500 ng/ml) for 3 h. Cells were then stained with appropriate extracellular markers, fixed, permeabilized (BD Cytofix/cytoperm, BD Bioscience), and incubated with PE-conjugated mAb against IL-22 (eBiosciences) and Alexa Fluor 647-conjugated mAb against IL-17 (Biolegend), or control rat IgG1 mAb. Cells were acquired on a Fortessa cytometer (Becton Dickinson), and analyzed using the FlowJo software.
Cytokine production was analyzed in total lung cells. For this, 5 × 105 lung cells were seeded on 96-well plates coated or not with anti-CD3 Ab (eBiosciences). Forty-eight hours later, supernatants were collected and analyzed for IFN-γ, IL-17, and IL-22 concentration by ELISA (R&D Systems).
2.7. Cell sorting and cocultures
Pulmonary cells from Air or CS-exposed mice were prepared as previously described (van der Poll and Opal, 2009) and were analyzed by flow cytometry on FACS Aria (Becton Dickinson). CD45+ Siglec F+ alveolar macrophages (AM) and CD45+ Siglec F− Ia+ CD64+ DC were sorted (purity > 98%). Splenic CD4+ T cells were purified from Air mice using magnetic microbeads (Myltenyi Biotech).
Sorted AM and DC were cultured with CD4+ T cells in RPMI 10% FCS, with the ratio 1/10. Supernatants were collected 48 h later to evaluate IL-17 and IL-22 levels by ELISA.
2.8. Statistical Analysis
All the experiments were repeated at least 3 times with 6–10 mice per group. Results are expressed as the mean ± SEM. Samples were simply randomized and blindly assigned to the different groups. No data have been excluded. The statistical significance of differences between experimental groups was calculated by a one-way ANOVA with a Bonferroni post-test or an unpaired Student t test (GraphPad, San Diego, CA). The possibility to use these parametric tests was assessed by checking if the population is Gaussian and the variance is equal (Bartlett's test). Results with a p value < 0.05 were considered significant.
This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), the Centre National de la Recherche Scientifique (CNRS) and the Conseil Régional du Nord-Pas de Calais [StreptoCOPD project; grant number # 13005300]. Funders had no role in study design, data collection, data analysis, interpretation, writing of the report.
3. Results
3.1. CS-exposed mice display delayed bacterial clearance and exacerbated inflammation upon S. pneumoniae challenge
An experimental model of COPD exacerbation was established in mice chronically exposed to CS using Sp as the trigger (see Fig. 1a). Whereas all air mice survived when infected with 5 × 105 CFU, all CS-exposed mice died within a week. Thus, CS-exposed mice are more susceptible to pneumococcal infection. In contrast, after administration of 5 × 104 CFU per mouse, both air and CS-exposed mice survived (Fig. 1b) and allowed the analysis of airway inflammation, remodeling and immune response.
Fig. 1.

CS-exposed mice are more susceptible to S. pneumoniae. (2 columns).
Mice were chronically exposed to air or CS (5 cigarettes/day, 5 days/week) over a period of 12 weeks to develop symptoms associated to COPD. Mice were then intranasally challenged with S. pneumoniae (Sp) to induce COPD exacerbation or with PBS (Mock) as a control (a). Survival of air and CS-exposed mice was monitored for a week after challenge with 5 × 104 or 5 × 105 CFU of Sp or Mock (b). Absolute numbers of total cells, macrophages, lymphocytes and neutrophils were determined in BAL fluids (c). Neutrophils identified as CD45+ F4/80− CD11b+ Ly6G+ were analyzed in lung tissues 24 h after infection (d). Histological changes were evaluated on lung sections either at 1 or 3 dpi (5 × 104 CFU) (e). CFU counts were evaluated in the BAL, lung tissues and blood at 1, 3 and 7 dpi (f). Data represent as mean ± SEM (n = 48–80 mice per group). *: p < 0.05 or **: p < 0.01 (one-way ANOVA test).
Relative to air mice, infection was associated with enhanced cellular recruitment (particularly neutrophils) in the BAL and the lungs of CS-exposed mice (Fig. 1c–d). Histological examination of lung tissues showed that inflammatory infiltrates mainly located in peribronchial areas and alveolar spaces (Fig. 1e). Moreover, a large thickening of the alveolar walls was observed in CS-exposed animal mice. In air mice challenged with bacteria, lung inflammation was nearly resolved 3 days post-infection (dpi). Enhanced pulmonary inflammatory response in CS-exposed mice was associated with a higher bacterial load that peaked at 3 dpi whilst no bacteria were detected in air mice (Fig. 1e). No bacteria were found in the lungs at 7 dpi. Finally, systemic pneumococcal dissemination peaked at 3 dpi to decline at 7 dpi. This enhanced susceptibility of CS-exposed to Sp was associated to a restricted defect in anti-microbial peptides, namely for cathelicidin, whereas levels of DefB-2 and -3 and s100a9 transcripts were similarly increased in air and CS-exposed (Supplemental Fig. 1).
Supplemental Fig. 1.

Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 5 × l04 CFU of S. pneumoniae (Sp) or with PBS (Mock). Anti-microbial peptide mRNA levels were analyzed in lung tissues 24 h post-infection. Results were expressed as mean ± SEM of fold increase compared to GAPH. *: p < 0.05 vs controls.
These data demonstrated that chronic exposure to CS leads to delayed clearance of Sp, an effect associated with enhanced pulmonary inflammation.
3.2. CS-exposed mice display a reduced production of Th17 cytokines in response to S. pneumoniae
To investigate mechanisms involved in enhanced susceptibility to pneumococcal infection in CS-exposed mice, IL-17 and IL-22 production was quantified. Challenge with Sp significantly enhanced IL-17 and IL-22 levels in BAL (Fig. 2a) and lung lysates (data not shown) of air mice. In marked contrast, Sp infection failed to do so in mice previously exposed to CS (Fig. 2a). Whilst IL-17 remained undetectable in the serum after Sp challenge, IL-22 increase was detected in air, but not CS-exposed, animals (Fig. 2b). IFN-γ levels failed also to increase in the BAL of CS-exposed mice after Sp challenge (Supplemental Fig. 2a). Anti-CD3 restimulation of pulmonary cells induced large amounts of IL-17 whatever the animal group and a tendency to higher concentration was observed in CS-exposed mice (Fig. 2c), as well as IFN-γ (Supplemental Fig. 2b). In contrast, upon CD3 restimulation, pulmonary cells from CS-exposed animals had a much lower ability to release IL-22. Collectively, CS-exposed mice have a lower capacity to produce Th17-type cytokines upon pneumococcal challenge.
Fig. 2.

CS-exposed mice exhibit a defective immune response to S. pneumoniae. (2 columns).
Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 5 × 104 CFU of S. pneumoniae (Sp) or with PBS (Mock). IL-17 and IL-22 levels were evaluated in BAL fluids (a), blood (b) and in the supernatants of restimulated pulmonary cells without (NS) or with anti-CD3 Ab (c). IL-17 and IL-22 producing cells were identified by intracellular staining among pulmonary NK cells (CD45+ TCRβ− NK1.1+), NKT-like cells (CD45+ TCRβ+ NK1.1+), T cells (CD45+ TCRβ+ NK1.1−) and Lin-negative cells (CD45+ CD3− CD11c− CD11b− CD45Rb− NK1.1− CD90.2+ CCR6+) cells (d and e). We have reported representative dot blot of the selected sub-populations and percentages of cytokine+ cells among the respective cell population are represented (c). The mean percentage of IL-17+ and IL-22+ cells was calculated for NK, NKT and Lin− cells (d). Results are expressed as mean ± SEM (n = 48–80 mice per group). One representative experiment out of three independent ones is shown concerning intracellular staining. *: p < 0.05 vs controls (one-way ANOVA test).
Supplemental Fig. 2.
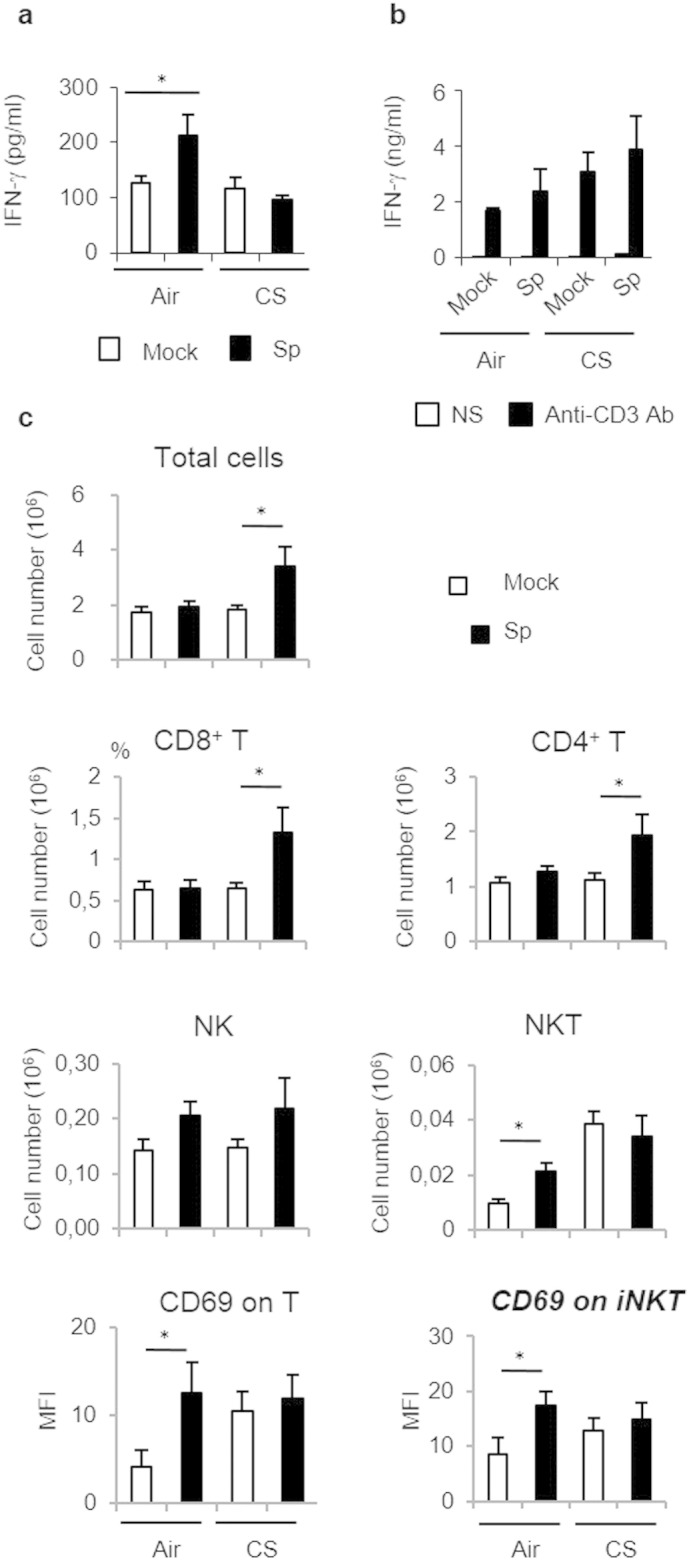
Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 5 × l04 CFU of S. pneumoniae (Sp) or PBS (Mock). IFN-g levels were measured in the BAL 24 h post-infection (a), and restimulated lung cells. (b). Total cell numbers, and absolute numbers (× 106) of NK cells (CD45+ NK1,1+ TCRb−), NKT cells (CD45+ PBS-57 CD1d tetramer+ TCRb+), conventional T cells (CD45+ PBS-57 CD1d tetramer+ TCRb+ CD4+ and/or CD8+) were evaluated in lung tissues 24 h post-infection. Activation of NKT and T cells was estimated by CD69 MFI. (c) Results were expressed as mean ± SEM. *: p < 0.05.
We next focused on Th17 cytokine producing cells. Infection with Sp enhanced the number, as well as the activation status (CD69 expression), of conventional T lymphocytes and NKT cells within lung tissues of air, but not CS-exposed mice (Supplementary Fig. 2c). A tendency towards an enhanced number of NK cells was noticed in both animals groups upon pneumococcal challenge whilst the number of γδ T cells and Lin-negative cells remained constant (Supplemental Fig. 2c and data not shown). Pneumococcal challenge of air mice resulted in higher frequencies of IL-17- and IL-22-producing conventional T cells, NK cells, NKT-like cells, γδ T cells and Lin-negative cells (Fig. 2d–e and not shown). In contrast, chronic exposure to CS dramatically reduced the percentages of pulmonary IL-17-producing NK and NKT-like cells, but not conventional T cells, γδ T cells and Lin-negative cells observed after Sp challenge (Fig. 2d–e and data not shown). Percentages of IL-22-producing conventional T cells, NK cells, NKT-like cells and Lin-negative cells were also significantly diminished in infected CS-exposed mice, relative to air mice.
Hence, upon pneumococcal challenge, chronic exposure to CS leads to defective production of Th17-type cytokines by conventional T cells and innate immune cells.
3.3. CS exposure alters the function of pulmonary APC
We next hypothesized that pulmonary APC could be impacted by CS exposure to lower the activation of innate and conventional T cells to bacteria. Indeed, we and others previously reported that chronic exposure to CS alters pulmonary APC phenotype and functions (Pichavant et al., 2014, Tsoumakidou et al., 2008). Exposure to a sub-lethal dose of Sp triggered phenotypic maturation of pulmonary APC, including alveolar macrophages (AM) and dendritic cells (DC) (CD86 and II+ MHC; data not shown). To analyze their ability to promote Th17 cytokine production, levels of polarizing cytokines including IL-1β and IL-23 were evaluated first in lung lysates (Fig. 3a) and secondary in isolated pulmonary APC (Fig. 3b and Supplemental Fig. 3). Pneumococcal challenge strongly induced mRNA levels of Il-1b and Il-23p19 in air but not in CS-exposed mice (Fig. 3a). Sp infection also induced mRNA levels of Il-1b, Il-6, Il-23p19, but not Il-12p40, in sorted lung DC and slightly increased the expression of Il-1b and Il-12p40 transcripts in sorted AM from air mice (Supplemental Fig. 3). In CS-exposed mice, a defect in Il-1b and Il-12p40 expression was observed in AM, whereas the expression of Il-23p19 mRNA was undetectable in lung DC. At the protein level, IL-1β and IL-23 secretion was increased in supernatants of AM and DC from infected air mice, but not in cells from CS-exposed mice (Fig. 3b).
Fig. 3.

CS-exposed mice exhibited a defective response of pulmonary APC to S. pneumoniae. (1.5 columns).
Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 5 × 104 CFU of S. pneumoniae (Sp) or with PBS (Mock). Il-1b and Il-23 mRNA levels were measured in lung tissues 24 h post-infection (a). CD45+ Siglec F+ AM and CD11c+ Ia+ CD64+ DC were sorted by flow cytometry 24 h post-infection. IL-1β and IL-23 levels were evaluated by ELISA in supernatants 24 h later (b). Cocultures were performed between sorted AM (c) or DC (d) and splenic CD4+ T cells purified from air mice. Supernatants were collected 48 h later and levels of IL-17 were evaluated by ELISA. Data represent mean ± SEM (n = 6–10 mice per group per experiment). One representative experiment out of three ones is shown concerning cell sorting and cocultures with T cells.
Supplemental Fig. 3.

Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 4 × 104 CFU of S. pneumoniae (Sp — black bars) or with PBS (Mock — white bars). Lung tissues were collected 1 day post-infection to sort alveolar macrophages (AM; CD45+ Siglec F+) and dendritic cells (DC; CD45+ Siglec F− la+ CD64+). mRNA levels of ll-1b, ll-6, Il-12p40 and Il-23pl9 were evaluated in AM (a) and DC (b) compared to GAPDH. One representative experiment out of three independent ones is represented.
To evaluate the capacity of these sorted APC to activate T cells, lung DC and AM were cultured with isolated splenic CD4+ T cells from air mice. In these conditions, IL-22 was always undetectable (data not shown). AM (Fig. 3c) and DC (Fig. 3d) sorted from infected air mice induced significant increase in IL-17 production by CD4+ T cells, whereas antigen-presenting cells from CS-exposed mice were unable to do so.
These data suggested that the defect in the Th17 response to Sp is associated with an altered function of pulmonary APC.
3.4. Exogenous IL-22 protects CS-exposed mice from S. pneumoniae infection
Since IL-17 and IL-22 production is ablated in CS-exposed mice, we questioned whether IL-17 and IL-22 supplementation by means of intranasal treatment could improve the outcome of pneumococcal infection in CS-exposed mice. We have previously demonstrated that IL-17 was critical in the development of COPD in a mouse model and higher in COPD patients than in controls (Pichavant et al., 2014). Therefore, exogenous IL-17 could have some deleterious effects during COPD exacerbations. We therefore tested the effect of exogenous IL-22 in the early control of Sp outgrowth and lung inflammation. Administration of recombinant IL-22 prior to bacterial challenge strongly reduced bacterial outgrowth in the lungs and dissemination outside the lungs (Fig. 4a). While treatment with IL-22 had no effect on neutrophil recruitment within the lungs, it enhanced the number of activated AM and DC in CS-exposed mice (Fig. 4b and Supplemental Fig. 4a), other well-known effector cells against pneumococcus. This process was also associated with an increased mobilization of NK and NKT cells in the lungs of infected COPD mice (Supplemental Fig. 4b). Of interest, administration of IL-22 resulted in enhanced levels of transcripts encoding defensin β2 (Defb2) and defensin β3 (Defb3) (Fig. 4c), both anti-microbial peptides playing a role in Sp clearance. Finally, IL-22 treatment also strongly reduced the lung lesions associated with Sp infection, namely the thickening of the alveolar walls and the inflammatory infiltrate in CS-exposed animals (Fig. 4d). This improved clearance of Sp in CS-exposed mice was also associated with higher IL-17 and IFN-γ production by lung cells (Supplemental Fig. 4c).
Fig. 4.

Exogenous IL-22 improves the clearance of S. pneumoniae in CS-exposed mice. (1.5 columns).
Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 5 × 104 CFU of S. pneumoniae (Sp) or with PBS (Mock). Recombinant murine IL-22 was intranasally administered the day before Sp infection. CFU counts were evaluated in BAL, lung tissues and blood (a). Percentages of neutrophils (identified as CD45+ F4/80− CD11c− Ly6G+ CD11b+), AM and DC among CD45+ cells were analyzed in lung tissues from infected CS-exposed mice treated or not with rmIL-22 (b). Anti-microbial peptide mRNA levels were analyzed in lung tissues 3 dpi (c). Histological changes were evaluated at 3 dpi (d). Data represent mean ± SEM (n = 24–40 mice per group). *: p < 0.05 vs controls (one-way ANOVA test).
Supplemental Fig. 4.

Mice were chronically exposed to air or CS over a period of 12 weeks and then intranasally challenged with 4 × l04 CFU of S. pneumoniae (Sp). Recombinant IL-22 was intranasally given to mice the day before Sp infection. Activation status of AM and DC was assessed by CD86 expression (a). Percentages of NK cells and NKT cells were analyzed among CD45+ cells in lung tissues one day post-infection (b). IL-17 and IFN-g levels were evaluated in restimulated lung cells (c). Results were expressed as mean ± SEM. *: p < 0.05 vs controls.
Together, recombinant IL-22 administration can compensate for the lack of Th17-associated cytokines in CS-exposed mice to restore anti-pneumococcal defenses.
3.5. COPD patients showed impaired Th17 cytokine production in response to S. pneumoniae
To evaluate whether COPD status modulates the response to Sp, PBMCs were isolated from 3 different groups: non-smoker healthy controls, smokers and COPD patients, and were stimulated with Sp. Levels of IL-17 and IL-22 were evaluated in PBMC supernatants. Resting PBMC exhibited similar levels of cytokines in the three groups (Fig. 5a). Exposure to Sp expectedly increased IL-22 and, to a lesser extent, IL-17 production in the non-smoker and smoker groups, but had no effect on cytokine production in COPD patients. The response to PHA was also partially altered in COPD patients, in contrast to the other two groups (data not shown).
Fig. 5.

COPD patients have a defective cytokine response to S. pneumoniae. (1.5 columns).
Levels of IL-17 and IL-22 was quantified by ELISA in supernatants from PBMC from healthy non-smoker subjects (n = 14), healthy smokers (n = 14) and COPD patients (n = 12) (a). In parallel, percentages of IL-17 and IL-22 producing cells were measured by intracellular staining in Lin− (CD11c− CD14− CD19− CD20−) cells, NK cells and NKT cells (b). Levels of IL-1β and IL-23 were quantified by ELISA in supernatants from PBMC from healthy non-smoker subjects, healthy smokers and COPD patients (c). Data represent mean ± SEM. *: p < 0.05 versus medium in the different groups (one-way ANOVA test).
We next looked at the cellular sources of IL-17 and IL-22, focusing on conventional T cells, NK cells, NKT cells, γδ T cells, mucosal-associated invariant T (MAIT) cells and Lineage-negative cells (Fig. 5b and Supplementary Fig. 5). In the non-smoker group, bacteria increased the proportion of IL-17-producing cells (in particular Lin−, NK and NKT cells), IL-22-producing cells (mainly NK and NKT cells) and MAIT cells (not shown). In contrast, the stimulation with Sp did not significantly amplify the proportion of cells producing these cytokines in NK, NKT and Lin− cells (Fig. 5b) as well as in MAIT cells (data not shown) from COPD patients. In smokers, IL-17 production induced by Sp was also impaired in these three cell types, whereas IL-22 expression was only reduced in NK cells. No modification of the percentage of cytokine+ cells was detected among the three groups of patients for γδT and CD4/CD8+ T cells (data not shown). This defective production of IL17 and IL-22 was probably linked to the lower production of IL-1β and IL-23 by PBMC from COPD patients (Fig. 5C).These data showed that the blood innate immune cells from COPD patients displayed an altered Th17 cytokine response to Sp.
4. Discussion
Infection with Sp is one of the main factors responsible for COPD exacerbation (Gaschler et al., 2007, van der Poll and Opal, 2009). In our mouse model mimicking COPD, Sp challenge resulted in greater lung inflammation and tissue remodeling, and therefore an exacerbation of the disease. Combined exposure to CS and SEB resulted in a raised number of lymphocytes and neutrophils, epithelial remodeling and over-production of IL-17 (Huvenne et al., 2011). Gaschler et al. used H. influenza to exacerbate COPD and demonstrated that the bacterial burden observed in COPD mice was mainly due to a skewed inflammatory mediator expression, probably in AM (Gaschler et al., 2009). Innate immunity associated with the recruitment of competent AM and neutrophils is crucial in the early phase of natural anti-pneumococcal host defense and particularly in bacterial clearance (Clement et al., 2008). Such a pattern was observed in our model despite a defective clearance of the pathogen in CS-exposed mice.
Indeed, we observed some important modifications in the activation of APC from CS-exposed mice, but also in not conventional lymphocytes. Major cellular sources of IL-17 and IL-22, described as NK, NKT, ILC in mice and humans (Colonna, 2009, Liang et al., 2006, Sonnenberg et al., 2012, Van Maele et al., 2010), failed to produce higher levels of Th17 cytokines in response to Sp in CS-exposed mice whereas only the production of IL-22 was altered in conventional T cells. According to the implication of such cells in the protection against Sp (Marques et al., 2012, Clement et al., 2008, Van Maele et al., 2010), this suggest that this defect might be an important determinant of bacterial susceptibility during COPD. In contrast, the activation of conventional T cells, MAIT and γδT cells was not clearly modified during COPD.
The defective activation of conventional T cells and some innate populations could be explained by the alteration of pulmonary APC, as previously reported in CS-exposed mice (Pichavant et al., 2014, Kroening et al., 2008). Indeed expression of pro-Th17 cytokines, such as IL-1β and IL-23 (Mucida and Salek-Ardakani, 2009), was decreased both in lung AM and DC from infected CS-exposed mice. IL-23 plays a key role in the clearance of the bacteria and the production of Th17 cytokines by ILC (Van Maele et al., 2014). IL-23 is also needed for IL-17 expression by others immune cells like NKT and γδ T-cells (Clement et al., 2008). Therefore, APC from CS-exposed mice cannot correctly educate the T cells and other innate cells to respond to Sp. Such a defective production of IL-1β and IL-23 was also observed in COPD patients in response to Sp. (Kroening et al., 2008).Altogether, during COPD, the reduced production of IL-17 and IL-22 by conventional T cells and other innate cells might result from a deficient IL-1β and IL-23 synthesis by DC in response to Sp.
Functionally, IL-17 and IL-22 have been reported as essential factors in anti-bacterial defenses. During infection, the early production of IL-22 by innate immune cells is crucial for host protective immunity (Aujla et al., 2008, Graham et al., 2011, Zheng et al., 2008) including a role in chemotaxis and tissue repair (Wolk et al., 2004, Zheng et al., 2008, Eyerich et al., 2010, Kolls et al., 2008, Sonnenberg et al., 2010, Witte et al., 2010). It has been recently shown that IL-17A is required for NTHi-exacerbated pulmonary neutrophilia induced by cigarette smoke although the role of IL-22 was not evaluated (Roos et al., 2015). Moreover, IL-17 and IL-22 induced the production of anti-microbial peptides (including β-defensins, S100A7-9, Reg3β and Reg3γ) important in the containment of pathogens (Zheng et al., 2008, Kolls et al., 2008, Sonnenberg et al., 2010, Cash et al., 2006). In our report, the defective production of IL-22 in CS-exposed mice to Sp was not associated to an impaired production of these antimicrobial peptides as previously reported in COPD patients (Pace et al., 2012). Nevertheless, opposite results have been reported suggesting that the levels of antimicrobial peptide expression were insufficient to control the higher bacterial load both in COPD patients and mice. We can also suspect that bacterial susceptibility in infected CS-exposed mice was not solely related with the defective production of cathelicidin, an IL-22 independent peptide involved in defense against Sp (Felgentreff et al., 2006). Interestingly, local administration of rmIL-22 amplified β-defensin levels in the lungs and a competent immune response, allowing Sp clearance in CS-exposed mice. No impact on neutrophil influx was observed in infected CS-exposed mice, suggesting that the effect of this cytokine is mostly related with cell priming to efficiently kill the bacteria and/or the release of anti-microbial peptides. In these settings, the preventive role of IL-22 on lung injury is potentially linked to its complementary action on the induction of antimicrobial peptides, the activation of immune cells (including neutrophils) and in the maintenance of the epithelial barrier (Kumar et al., 2013). In infected CS-exposed animals, treatment with rmIL-22 results in an efficient resolution and to the preservation of lung tissue after infection, as previously reported in inflammatory models (Liu et al., 2009). The balance between IL-17 and/or IL-22 expression has been found to contribute to either the pro-inflammatory or tissue-protective phases of lung defense, depending on the context (Liang et al., 2006, Eyerich et al., 2010, Sonnenberg et al., 2010). In our model, a protective role for IL-17 cannot be excluded (Lu et al., 2008), since the production of IL-17 was also defective in CS-exposed mice and patients in response to Sp. However, IL-17 is implicated in COPD pathogenesis. During COPD exacerbation, this cytokine has no effect on lung bacterial load and promotes the neutrophil recruitment which is potentially deleterious (Roos et al., 2015).
Alteration of the innate immune response to bacterial infection is a key determinant in the COPD course. It is now well recognized that respiratory infections are important in the induction, progression and exacerbation of the disease. Here we identified the IL-22 defect as a key factor in COPD exacerbations, both in patients and in the murine model. This alteration related to deficient activation of lymphocytes by APC offers hints for the development of novel therapeutic strategies in COPD exacerbations. Thereby, we propose IL-22 as a promising target in the treatment and/or the prevention of COPD exacerbations. Restoring this defective cytokine response could represent an ideal therapy to build a competent immune response against pathogens in COPD patients, and to limit the consequences of exacerbation of the disease.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Author contributions
Conception and design: MP and PG; analysis and interpretation: RS, OLR, CO, FH, GR, MP, BK, and PG; drafting the manuscript for important intellectual content: MP and PG. All authors gave their agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The following are the supplementary data related to this article
Acknowledgements
We thank the NIAID Tetramer Facility (Emory University, Atlanta, GA) for supplying CD1d tetramer and Dr. T. Camou (National Reference Laboratory, Ministry of Health, Montevideo, Uruguay) for the gift of the S. pneumoniae serotype 1 clinical isolate E1586. We also thank Hélène Bauderlique for her help for cell sorting (BICel Cytométrie Plateform, Institut Pasteur de Lille, France). A special thank to Gwénola Kervoaze and Eva Vilain for their technical support, and Jean-Claude Sirard, François Trottein, Isabelle Wolowczuk and Sandra Weller for their critical reviewing of the paper.
This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), the Centre National de la Recherche Scientifique (CNRS) and the Conseil Régional du Nord-Pas de Calais [StreptoCOPD project; grant number # 13005300]. Funders had no role in study design, data collection, data analysis, interpretation, writing of the report.
References
- Agusti A.G., Noguera A., Sauleda J., Sala E., Pons J., Busquets X. Systemic effects of chronic obstructive pulmonary disease. Eur. Respir. J. 2003;21:347–360. doi: 10.1183/09031936.03.00405703. [DOI] [PubMed] [Google Scholar]
- Aujla S.J., Chan Y.R., Zheng M., Fei M., Askew D.J., Pociask D.A., Reinhart T.A., McAllister F., Edeal J., Gaus K., Husain S., Kreindler J.L., Dubin P.J., Pilewski J.M., Myerburg M.M., Mason C.A., Iwakura Y., Kolls J.K. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P.J., Stockley R.A. COPD: current therapeutic interventions and future approaches. Eur. Respir. J. 2005;25:1084–1106. doi: 10.1183/09031936.05.00139104. [DOI] [PubMed] [Google Scholar]
- Cash H.L., Whitham C.V., Behrendt C.L., Hooper L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement C.G., Evans S.E., Evans C.M., Hawke D., Kobayashi R., Reynolds P.R., Moghaddam S.J., Scott B.L., Melicoff E., Adachi R., Dickey B.F., Tuvim M.J. Stimulation of lung innate immunity protects against lethal pneumococcal pneumonia in mice. Am. J. Respir. Crit. Care Med. 2008;177:1322–1330. doi: 10.1164/rccm.200607-1038OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Doisne J.M., Soulard V., Becourt C., Amniai L., Henrot P., Havenar-Daughton C., Blanchet C., Zitvogel L., Ryffel B., Cavaillon J.M., Marie J.C., Couillin I., Benlagha K. Cutting edge: crucial role of IL-1 and IL-23 in the innate IL-17 response of peripheral lymph node NK1.1− invariant NKT cells to bacteria. J. Immunol. 2011;186:662–666. doi: 10.4049/jimmunol.1002725. [DOI] [PubMed] [Google Scholar]
- Drannik A.G., Pouladi M.A., Robbins C.S., Goncharova S.I., Kianpour S., Stampfli M.R. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2004;170:1164–1171. doi: 10.1164/rccm.200311-1521OC. [DOI] [PubMed] [Google Scholar]
- Eidenschenk C., Rutz S., Liesenfeld O., Ouyang W. role of il-22 in microbial host defense. Curr. Top. Microbiol. Immunol. 2014;380:213–236. doi: 10.1007/978-3-662-43492-5_10. [DOI] [PubMed] [Google Scholar]
- Eyerich S., Eyerich K., Cavani A., Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol. 2010;31:354–361. doi: 10.1016/j.it.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Felgentreff K., Beisswenger C., Griese M., Gulder T., Bringmann G., Bals R. The antimicrobial peptide cathelicidin interacts with airway mucus. Peptides. 2006;27:3100–3106. doi: 10.1016/j.peptides.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Fletcher C., Peto R. The natural history of chronic airflow obstruction. Br. Med. J. 1977;1:1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler G.J., Bauer C.M., Zavitz C.C., Stampfli M.R. Animal models of chronic obstructive pulmonary disease exacerbations. Contrib. Microbiol. 2007;14:126–141. doi: 10.1159/000107059. [DOI] [PubMed] [Google Scholar]
- Gaschler G.J., Skrtic M., Zavitz C.C., Lindahl M., Onnervik P.O., Murphy T.F., Sethi S., Stampfli M.R. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am. J. Respir. Crit. Care Med. 2009;179:666–675. doi: 10.1164/rccm.200808-1306OC. [DOI] [PubMed] [Google Scholar]
- Graham A.C., Carr K.D., Sieve A.N., Indramohan M., Break T.J., Berg R.E. IL-22 production is regulated by il-23 during Listeria monocytogenes infection but is not required for bacterial clearance or tissue protection. PLoS One. 2011;6:e17171. doi: 10.1371/journal.pone.0017171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvenne W., Lanckacker E.A., Krysko O., Bracke K.R., Demoor T., Hellings P.W., Brusselle G.G., Joos G.F., Bachert C., Maes T. Exacerbation of cigarette smoke-induced pulmonary inflammation by Staphylococcus aureus enterotoxin B in mice. Respir. Res. 2011;12:69. doi: 10.1186/1465-9921-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov S., Paget C., Trottein F. Role of non-conventional t lymphocytes in respiratory infections: the case of the Pneumococcus. PLoS Pathog. 2014;10:e1004300. doi: 10.1371/journal.ppat.1004300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov S., Renneson J., Fontaine J., Barthelemy A., Paget C., Fernandez E.M., Blanc F., De Trez C., Van Maele L., Dumoutier L., Huerre M.R., Eberl G., Si-Tahar M., Gosset P., Renauld J.C., Sirard J.C., Faveeuw C., Trottein F. Interleukin-22 reduces lung inflammation during influenza a virus infection and protects against secondary bacterial infection. J. Virol. 2013;87:6911–6924. doi: 10.1128/JVI.02943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolls J.K., McCray P.B., Jr., Chan Y.R. Cytokine-mediated regulation of antimicrobial proteins. Nat. Rev. Immunol. 2008;8:829–835. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroening P.R., Barnes T.W., Pease L., Limper A., Kita H., Vassallo R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through erk-dependent pathways. J. Immunol. 2008;181:1536–1547. doi: 10.4049/jimmunol.181.2.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Thakar M.S., Ouyang W., Malarkannan S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013;6:69–82. doi: 10.1038/mi.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S.C., Tan X.Y., Luxenberg D.P., Karim R., Dunussi-Joannopoulos K., Collins M., Fouser L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.Z., Pezeshki M., Raffatellu M. Th17 cytokines and host-pathogen interactions at the mucosa: dichotomies of help and harm. Cytokine. 2009;48:156–160. doi: 10.1016/j.cyto.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.J., Gross J., Bogaert D., Finn A., Bagrade L., Zhang Q., Kolls J.K., Srivastava A., Lundgren A., Forte S., Thompson C.M., Harney K.F., Anderson P.W., Lipsitch M., Malley R. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 2008;4:e1000159. doi: 10.1371/journal.ppat.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques J.M., Rial A., Munoz N., Pellay F.X., Van Maele L., Leger H., Camou T., Sirard J.C., Benecke A., Chabalgoity J.A. Protection against Streptococcus pneumoniae serotype 1 acute infection shows a signature of Th17- and IFN-gamma-mediated immunity. Immunobiology. 2012;217:420–429. doi: 10.1016/j.imbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- Mucida D., Salek-Ardakani S. Regulation of TH17 cells in the mucosal surfaces. J. Allergy Clin. Immunol. 2009;123:997–1003. doi: 10.1016/j.jaci.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace E., Ferraro M., Minervini M.I., Vitulo P., Pipitone L., Chiappara G., Siena L., Montalbano A.M., Johnson M., Gjomarkaj M. Beta defensin-2 is reduced in central but not in distal airways of smoker COPD patients. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichavant M., Remy G., Bekaert S., Le Rouzic O., Kervoaze G., Vilain E., Just N., Tillie-Leblond I., Trottein F., Cataldo D., Gosset P. Oxidative stress-mediated iNKT-cell activation is involved in COPD pathogenesis. Mucosal Immunol. 2014;7:568–578. doi: 10.1038/mi.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T., Opal S.M. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. 2009;374:1543–1556. doi: 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- Roos A.B., Sethi S., Nikota J., Wrona C.T., Dorrington M.G., Sanden C., Bauer C.M., Shen P., Bowdish D., Stevenson C.S., Erjefalt J.S., Stampfli M.R. IL-17A and the Promotion of Neutrophilia in Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015;192:428–437. doi: 10.1164/rccm.201409-1689OC. [DOI] [PubMed] [Google Scholar]
- Sethi S., Murphy T.F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- Soler N., Ewig S., Torres A., Filella X., Gonzalez J., Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur. Respir. J. 1999;14:1015–1022. doi: 10.1183/09031936.99.14510159. [DOI] [PubMed] [Google Scholar]
- Soler-Cataluna J.J., Martinez-Garcia M.A., Roman Sanchez P., Salcedo E., Navarro M., Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:925–931. doi: 10.1136/thx.2005.040527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg G.F., Fouser L.A., Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- Sonnenberg G.F., Monticelli L.A., Alenghat T., Fung T.C., Hutnick N.A., Kunisawa J., Shibata N., Grunberg S., Sinha R., Zahm A.M., Tardif M.R., Sathaliyawala T., Kubota M., Farber D.L., Collman R.G., Shaked A., Fouser L.A., Weiner D.B., Tessier P.A., Friedman J.R., Kiyono H., Bushman F.D., Chang K.M., Artis D. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg G.F., Nair M.G., Kirn T.J., Zaph C., Fouser L.A., Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 2010;207:1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoumakidou M., Demedts I.K., Brusselle G.G., Jeffery P.K. Dendritic cells in chronic obstructive pulmonary disease: new players in an old game. Am. J. Respir. Crit. Care Med. 2008;177:1180–1186. doi: 10.1164/rccm.200711-1727PP. [DOI] [PubMed] [Google Scholar]
- Van Maele L.C., Carnoy D., Cayet S., Ivanov R., Porte E., Deruy J.A., Chabalgoity J.C., Renauld G., Eberl A.G., Benecke F., Trottein C., Faveeuw, Sirard J.C. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J. Infect. Dis. 2014;210:493–503. doi: 10.1093/infdis/jiu106. [DOI] [PubMed] [Google Scholar]
- Van Maele L., Carnoy C., Cayet D., Songhet P., Dumoutier L., Ferrero I., Janot L., Erard F., Bertout J., Leger H., Sebbane F., Benecke A., Renauld J.C., Hardt W.D., Ryffel B., Sirard J.C. TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127 + immune cells in spleen and mucosa. J. Immunol. 2010;185:1177–1185. doi: 10.4049/jimmunol.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte E., Witte K., Warszawska K., Sabat R., Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365–379. doi: 10.1016/j.cytogfr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Wolk K., Kunz S., Witte E., Friedrich M., Asadullah K., Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Zheng Y., Valdez P.A., Danilenko D.M., Hu Y., Sa S.M., Gong Q., Abbas A.R., Modrusan Z., Ghilardi N., de Sauvage F.J., Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]