

Over the past five years, the genome-wide association study (GWAS) method has produced significant findings that are providing insights into the biological pathways involved in disease susceptibility for both schizophrenia and bipolar disorder. Yet we have no such findings for major depressive disorder (MDD) (1) -- the most common of these disorders, causing the greatest disability in the world population. We were asked by the editor of this journal to comment on why progress has been so difficult for MDD, and how we can do better.
Consider, by contrast, the remarkable GWAS results achieved for schizophrenia.(2) First, some background: GWAS arrays assay common single nucleotide polymorphisms (SNPs) and copy number variants (CNVs). (Strong associations have been observed between rare CNVs and schizophrenia but not mood disorders, with larger analyses of the latter now underway. Thus, CNVs are not considered further here.) “Common” SNPs are variations at one nucleic acid position in 5% or more of chromosomes in a population -- a frequency achieved, over thousands of generations, by variants that are typically neutral with respect to survival and have small or no effects on serious disease. GWAS arrays of 250,000 to 1,000,000 “tag” SNPs capture most of the effects of common SNPs on diseases or traits, because each genotyped SNP is strongly correlated with many nearby SNPs. The statistical power to identify significant association depends on the effect size of each SNP and the sample size.
Figure 1 illustrates the relationship between the number of cases studied (plus adequate controls) and the number of significant independent associations for several disorders or traits. (By independent, we mean that a region with significant association to a set of inter-correlated SNPs is counted only once.) For schizophrenia, analyses of multiple GWAS datasets by the Psychiatric Genomics Consortium (PGC) detected 5 significant associations with ~9,000 cases (3), increasing to 108 associations with ~35,000 cases. (2) Above an “inflection point” (a critical sample size), the number of significant associations increases linearly with N -- e.g., for schizophrenia, above ~13-18,000 cases, there are ~4 new “hits” per 1,000 additional cases. This kind of relationship was previously described for height, lipid levels and blood pressure as examples, showing a linear or more rapid increase in “hits” above the inflection point. (4) The likely explanation is that the genetic contributions to these traits are predominantly polygenic, with each associated SNP making a small additive contribution to risk. Below the inflection point, samples are underpowered to reliably detect even the strongest (individually small) common SNP effects; above the inflection point, numerous weaker effects are detected in a more or less linear fashion.
Figure 1. The GWAS “inflection point”: discoveries in relation to sample size.
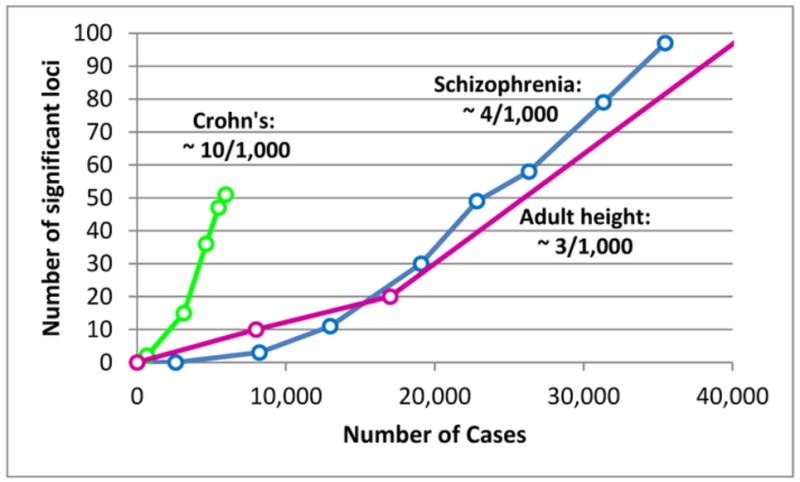
Each point represents the number of independent chromosomal regions (loci) containing SNPs associated with the disease or trait at a genome-wide level of significance in a GWAS analysis with a given number of cases (and a control group of similar or greater size). The curve for adult height is truncated but the next datapoint is 180 loci with the equivalent of ~65,000 cases. The data points are results of unpublished meta-analyses carried out by S.R. on roughly chronological subsets of each dataset in order to demonstrate the linear relationship between sample size and discoveries once a minimum N (“inflection point”) has been achieved. The ratios for each trait are the approximate number of discoveries per 1,000 additional cases (plus controls). Each ratio results from a combination of the number of susceptibility loci for the trait and the size of their genetic effects. Large Ns would discover additional loci for each trait. (See Figure S1 for references.)
MDD stands out as a difficult problem for GWAS: the first PGC MDD analysis (1), with ~9,500 cases, is probably the largest common-disease GWAS that detected no significant associations (http://www.genome.gov/gwastudies/ lists findings for hundreds of diseases/traits). Larger preliminary PGC MDD analyses have also been negative for single SNP associations (unpublished data).
Yet, two types of GWAS analysis show that there is a signal buried in the noise, by demonstrating an aggregate effect of common SNPs on MDD. GREML analysis (genomic- relatedness-matrix restricted maximum likelihood) (5) tests whether cases are genetically more similar to each other than to controls, and estimates the total proportion of variance (“SNP heritability”) attributable to common SNPs: 0.21 for MDD, similar to the value for schizophrenia (0.23), with a larger standard error for MDD. (6) (These values are lower bounds, because the analysis uses a subset of SNPs which do not perfectly tag the true susceptibility SNPs.) These results are consistent with a multifactorial etiology: everyone carries genetic risk variants for each disorder; disease risk increases with the burden of genetic and non-genetic risk factors.
A second, statistically related approach uses the association test result for each SNP in a “training” dataset to weight the genotypes of individuals in an independent “test” dataset to compute genomic polygenic risk scores, and then tests whether these scores predict case vs. control status. (7) Split-sample analysis of the first PGC MDD dataset (1), as well as unpublished analyses predicting single datasets from the remaining datasets, demonstrated modest but significant cross-dataset predictions. Thus, SNP heritability and polygenic score analyses show that individual SNP effects are there. Why can’t we detect them?
The answers are probably: sample size and heterogeneity. Compared with schizophrenia, 3-5 times as many cases are required to detect the same number of single-SNP associations, because MDD is more frequent (10-20% lifetime risk vs. <1%) and (making the plausible assumption that there are a similar number of common causal variants for the two disorders) effect sizes are smaller because MDD is less heritable (<40% vs. 65-85%). (1) Extrapolating from the schizophrenia inflection point (Figure 1), we expect to detect multiple MDD associations when N exceeds 75,000-100,000 cases. (To the skeptical reader: until recently, there were strong doubts about GWAS as an approach for schizophrenia, but appropriate Ns produced dramatic success.)
The required sample size could be higher if MDD is more genetically heterogeneous than schizophrenia. There are many types of heterogeneity. The multifactorial-polygenic model assumes that each MDD case will have a unique set (and proportion) of genetic and non- genetic risk factors. GWAS overcomes this problem with large sample sizes. Misdiagnosis is a form of heterogeneity: power is greatly reduced if 10-20% of cases do not have the disorder of interest (8), or if genetically quite different disorders are included. (9) For MDD, there are many potential sources of heterogeneity due to non-genetic factors, ranging from childhood psychological trauma to the depressogenic effects of medications to cardiovascular disease. Unfortunately we do not know how to differentiate “misdiagnosis” (e.g., from measurement error due to unreliability of research methods or subjects’ reports) from phenotypic similarities that are due to genetic correlations among disorders with overlapping phenotypes (i.e., shared genetic factors explaining a proportion of heritability). For example, substantial genetic correlations are observed between MDD and bipolar disorder (0.47), schizophrenia (0.43) and attention deficit- hyperactivity disorder (0.32) (6); and twin studies demonstrate substantial shared heritability for depressive and anxiety disorders. (10)
PGC carries out combined analyses of multiple GWAS datasets for diverse psychiatric disorders and combinations of disorders under an open, rapid progress model (http://pgc.unc.edu). PGC’s MDD Working Group is attempting to (1) dramatically increase the overall GWAS sample size, and (2) define more homogeneous subsets of cases. Increased sample size alone could lead to progress, or we may need to increase power by identifying less heterogeneous subsets of cases. Both approaches are needed, because much larger samples will be required to test models of heterogeneity, and phenotypic information will help us to understand associations.
Five years ago, genetic samples of thousands of cases seemed massive. How can we get to 100,000 and beyond at a feasible cost? The most promising approach is to obtain phenotypic information from questionnaires, health registries or electronic medical records for tens or hundreds of thousands of individuals in large-scale “biobanks” or other population-based datasets, expecting that 10-20% of individuals will have MDD. A U.S. example is the Genetic Epidemiology Research on Aging study of 78,000 Kaiser Permanente health plan members with 12,000 antidepressant-treated cases (dbGAP dataset phs000674.v1.p1, http://www.ncbi.nlm.nih.gov/gap). Relevant studies are underway in the UK, Scotland, Denmark, Germany and the Netherlands. Ideally, assessments should be sufficiently brief to permit collecting large samples, while querying critical variables that can later be used to index or model heterogeneity. (A caveat is that these samples will not be enriched for more severe cases.) The cost of genotyping has decreased substantially (to under $100 per sample) with the development of the “PsychChip” by PGC and Illumina, Inc. (http://www.illumina.com/products/psycharray.ilmn). Genotyping of such datasets is particularly cost-effective because many health-related phenotypes can be analyzed.
Many variables that could prove useful in modeling heterogeneity can be collected in large samples, such as: sex; recurrence (vs. single episodes); age at onset; the interaction between genetic factors and childhood abuse and neglect; symptom patterns (e.g., “atypical” depression); perinatal depression; comorbidity with psychiatric disorders (e.g., anxiety) and medical disorders (e.g., migraine); longitudinal course of MDD, its relationship to comorbidities, and treatment response; and polygenic relatedness to disorders such as schizophrenia and bipolar disorder. PGC-affiliated investigators are analyzing these and other variables.
In conclusion, MDD has been challenging for GWAS. Major obstacles include moderate heritability, high prevalence, heterogeneity of genetic and non-genetic factors and limited knowledge about genotype-phenotype relationships. But for common multifactorial disorders, GWAS has been the most successful method for genetic discovery in history. SNP heritability analyses suggest that common SNPs contribute to genetic risk of MDD; therefore, GWAS should be successful. Much larger sample sizes will be needed to achieve adequate power to detect multiple associations to MDD and to identify and replicate predictors of heterogeneity, which can then be modeled to improve overall power. Achieving such a large N will require coordinated and cost-effective phenotyping by investigators worldwide, with multiple sources of funding for phenotyping and genotyping. Half of the battle is knowing what needs to be done. Now, we need to do it.
Supplementary Material
Acknowledgments
Financial Disclosures
The authors acknowledge the assistant of multiple members of the Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium in preparing this commentary. This work was supported in part by NIH grants U01 MH094411 and U01 MH094421.
Footnotes
The authors have no competing interests to declare.
References
- 1.Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, et al. A mega- analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panagiotou OA, Willer CJ, Hirschhorn JN, Ioannidis JP. The power of meta- analysis in genome-wide association studies. Annual review of genomics and human genetics. 2013;14:441–465. doi: 10.1146/annurev-genom-091212-153520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross-Disorder Group of the Psychiatric Genomics C. Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wray NR, Lee SH, Kendler KS. Impact of diagnostic misclassification on estimation of genetic correlations using genome-wide genotypes. Eur J Hum Genet. 2012;20:668–674. doi: 10.1038/ejhg.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manchia M, Cullis J, Turecki G, Rouleau GA, Uher R, Alda M. The impact of phenotypic and genetic heterogeneity on results of genome wide association studies of complex diseases. PloS one. 2013;8:e76295. doi: 10.1371/journal.pone.0076295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry. 2003;60:929–937. doi: 10.1001/archpsyc.60.9.929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.