

Abstract
Background & Aims
Enterohemorrhagic Escherichia coli (EHEC) causes over 70,000 episodes of foodborne diarrhea annually in the United States. The early sequence of events that precede life-threatening hemorrhagic colitis and hemolytic uremic syndrome is not fully understood due to the initial asymptomatic phase of the disease and the lack of a suitable animal model. We determined the initial molecular events in the interaction between EHEC and human colonic epithelium.
Methods
Human colonoids derived from adult proximal colonic stem cells were developed into monolayers to study EHEC-epithelial interactions. Monolayer confluency and differentiation were monitored by transepithelial electrical resistance measurements. The monolayers were apically infected with EHEC, and the progression of epithelial damage over time was assessed using biochemical and imaging approaches.
Results
Human colonoid cultures recapitulate the differential protein expression patterns characteristic of the crypt and surface colonocytes. Mucus-producing differentiated colonoid monolayers are preferentially colonized by EHEC. Upon colonization, EHEC forms characteristic attaching and effacing lesions on the apical surface of colonoid monolayers. Mucin 2, a main component of colonic mucus, and protocadherin 24 (PCDH24), a microvillar resident protein, are targeted by EHEC at early stages of infection. The EHEC-secreted serine protease EspP initiates brush border damage through PCDH24 reduction.
Conclusions
Human colonoid monolayers are a relevant pathophysiologic model that allow the study of early molecular events during enteric infections. Colonoid monolayers provide access to both apical and basolateral surfaces, thus providing an advantage over three-dimensional cultures to study host–pathogen interactions in a controllable and tractable manner. EHEC reduces colonic mucus and affects the brush border cytoskeleton in the absence of commensal bacteria.
Keywords: Human Colonoid Monolayers, Intestinal Organoids, Microvillar Effacement, Serine Protease EspP
Abbreviations used in this paper: A/E, attaching and effacing; BB, brush border; CCS, cold chelating solution; CM, complete medium; 3D, three dimensional; EHEC, enterohemorrhagic Escherichia coli; EM, expansion medium; HCM, human colonoid monolayers; HUS, hemolytic uremic syndrome; IEC, intestinal epithelial cell; LGR5, leucine-rich repeat containing G protein-coupled receptor 5; MLPCDH, mucin-like protocadherin; MUC2, extracellular mucin 2; NHE2, sodium-hydrogen exchanger isoform 2; NHERF3, sodium-hydrogen exchanger regulatory factor 3; PBS, phosphate-buffered saline; PCDH24, protocadherin 24; PCR, polymerase chain reaction; SPATE, serine protease autotransporters of Enterobacteriaceae; Stx, Shiga toxins; TBS, Tris-buffered saline; TER, transepithelial electrical resistance; TJ, tight junction
Summary.
Using a novel human colonoid monolayer model, the earliest targets of enterohemorragic Escherichia coli infection by the serine protease EspP have been identified. Mucin-2 and protocadherin-24 are targeted sequentially, leading to bacterial attachment to the epithelium and microvillar effacement.
Shiga toxin–producing enterohemorrhagic Escherichia coli (EHEC) is the major disease-causing food borne E. coli, with ∼73,000 illnesses, ∼3000 hospitalizations, and ∼500 deaths annually in the United States.1 In humans, EHEC colonizes the ascending colon and causes watery diarrhea that can progress into hemorrhagic colitis, and in ∼10% of patients causes life-threatening extraintestinal complications, including hemolytic uremic syndrome (HUS).2 There is currently no treatment available for EHEC infections3 because drugs commonly used to treat bacterial infections, such as antibiotics, antimotility agents, and narcotics, promote HUS development.4
The absence of a specific treatment is partially due to the lack of animal or cell culture models that fully recapitulate the disease. Thus far, studies that have used carcinoma-derived human intestinal epithelial cell lines have had low impact due to their transformed phenotype.5 Animal models have been developed to study the pathogenesis of EHEC infection in vivo but do not mimic all aspects of EHEC-induced disease in humans. For example, rabbits exhibit some of the gastrointestinal characteristics of human EHEC-induced disease, including bloody diarrhea, but do not develop HUS.6
Upon interaction with the human intestinal epithelium, EHEC implements two major virulence strategies: production of Shiga toxins (Stx) and formation of attaching and effacing (A/E) lesions on enterocytes.7 A/E lesions are characterized by extensive actin remodeling of the host cell cytoskeleton, leading to effacement of the microvilli and formation of an F-actin pedestal-like structure beneath the bacteria.8 Improved understanding of the basis for EHEC pathogenicity is of increasing importance given the growing number of outbreaks worldwide. An infection model that recapitulates the human pathophysiology of EHEC infection is necessary for the development of therapeutic strategies for these currently untreatable conditions.
Stx is the main virulence factor in EHEC infection; however, Stx’s effects are most potent in the circulatory and renal systems. Other known EHEC virulence factors that act upon the intestinal epithelium include intimin, tir, and the serine protease autotransporters of Enterobacteriaceae (SPATE) family. EspP is a major SPATE secreted by EHEC via the type V secretion system at the early stage of infection.9, 10 Although EspP’s primary functions in EHEC-induced disease are not well understood, previous studies on the SPATE family have reported that they cause host cytotoxicity and cleave actin-bound cytoskeletal proteins, causing massive actin rearrangement.11 Using the intestinal epithelial T84 cell model, we have previously shown that recombinant EspP is sufficient to trigger the described actin remodeling.12 Therefore, we hypothesized that EspP plays a major role in promoting EHEC pathogenicity.
Recent progress in human stem cell biology, particularly the technology to establish and indefinitely propagate an intestinal epithelial culture,13 opens new possibilities for studying EHEC interaction with human intestinal epithelium, the first step in disease development. These cultures, termed enteroids or colonoids, typically grow as three-dimensional (3D) spheres with the apical surface facing inward. They are not ideal for studying the interaction between luminal gut bacteria and epithelial cells because the lumen is not easily accessible. We and others have recently pioneered human enteroid monolayer cultures grown on Transwell filters in which the apical surface faces outward and the basolateral surface is attached to the filter.14, 15 These human monolayer cultures provide an advantage in studying luminal infections and testing strategies for luminally delivered pharmacologic agents to interfere with intestinal epithelial infections.
Here we report the method for establishing colonoid monolayers derived from the human proximal colon as a model of EHEC infection. We show that extracellular mucin 2 (MUC2) and the brush border (BB) resident protein protocadherin 24 (PCDH24) are initial targets of EHEC during infection. We determined that the EHEC virulence factor EspP, specifically its protease activity, is responsible for PCDH24 reduction. We conclude that human colonoid monolayers (HCM) are a suitable model to study EHEC intestinal colonization and to characterize the molecular mechanisms of host-EHEC interactions.
Materials and Methods
All authors had access to the study data and reviewed and approved the final manuscript. All human tissue was obtained with informed consent from healthy individuals at the Johns Hopkins Hospital and coded with no patient identifiers. This study was approved by the Johns Hopkins institutional review board protocol (NA_0038329).
Reagents and Antibodies
Advanced Dulbecco’s modified Eagle medium/Ham’s F12, HEPES, GlutaMAX, B27 supplement minus vitamin A, N2 supplement, epidermal growth factor, Alexa Fluor 568 Phalloidin, 4′,6-diamidino-2-phenylindole, Tris-acetate gradient gels, Tris-glycine gradient gels, and monoclonal antibody against occludin were purchased from Life Technologies (Carlsbad, CA). Penicillin/streptomycin was purchased from Quality Biological (Gaithersburg, MD). Matrigel, Cell Recovery Solution, and the Transwell filter inserts were purchased from Corning (Tewksbury, MA). Jagged-1 and [Leu-15] gastrin were purchased from AnaSpec (Fremont, CA). N-acetylcysteine, prostaglandin E2, collagen IV from human placenta, protease inhibitor cocktail, and polyclonal antibodies against PCDH24, GAPDH, and sodium-hydrogen exchanger regulatory factor 3 (NHERF3) were purchased from Sigma-Aldrich (St. Louis, MO). A83-01 [3-(6-methylpyridin-2-yl)-N-phenyl-4-quinolin-4-ylpyrazole-1-carbothioamide], Y-27632 [4-[(1R)-1-aminoethyl]-N-pyridin-4-ylcyclohexane-1-carboxamide], and CHIR99021 [6-[2-[[4-(2,4-dichlorophenyl)-5-(5-methyl-1H-imidazol-2-yl)pyrimidin-2-yl]amino]ethylamino]pyridine-3-carbonitrile] were purchased from Tocris (Bristol, UK). Primocin was purchased from Invivogen (San Diego, CA). Polyclonal antibody against sodium-hydrogen exchanger isoform 2 (NHE2) was kindly provided by Dr. C. Ming Tse (Johns Hopkins University, Baltimore, MD). Polyclonal antibody against EspP was kindly provided by Dr. Harris Bernstein (National Institutes of Health, Bethesda, MD). Monoclonal antibody against Muc2 and polyclonal antibody against phosphorylated ezrin were purchased from Abcam (Cambridge, MA). IRDye-conjugated secondary antibodies were purchased from Rockland (Limerick, PA). Wnt3A (American Type Culture Collection, Manassas, VA), R-spondin1 (kindly provided by Dr. Calvin Kuo, Stanford University, Stanford, CA), and Noggin16 (kindly provided by Dr. Marcel Bijvelds, Erasmus University, Rotterdam, the Netherlands) cell lines were maintained to produce conditioned media.
Generation and Culturing of Colonoid Monolayers
Proximal colonic crypts isolated from tissue resections or biopsies were processed for colonoid generation as described for small intestinal enteroids.13, 14 Briefly, colonic tissue was collected in sterile phosphate-buffered saline (PBS) and kept on ice. The tissue was then transferred into cold chelating solution (CCS; 5.6 mM Na2HPO4, 8 mM KH2PO4, 96.2 mM NaCl, 1.6 mM KCl, 43.4 mM sucrose, 54.9 mM d-sorbitol, and 0.5 mM DL-dithiothreitol) and finely minced to approximately 0.5-mm pieces using curved surgical scissors. The tissue pieces were transferred to a 15-mL conical tube with fresh CCS and allowed to settle by gravity. The supernatant was removed, fresh CCS was added, and the tissue pieces triturated to simulate washing. This process was performed five times or until the supernatant was completely cleared of debris.
The tissue pieces were then incubated in 10 mM EDTA in CCS for 1 hour at 4°C, during which they were vigorously shaken on an orbital shaker. Afterward, the tissue pieces and supernatant were collected in a 15-mL conical tube, and fetal bovine serum (final concentration of 20%) was added to create buoyancy and allow the released crypts to remain in the supernatant while the tissue pieces settled to the bottom. The crypt-rich supernatant was collected and spun at 300g for 10 minutes to pellet the crypts. The crypt pellet was washed with complete medium (CM; Advanced Dulbecco’s modified Eagle medium/Ham’s F-12, 100 U penicillin/streptomycin, 10 mM HEPES, and 0.2 mM GlutaMAX) and spun down. The crypt pellet was resuspended in Matrigel containing 1 μM Jagged-1 peptide.
Approximately 100 crypts per 50 μL Matrigel were plated into an individual well of a 24-well plate and placed at 37°C for 10–15 minutes to polymerize. Each well received 500 μL of expansion medium (EM) The EM was CM containing 50% v/v Wnt3a conditioned medium, 20% v/v R-spondin-1 conditioned medium, 10% v/v Noggin conditioned medium, 1x B27 supplement minus vitamin A, 1x N2 supplement, 1 mM N-acetylcysteine, 50 ng/mL human epidermal growth factor, 1 μg/mL [Leu-15] gastrin, 500 nM A83-01, 10 μM SB202190 [4-[4-(4-fluorophenyl)-5-pyridin-4-yl-1,3-dihydroimidazol-2-ylidene]cyclohexa-2,5-dien-1-one], 10 nM prostaglandin E2, 100 μg/mL primocin, 10 μM CHIR99021, and 10 μM Y-27632. The EM (without CHIR99021 and Y-27632) was replaced every other day.
Colonoid Expansion, Monolayer Formation and Differentiation
Colonoids were passaged approximately every 7 to 10 days by incubation in Cell Recovery Solution (nonenzymatic proprietary solution purchased from Corning) for 15 to 20 minutes at 4°C with vigorous shaking on an orbital shaker. The colonoids in Matrigel were scraped using a mini-cell-scraper and triturated with a P200 pipet 20–30 times to break colonoids apart into fragments. They were then collected and centrifuged at 300g for 10 minutes, then the pellet was resuspended in Matrigel containing 1 μM Jagged-1 peptide. The pellet was split to generate a minimum of 50 colonoids per well post-split. Each well received 500 μL of EM with CHIR99021 and Y-27632 on the day they were split, but they were refreshed with EM without CHIR99021 and Y-27632 on the following days.
To form colonoid monolayers, Transwell filters (24-well inserts, 0.33 cm2 surface area, 0.4 μM pore polyester membrane) were coated with human collagen IV solution (final concentration of 10 μg/cm2) and incubated at 37°C for a minimum of 2 hours. Per manufacturer’s suggestion, the lyophilized collagen IV was reconstituted with sterile 0.25% acetic acid for a stock concentration of 1 mg/mL. This solution was diluted in sterile, DNAse- and RNAse-free water for plating at a final concentration of 10 μg/cm2. Before plating of colonoid fragments, the collagen-coated Transwell filters were washed three times with CM. Colonoid fragments were obtained by following the passaging procedure. After centrifugation, the pellet was resuspended in EM at a ratio of approximately 50 colonoid fragments per 100 μL EM. We added 100 μL EM/colonoid fragment solution to each Transwell filter and allowed it to settle at 37°C. EM was also added to the well of each filter, representing the basolateral media. Both media were exchanged every other day, and spreading of the colonoids into a two-dimensional monolayer was monitored with a Zeiss AXIO light microscope (Zeiss, Oberkochen, Germany). Confluency of monolayers was determined by transepithelial electrical resistance (TER) measured by the EVOM2 epithelial volt ohmmeter (World Precision Instruments, Sarasota, FL).
Infection of Colonoids
Infection of colonoids was performed with the following strains: 1) EHEC O157:H7 strain EDL933 modified to be Stx-negative, 2) EDL933 espP-deletion mutant (EHECΔespP), 3) EHECΔespP complemented with an espP-expressing plasmid (EHEC + pespP), and 4) EDL933 stcE-deletion mutant (EHECΔstcE). Bacterial strains were grown by shaking in Lysogeny broth (LB) broth at 37°C for 12 hours to the stationary phase, then centrifuging at 200g and resuspending in antibiotic-free EM. The colonoids were infected apically at bacterial concentrations of 106 colony-forming units/mL and incubated at 37°C in 5% CO2 for 4, 8, or 18 hours, during which period the medium was not changed. After the predetermined time point, the colonoids were washed three times with cold PBS and fixed for immunofluorescence or lysed for immunoblotting.
Bacterial Strains, Plasmids, and Factors
In-frame deletion mutants were constructed in the EHEC O157:H7 strain EDL933 Shiga toxin-deleted derivative TUV93-017 by Lambda Red-mediated recombination using linear DNA fragments as described elsewhere.37 Briefly, DNA fragments encoding a kanamycin resistance cassette flanked by Flipase Recognition Target site-inverted repeats and sequences homologous to regions flanking espP or stcE were polymerase chain reaction (PCR) amplified from, respectively, pKD13 and pKD4 using primer sets AH1253/AH1254 and AH1322/AH1323 by the high-fidelity DNA polymerase EasyA (Agilent Technologies, Santa Clara, CA). DNA fragments were electroporated into TUV93-0, where the Red recombinase system was expressed from pKD46. Recombinants were selected and purified on l-agar plates containing 50 μg/mL kanamycin. The kanamycin resistance marker was eliminated from EHEC ΔespP::kan and EHEC ΔstcE::kan using a pCP20-encoded FLP flipase,18 resulting in EHEC ΔespP (AMH 194) and EHEC ΔstcE (AMH 196). Gene deletions of espP and stcE were verified by PCR amplification analysis using primer sets AH1255/AH1256 and AH1324/AH1325, respectively. The complementation plasmid pAMH351 expressing espP from its native promoter was generated by cloning a DNA fragment, which was PCR amplified from a TUV93-0 genomic DNA preparation with high-fidelity DNA polymerase Phusion Flash (Thermo Scientific, Waltham, MA) using primers AH1256/AH1327, into the BamHI/HindIII sites of the low copy number plasmid pSec10. EDL933 T3SS deletion mutant of E. coli–secreted protein A (ΔespA) was constructed by in-frame deletion as we previously described elsewhere.12
The oligonucleotide sequences used were AH1253: 5′-CATTAGAAAAACCAATGTTTCCCTTAAAAATGGAGCTTATGTCCGGGGATCCGTCG ACCT-3′; AH1254: 5′-GAGCGTAAAGGGCCCGCAGGCCCTTTTGAATACGGAGTAGTGTAGGCTGGAGCTG CTTCG-3′; AH1255: 5′-CACTGCGGCCGCTTATTATGCTTCCATCAGAAACGATG-3′; AH1256: 5′-CACTGGATCCTGCCAGCTTTAGTCATCGCAGTTAAG-3′; AH1322: 5′-AATAAAATATAGAAATACTGTTATATCCGGCTGCATGTTGGTGTAGGCTGGAGCTG CTTC-3′; AH1323: 5′-TTATTTATATACAACCCTCATTGACCTAGGTTTACTGAAGCATATGAATATCCTCCT TAG-3′; AH1324: 5′-CATGTAAGCTTGAATGTATCATAATGCAATTGTTTGATGTGTTAAACG-3′; AH1325: 5′-CATGTAAGCTTACGAATGTGTTACTAATGCGGCCGA-3′; and AH1327: 5′-CAGAAAAAGCTTATTATGCTTCCATCAGAAAC-3′.
Genes encoding full-length EspP and serine protease mutant EspP S263A were cloned into the pRLS5 plasmid under the control of an isopropyl β-d-1-thiogalactopyranoside–inducible promoter as previously described elsewhere.19 The plasmids were transduced into the E. coli strain AD202. EspP or EspP S263A was expressed upon isopropyl β-d-1-thiogalactopyranoside induction and precipitated from the supernatant with 60% ammonium sulfate. The recombinant proteins were concentrated by centrifugation, solubilized in PBS, and purified on a gel filtration column. Recombinant EspP and EspP S263A were verified by sequencing and Coomassie blue staining. The final protein concentration was determined by BCA colorimetric assay. Colonoids were treated apically with recombinant EspP or EspP S263A at a maximum concentration of 10 μM for 5 hours. Afterward, cells were washed three times with cold PBS and lysed for immunoblotting.
Immunofluorescence
Colonoid monolayers on filters were fixed with 3% formaldehyde/PBS for 10 minutes, washed three to four times with PBS, permeabilized with 0.1% saponin, and blocked with 2% bovine serum albumin/fetal bovine serum for 30 minutes. For MUC2 labeling, colonoid monolayers were fixed in Carnoy’s solution (90% [v/v] methanol, 10% [v/v] glacial acetic acid). The cells were then immunostained with the following antibodies: MUC2 (1:100), PCDH24 (1:100), fluorescence Alexa Fluor secondary antibody, Hoechst (1 μg/mL), Alexa Fluor 568 Phalloidin (1:200), and fluorescein labeled EHEC antigen (1:100).
The filters were carefully removed by cutting the filter away from the insert, then immersed in gel mount and mounted onto glass slides. Fluorescence confocal imaging was performed using a Zeiss 510 LSM (Zeiss, Oberkochen, Germany). The images were quantitatively and qualitatively analyzed using MetaMorph software (Molecular Devices, Sunnyvale, CA).
Western Blot Analysis
Colonoid monolayers were washed three to four times with cold PBS then carefully dried by inverting the filter insert and tapping to remove excess fluid and to preserve the MUC2-positive mucus layer. The cells were harvested in lysis buffer (60 mM HEPES pH 7.4, 150 mM KCl, 5 mM Na3EDTA, 5 mM EGTA, 1 mM Na3VO4, 50 mM NaF, 1% Triton X-100) with protease inhibitor cocktail (1:100) and disrupted by vigorous pipetting with a P200 tip, followed by flash freeze in liquid N2, then end-over-end rotation at 4°C for 2 hours. One confluent colonoid monolayer consisted of approximately 120 μg of protein. Lysates were further solubilized in sodium dodecyl sulfate gel-loading buffer with β-mercaptoethanol or with dithiothreitol (for MUC2). Lysates were heated at 70°C for 10 minutes, and then the proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on Tris-glycine or Tris-acetate (for MUC2) gels and transferred to nitrocellulose membranes.
After transfer, membranes were blocked in 5% milk/Tris-buffered saline (TBS)-Tween (0.1%) at 25°C for 1 hour, then incubated with primary antibodies at 4°C overnight. The primary antibodies used were occludin (1:250), NHE2 (1:500), NHERF3 (1:250), PCDH24 (1:500), MUC2 (1:500), GAPDH (1:1000), and EspP (1:500). Membranes were washed five times for 5 minutes each with TBS-Tween (0.1%) then incubated with IRDye-conjugated secondary antibodies at 25°C for 1 hour. Membranes were again washed five times for 5 minutes each with TBS-Tween (0.1%) then imaged using a Li-Cor Odyssey system infrared imaging scanner (Li-Cor, Lincoln, NE). Relative fluorescence intensity of the target protein was normalized to the fluorescence intensity of GAPDH, as previously described elsewhere.20
Real-Time Polymerase Chain Reaction
Two colonoid monolayers grown on Transwells were pooled for total RNA extraction. RNA extraction was performed using the PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA). SuperScript III Reverse Transcriptase (Life Technologies) was used to synthesize cDNA. The LGR5 (leucine-rich repeat containing G protein-coupled receptor 5) primer (purchased from Life Technologies) was previously described elsewhere.21 The PCR reactions were performed with Power Sybr Green master mix (Life Technologies) on a QuantStudio 12K Flex (Life Technologies). Relative expression for both undifferentiated and differentiated conditions was normalized to the internal control, RN18S, and averaged over three independent experiments and three separate colonoid lines.
Scanning and Transmission Electron Microscopy
Monolayers were fixed overnight at 4°C and processed for scanning electron microscopy as described elsewhere.20 Briefly, filters were postfixed in 2% osmium tetroxide and 0.1 M sodium cacodylate at 4°C for 1 hour. Filters were dehydrated through a graded series of ethanol and passed through pure hexamethyldisilazane. The drying agent was decanted, and filters were desiccated overnight. Filters were attached to aluminum stubs via carbon sticky tabs and coated with 20-nm AuPd. The stubs were viewed and images captured on a LEO 1530 field emission scanning electron microscope (LEO Electron Microscopy/Carl Zeiss, Thornwood, NY) operating at 1–3 kV.
Monolayers were processed for transmission electron microscopy as previously described elsewhere.12 Briefly, filters were fixed for 2 hours in solution containing 2% glutaraldehyde, 2% paraformaldehyde, 0.1 M Na-cacodylate, 3 mM CaCl2, pH 7.4 at room temperature. Filters were incubated in 1% H2O2 for 1 hour then post-fixed in 2% osmium tetroxide in 0.1 M Na-cacodylate for 1 hours then placed in 2% uranyl acetate for 1 hour. After en-bloc staining, filters were dehydrated through a graded series of ethanol to 100%, transferred through propylene oxide, embedded in Eponate 12 (Ted Pella, Redding, CA) and cured at 60°C for 2 days. Sections of 80 nm were cut on a Reichert Ultracut E (Leica Microsystems, Buffalo Grove, IL) with a Diatome Diamond knife (Diatome, Hatfield, PA) then collected on Formvar coated 1 × 2 mm copper slot grids and stained with uranyl acetate followed by lead citrate. Grids were viewed and captured on a Hitachi 7600 transmission electron microscope (Hitachi High Technologies America, Schaumburg, IL) operating at 80 kV.
Statistical Analysis
Values are presented as mean ± standard error of the mean, with n representing number of independent preparations. Statistical significance was determined using the Student unpaired t test, and P < .05 was considered statistically significant.
Results
Establishment of Human Proximal Colonoid Monolayer Cultures
Cultures of 3D Matrigel-grown colonoids (cultures obtained from five separate donors) were converted into monolayers. Approximately 200 colonoids were recovered from the Matrigel culture and were seeded on Transwell filters (0.33 cm2) coated with human collagen IV (10 μg/cm2). Confluent HCM were achieved approximately 2 weeks after seeding. HCM were differentiated to produce a culture consisting of surface-like colonocytes and goblet cells by withdrawal of WNT3A from the growth medium for 5 days, as has been previously described elsewhere.13, 22 The cell polarization and formation of confluent undifferentiated monolayers were documented by a significant and sustainable increase in transepithelial electrical resistance (TER), while differentiation induced an even greater increase in TER (Table 1). Differentiation led to a significant increase in a number of proteins that are enriched in surface colonocytes and at the top of crypts,23 including occludin, NHE2, NHERF3, and the goblet cell marker MUC2, as shown by immunoblots (Figure 1A). In contrast, there was a significant decrease in the stem cell marker LGR5 (Supplementary Figure 1). The differential expression of the surface and crypt markers were verified in all five proximal colonoid lines generated from different donors throughout the course of this study.
Table 1.
Transepithelial Electrical Resistance of Human Colonoid Monolayers (Ω·cm2)
| Monolayers | TER | No. |
|---|---|---|
| Undifferentiated uninfected | 213 ± 7a | 12 |
| Differentiated uninfected | 1368 ± 61a | 15 |
| Differentiated infected | ||
| 4 h | 1354 ± 86 | 12 |
| 8 h | 1049 ± 118 | 12 |
P < .05.
Figure 1.

Removal of Wnt3A induces differentiation of colonoids. (A) The relative amount of each marker in both undifferentiated and differentiated colonoids normalized to GAPDH as shown in Western blots. *Statistically significant increase, P < .05, n = 3. Protein expression changes show transition to surface colonocyte population upon differentiation. (B) Scanning electron microscope (SEM) images of undifferentiated and differentiated colonoid monolayers at low magnification show the uniform mucus-covered brush border (BB) in differentiated monolayers compared with the sparse and uneven BB of the undifferentiated monolayers. (C) Scanning electron microscope images of undifferentiated and differentiated colonoid monolayers at higher magnification show distinct changes in the BB and microvillar height (lower panel) induced by differentiation.
Supplementary Figure 1.

Differentiation results in a decrease of the stem cell marker leucine-rich repeat containing G protein-coupled receptor 5 (LGR5). Relative LGR5 expression was measured by semiquantitative polymerase chain reaction in three different human colonoid lines (undifferentiated and differentiated for 5 days). LGR5 was normalized to RN18S and relative expression levels from the three colonoid lines were averaged. Average LGR5 expression was 877.4 ± 157.9 and 9.9 ± 3.5 in undifferentiated and differentiated, respectively. *Statistically significant decrease of LGR5 expression, P < .05, n = 5.
Colonoid differentiation was also characterized by significantly enhanced BB organization (see Figure 1B). In undifferentiated HCM, colonocyte microvilli were sparse and variable in height. After differentiation, HCM exhibited tightly packed microvilli (10–15 microvilli per 1 μm of membrane) that were ∼1 μm tall. Intestinal epithelial cell (IEC) differentiation and BB organization are accompanied by the formation of a regular network of intermicrovillar bridges.24 PCDH24 is an essential building block of these bridges and is necessary for the tight packing, clustering, and uniformity of microvillar length during BB assembly. Indeed, PCDH24 expression was significantly increased upon HCM differentiation (see Figure 1A), similar to the surface expression of PCDH24 in human tissue from the proximal colon. Numerous intermicrovillar bridges were detected in the BB of differentiated HCM (see Figure 1B), consistent with the important role of PCDH24 in BB structural organization described by Crawley et al.24 We conclude that HCM recapitulate essential features of the adult human colonic epithelium.
Enterohemorrhagic Escherichia coli Preferentially Colonizes the Differentiated Human Colonoid Monolayers
To determine whether colonic epithelial differentiation plays a role in initial EHEC colonization, we infected both undifferentiated and differentiated HCM with equal concentrations of EHEC (106) for 8 hours, then compared the number of EHEC bacteria associated with the apical surface of differentiated versus undifferentiated HCM. The number of EHEC associated with the surface of differentiated monolayers (Figure 2B) was ∼6-fold higher (see Figure 2D) compared with undifferentiated (see Figure 2A), indicating that differentiation might play an important role in EHEC interaction with host epithelium. To test the specificity of colonic differentiation in EHEC colonization, we infected differentiated human enteroid monolayers derived from jejunal stem cells with the same EHEC concentration for 8 hours and found few bacteria attached (see Figure 2C), indicating that colon-specific differentiation plays an important role in EHEC colonization.
Figure 2.

Enterohemorrhagic Escherichia coli (EHEC) preferentially colonize differentiated colonoid monolayers. (A) Representative confocal three-dimensional projections from images of undifferentiated and (B) differentiated human colonoid monolayers (HCM) infected with 106 EHEC for 8 hours. Phalloidin (red), DNA (blue), EHEC antigen (green). Bars: 10 μM. (C) Representative confocal three-dimensional projection from images of differentiated jejunal monolayers infected with 106 EHEC for 8 hours. Very few EHEC were found attached or associated. Phalloidin (red), EHEC antigen (green). Bar: 10 μM. (D) Quantification of EHEC or EHECΔespA bacteria associated with undifferentiated and differentiated colonic monolayers. Twenty separate fields (each measuring 50 × 50 μm) were counted for EHEC and averaged. EHECΔespA were counted in 12 separate fields (each measuring 50 × 50 μm) and averaged (n = three independent experiments). **P < .001; *P < .05.
One of the striking differences between surface colonic versus villus small intestinal epithelium is the presence of thick, nearly impenetrable luminal mucus.25 Thus, we tested the effect of EHEC infection on colonic mucus.
Human Colonoid Monolayers Produce a Mucus Layer That Is Destroyed by Enterohemorrhagic Escherichia coli Infection
To attach to human IEC, EHEC must penetrate the two layers of colonic mucus: the inner attached layer and the loose (gel) outer layer. Mucus consists primarily of a mixture of large, highly glycosylated proteins called mucins (MUCs), which are secreted by goblet cells. The inner layer consists of transmembrane mucins and the major colonic gel mucin MUC2, and the outer layer mainly contains MUC2.25, 26
Differentiated HCM expressed MUC2-producing goblet cells and formed a ≥25 μm thick layer of MUC2-containing apical mucus (Figure 3A and B), indicating active mucus secretion, comparable to that of the human colon. This mucus layer was absent from undifferentiated HCM or differentiated jejunal monolayers (data not shown), similar to what has been reported in human tissue.25, 26
Figure 3.

Enterohemorrhagic Escherichia coli (EHEC) infection targets the mucus layer in human colonoids. (A, B) Differentiated human colonoid monolayers (HCM) express a thick (>25 μm) inner mucus layer primarily composed of MUC2 (red). DNA (blue). (A) XY projection; (B) XZ projection. (C, D) EHEC infection for 4 hours reduces thickness of the extracellular mucin 2 (MUC2)–positive mucus layer. Remaining MUC2 (red) is intracellular in goblet cells. Arrows point to attached EHEC (green). (C) XY projection; (D) XZ projection . (E–G) Scanning electron microscope images of HCM (E) uninfected and (F, G) EHEC-infected after 6 hours. EHEC infection leads to microvillar effacement and apical membrane perturbation (F) and EHEC attachment to the remaining glycocalyx (G).
We tested the effect of EHEC infection on the amount and distribution of MUC2. Infection of colonoid monolayers with EHEC (106) for 4 hours caused significant reduction of the MUC2-positive mucus layer, with MUC2 restricted mainly to goblet cells (Figure 3C and D) and small patches at the cell surface. The decrease in mucus layer thickness allowed bacteria to reach the apical surface of the colonocytes and colonize the epithelium (see Figure 3C and D). These data suggest that EHEC reduces colonic mucus to increase its proximity to the colonocyte surface, which allows EHEC to directly interact with host IEC.
Scanning electron microscopy further showed that the HCM apical surface is covered with a thick layer of mucus (see Figure 3E). However, 4 hours after infection, the apical surface was perturbed, and EHEC were attached (see Figure 3F). In addition, we observed a tight association of EHEC with patches of the remaining mucus layer by hundreds of threadlike structures (see Figure 3G), likely corresponding to the reported bacterial fibrillar network.27 These data suggest that the colonic mucus layer may promote the initial colonization of the host epithelium.
We next investigated how EHEC destroys the mucus layer and gains access to the HCM apical surface. It was previously suggested that the zinc metalloprotease StcE, secreted by EHEC O157:H7, contributes to intimate adherence of bacteria to host cells by cleavage of heavily glycosylated mucin layer proteins, thereby allowing the pathogen to come into close proximity with the host cell membrane.28 However, StcE was shown to specifically target orally expressed MUC7,29 and its role in the reduction of MUC2-positive mucus in nontransformed human colonic epithelial cells is unknown.29 Therefore, we tested whether StcE contributes to EHEC-induced reduction of the MUC2-positive mucus layer in HCM.
Infection with an isogenic EHEC strain deleted for stcE (EHECΔstcE) resulted in a mucus layer reduction (Figure 4A), suggesting that StcE is not essential for MUC2 reduction or is functionally redundant. Numerous bacteria were associated with the mucus-free apical surface, and MUC2 was detected only in small patches at the cell surface (see Figure 4A), similar to what we observed upon infection with the parental EHEC strain (see Figure 3C and D).
Figure 4.
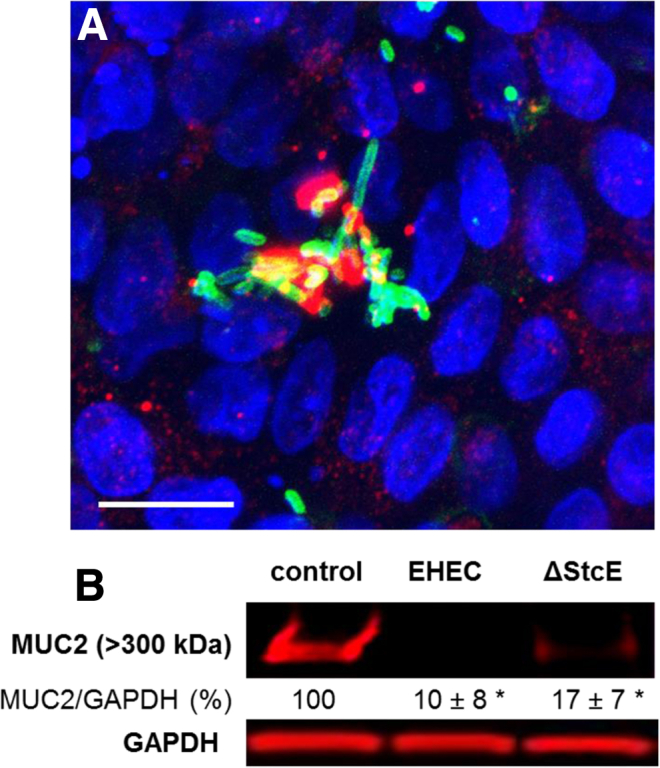
Enterohemorrhagic Escherichia coli (EHEC) ΔstcE is able to degrade the human colonoid monolayers (HCM) mucus layer. (A) Representative confocal three-dimensional projection from images of EHECΔstcE-infected HCM shows that EHECΔstcE infection does not prevent the extracellular mucin 2 (MUC2)–positive mucus layer reduction. Remaining MUC2 (red) resides intracellularly in goblet cells. EHEC antigen (green), DNA (blue). Bar: 10 μM. (B) MUC2 expression normalized to GAPDH is significantly decreased (*P < .05) in EHEC and EHECΔstcE infected colonoids compared with uninfected.
Enterohemorrhagic Escherichia coli Damages the Brush Border of Human Colonocytes Beyond the Areas of Attaching and Effacing Lesions
In areas of complete mucus reduction there was significant damage to BB organization, with a substantial decrease in the amount and height of microvilli (Figure 5A). With mucus reduction, EHEC is able to efface microvilli, forming characteristic A/E lesions (see Figure 5B) that are not seen on uninfected HCM (see Figure 5D). The sites of bacterial attachment were accompanied by characteristic F-actin pedestals (see Figure 5C), which were also detected by scanning electron microscopy (see Figure 5A, inset).
Figure 5.

Enterohemorrhagic Escherichia coli (EHEC) forms attaching and effacing (A/E) lesions and actin pedestals while also remodeling the basolateral membranes. (A) Scanning electron microscope and (B) transmission electron microscope (TEM) images of EHEC-infected differentiated human colonoid monolayers (HCM) after 8 hours. EHEC effaces the microvilli and attaches via actin pedestals (arrows, inset of A). (C) EHEC attaches to the apical surface of HCM via F-actin pedestals. Note the overlapping actin with EHEC (arrow). F-actin (red), EHEC antigen (green), DNA (blue). (D, E) TEM images of (D) uninfected differentiated HCM and (E) EHEC-infected differentiated HCM show that apical infection with EHEC causes membrane perturbation and actin remodeling of both apical and basolateral membranes. Note that the apical tight junctions were less affected by EHEC infection.
To show the significance of the T3SS in EHEC colonization of HCM, we used the EHECΔespA mutant, which lacks T3SS activity, as we have described elsewhere.12 EspA encodes the translocation filament that delivers multiple T3SS effectors, including the intimin receptor tir, into host epithelial cells.1 Infection of HCM with EHECΔespA significantly decreased the number of attached bacteria compared with infection with parental EHEC (see Figure 2D), similarly to EHEC infection of human colonic biopsies.30
Importantly, the damage to the epithelial monolayer was much broader and was not restricted to the sites of A/E lesions. Overall, the whole monolayer was significantly damaged, including the apical and lateral membranes (see Figure 5E), further confirming observations by ourselves and others that EHEC-induced damage of intestinal cells is not limited to cells that directly interact with EHEC through A/E lesions.31
Enterohemorrhagic Escherichia coli Targets the Protocadherin 24–Containing Intermicrovillar Bridges
A signature of EHEC infection is the loss of microvilli, which occurs throughout the epithelium and not only in places of bacterial A/E.31 However, the mechanism for BB effacement is poorly understood. It has been shown that loss of PCDH24, a major building block of intermicrovillar bridges, gives rise to BBs that appear disorganized, sparse, and have a highly variable microvillar length,24 similar to what we found with EHEC infection (see Figure 5A). We thus hypothesized that the loss of PCDH24 and intermicrovillar bridges may be an initial step in EHEC-induced global BB damage.
We first determined the expression pattern of PCDH24 in human colonic tissue and tested whether differentiated HCM express PCDH24 in a similar pattern. PCDH24 expression was very much restricted to terminally differentiated surface cells and was not detected in the upper crypts (Figure 6A). Similarly, PCDH24 expression in differentiated HCM was detected in patches of cells, likely representing the differentiated surface colonocytes in the monolayer (see Figure 6B) with the PCDH-negative cells representing the upper crypt cells. PCDH24 was expressed mainly at the apical surface of epithelial cells, including the cell perimeter.
Figure 6.

EspP significantly decreases the amount of protocadherin 24 (PCDH24) and remodels the brush border (BB). (A) Representative image of normal human colonic tissue immunostained for PCDH24 shows that PCDH24 is localized to the apical surface of colonocytes and to the brush border. Inset shows a zoomed-in section of the BB. PCDH24 (green), DNA (blue). (B, C) PCDH24 expression on (B) uninfected and (C) enterohemorrhagic Escherichia coli (EHEC)–infected (8 hours) human colonoid monolayers (HCM). PCDH24 is localized to the apical membrane of uninfected HCM and in intracellular puncta of infected HCM. PCDH24 (green), F-actin (red), EHEC antigen (pink), DNA (blue). XY projection (top panels), XZ projection (bottom panels).
We assessed the effect of EHEC infection on expression and distribution of PCDH24 in HCM. After infection with EHEC (106) for 8 or 18 hours, PCDH24 protein expression was significantly reduced (Figure 7B and Supplementary Figure 2). Immunostaining of EHEC-infected monolayers also showed significant rearrangement of BB and lateral membrane actin, and redistribution of PCDH24 from the BB and the subapical compartment into intracellular vesicles (see Figure 6C). However, expression levels of mucin-like protocadherin (MLPCDH), a PCDH24 binding partner, which together compose the intermicrovillar links,24 were unaffected by EHEC, even after 18 hours of infection (Supplementary Figure 3). Immunostaining of EHEC-infected monolayers showed that MLPCDH is partially redistributed from the BB to the cytosol in a disperse pattern (Supplementary Figure 3). These data indicate that EHEC specifically targets PCDH24 to cause microvillar damage.
Figure 7.

EspP significantly decreases the amount of protocadherin 24 (PCDH24) and remodels the BB. (A) EspP production was determined by Western blot analysis using a polyclonal EspP antiserum. EspP was detected in overnight cultures of wild-type enterohemorrhagic Escherichia coli (EHEC) (lane 1), and the EHECΔespP strain complemented with a plasmid expressing espP (lane 3), but not in the culture of the EHECΔespP strain (lane 2). (B) Expression of PCDH24 normalized to GAPDH is quantitated after infection by wild-type EHEC, mutant strain EHECΔespP, or complemented espP mutant strain (pespP), and after treatment (5 hours) by purified recombinant EspP or EspP S263A. *Statistically significant decrease compared with control (P < .05, n ≥ 3 per each condition). (C) Infection with wild-type EHEC, but not EHECΔespP, causes apical membrane perturbation and actin remodeling of the BB. F-actin (red), EHEC antigen (green), DNA (blue).
Supplementary Figure 2.

Confirmation of protocadherin 24 (PCDH24) reduction by EspP. PCDH24 expression is significantly decreased with enterohemorrhagic Escherichia coli (EHEC) or EHECΔstcE, but minimally with EHECΔespP infection. Lower molecular weight, or cleaved, forms of PCDH24 are not detected, possibly due to the lack of antigen recognition by the antibody used.
Supplementary Figure 3.

Mucin-like protocadherin (MLPCDH), a protocadherin 24 (PCDH24) binding partner, is mislocalized, but protein expression level does not change upon enterohemorrhagic Escherichia coli (EHEC) infection. Representative immunoblot of MLPCDH in control and infected human colonoid monolayers. The MLPCDH expression level does not change upon EHEC infection (up to 18 hours) or treatment with recombinant EspP (10 μM, 60 minutes). (A) Representative image of a differentiated colonoid monolayer immunostained for MLPCDH (red, left panel), actin (phalloidin, white, middle panel), and overlayed in the right panel; DNA (Hoechst, blue). Note that MLPCDH is mainly on the apical surface and overlaps with the actin pattern. (B) Representative image of an EHEC-infected differentiated colonoid monolayer immunostained for actin (phalloidin, white), MLPCDH (red), EHEC antigen (green), and DNA (Hoechst, blue). Overlay in the XY plane is shown on the left panel. XZ planes (right panels) show that MLPCDH becomes mislocalized from the BB into the cytosol.
Enterohemorrhagic Escherichia coli Serine Protease EspP Targets Protocadherin 24 and Causes Brush Border Effacement
Based on our previous study in which we found that the EHEC serine protease EspP is essential for colonic BB actin remodeling,12 we hypothesized that EspP is responsible for PCDH24 reduction and EHEC-induced microvillar effacement. Indeed, infection of HCM with an isogenic EHEC strain deleted for espP (EHECΔespP, 106) for 18 hours did not cause a decrease in the amount of PCDH24 (see Figure 7B). Moreover, although wild-type EHEC caused F-actin blebbing and severe damage to the BB and apical surface, infection with EHECΔespP did not cause any gross damage to the BB (see Figure 7C), and the monolayer remained virtually intact. The absence of severe damage to microvilli in EHECΔespP-infected monolayers was not due to fewer attached bacteria because bacterial counts from nine random fields of view from infected monolayers (n = 3) showed that there were 56 ± 12/580 μm2 and 51 ± 14/580 μm2 EHEC and EHECΔespP bacteria associated with the host cells, respectively. Complementation of the EHECΔespP strain with a plasmid expressing the espP gene (see Figure 7A) restored the bacterial EspP protein expression to levels detected in the parental EHEC strain and also restored the ability of the bacteria to target PCDH24 (see Figure 7B), supporting our hypothesis that EspP is involved in reduction of intermicrovillar bridges.
To further assess the role of EspP in PCDH24 reduction and BB effacement, HCM were treated apically with purified recombinant EspP. Recombinant EspP (added at a rate of 5 μg/hours) reduces PCDH24 in a time-dependent manner (data not shown) with complete PCDH24 reduction seen after 5 hours (see Figure 7B). Importantly, the serine protease activity of EspP was necessary for PCDH24 reduction because the serine protease deficient mutant, EspP S263A, was unable to cleave PCDH24 (see Figure 7B). These results show that the EHEC serine protease EspP contributes to BB effacement by promoting reduction of the intermicrovillar protein PCDH24.
Enterohemorrhagic Escherichia coli Infection Affects the Integrity of the Tight Junctions
It has been previously shown that EHEC infection caused significant decrease in TER and redistribution of several tight junction (TJ) proteins, including occludin.32 Thus, we analyzed the effect of EHEC infection on occludin distribution in colonoid monolayers. In differentiated uninfected HCM, occludin was mainly present at the TJ (Figure 8A–C). In contrast, occludin was redistributed from TJ into the cytosol of HCM infected with EHEC for 18 hours (see Figure 8D–F). EHEC infection also decreased the TER of colonoid monolayers (Table 1), although the TER decreases occurred 8 hours after infection; indicating that some claudins may be affected by EHEC, correlating with a previous study that found claudin-2 expression increases and causes increased permeability after EHEC infection.32 Thus, our data from EHEC-infected human colonoid monolayers are in good agreement with previously published in vitro and in vivo animal models.33, 34
Figure 8.

Enterohemorrhagic Escherichia coli (EHEC) causes redistribution of occludin from tight junctions to cytosolic puncta. (A–C) Occludin is localized to the lateral membrane of uninfected differentiated human colonoid monolayers (HCM). Occludin (red), DNA (blue). (A, B) XY projection; (C) XZ projection. (D–F) EHEC infection (18 hours) of HCM causes occludin redistribution to cytosolic puncta and significant loss from the tight junction. Occludin (red), EHEC antigen (green), DNA (blue). (D, E) XY projection; (F) XZ projection.
Discussion
We have described an ex vivo human model for studying intestinal host–pathogen interactions using stem cell–derived HCM established from the proximal colon, the site of EHEC disease in humans. These newly characterized HCM provide a great advantage for the study of luminal enteric infections compared to 3D Matrigel-embedded cultures because each 3D colonoid is composed of a variable number of cells and, owing to the topography, the apical side is not directly accessible. Additionally, each 3D colonoid lumen contains an unknown volume of fluid that is subject to sporadic expelling into the medium, jeopardizing controlled measurements of host–pathogen interactions or final luminal concentration of the microbes, toxins, or pharmacologic agents.
Monolayers allow for controlled access to both apical and basolateral surfaces, thus facilitating highly reproducible measurements of EHEC–colonic epithelial interactions. It was recently shown that human intestinal enteroid monolayers are a suitable model for EHEC infection.15 However, with the exception of EHEC association with the ileal and rectal cultures, no other aspects of EHEC-induced pathologies were analyzed. Here, we provide the analysis of EHEC-induced human proximal colonic disease at early times of infection (0 to 8 hours), which has never been described in patients due to the asymptomatic phase of disease at this stage.
We determined that HCM can be differentiated in a manner similar to the established protocols for 3D colonoid cultures.13, 22 Our data indicate that colonic differentiation significantly facilitates EHEC colonization. Impaired bacterial colonization in the absence of mucus, as observed with undifferentiated colonoids and differentiated jejunal monolayers, suggests that mucus may serve as an initial anchor and a vital source of energy for EHEC, contributing to survival in the colonic lumen.
Our data show that the MUC2-positive inner mucus layer, which creates a protective barrier between luminal content, commensal bacteria, and the colonic epithelium, is rapidly (<6 hours) reduced by EHEC. The role of human colonic mucus in EHEC colonization is just emerging. Recently it was shown that EHEC pathogenicity and metabolism is positively modulated by consumption of fucose derived from the mucus layer of transformed intestinal epithelial HT-29 cells.35 These data are supported by studies from the interaction of EHEC with bovine intestine. EHEC rapidly cleaves and consumes carbohydrates from bovine mucus, which promotes EHEC growth in the bovine intestine that enables EHEC to compete with commensal E. coli.36 It has yet to be determined whether EHEC glycoside hydrolases, which catabolize monosaccharides and disaccharides of bovine mucus,36 are similarly involved in human colonic mucus degradation. Interestingly, the EHEC O157:H7 flagella has been found to possess adhesive properties and to bind to bovine mucus, particularly the mucins MUC1 and MUC2.37 This interaction between the colon’s inner mucus layer and the bacterial H7 flagella may represent an early step of EHEC attachment to the human epithelium before intimate attachment by the T3SS and may contribute to colonization.
The most recognized feature of EHEC infection is A/E lesions of the microvilli. Here, using transmission electron microscopy, scanning electron microscopy, and confocal microscopy, we showed the formation of A/E lesions on infected HCM surfaces. However, the epithelial damage was not restricted solely to areas of EHEC attachment; rather, it was a common overall feature of infected HCM. This indicates that, in addition to the T3SS, EHEC use other mechanisms of epithelial damage, including damage to the BB and the TJ protein occludin. Our observations are in agreement with a recent discovery that the tips of adjacent microvilli are connected by bridges composed of PCDH24 and MLPCDH heterodimers, which play a critical role in assembly and organization of BB.24 These proteins are readily exposed to luminal contents, including bacterial virulence factors. Interestingly, the EHEC-secreted serine protease EspP is responsible for the reduction in PCDH24 but not MLPCDH, leading to destruction of the bridges and BB disorganization.
The importance of bacterial SPATEs secreted by the type V secretion system in promoting Shiga toxin-producing E. coli colonization is not well understood. Sequence analyses of the Shiga toxin-producing E. coli strain of enteroaggregative E. coli (EAEC) O104:H4 that caused the severe 2011 outbreak in Germany revealed that it encodes three serine proteases: SepA, SigA, and Pic.38 The importance of these proteases in enteroaggregative E. coli–induced disease is controversial, with one study suggesting that they are dispensable for intestinal colonization,39 while another reporting that these proteases are essential for IEC colonization and Stx translocation.40 We and others have previously shown that the EHEC serine protease EspP is a powerful virulence factor that plays an important role in EHEC pathogenesis9 by causing microvillar remodeling and formation of F-actin–coated macropinosomes.12 However, the mammalian targets of EspP that lead to BB remodeling are not well defined. We now show that active EspP is necessary and sufficient for initial microvillar effacement though the reduction in PCDH24. Whether EspP cleaves PCDH24 directly or acts by an indirect mechanism remains to be determined.
In conclusion, HCM provide an ideal, pathophysiologically relevant model to study the early molecular events in intestinal epithelial colonization by EHEC; an area of study that has been limited due to the use of transformed colonic lines or animal models. Mucins, particularly MUC2 and the BB protein PCDH24, are two initial targets of EHEC in host colonization and microvillar effacement. The use of HCM for further host–EHEC studies will likely provide new targets of EHEC infection.
Acknowledgments
The authors thank the Kudsi Imaging Facility and the Integrated Physiology Core of the Conte Digestive Disease Basic & Translational Research Core Center at Johns Hopkins University and the image processing assistance of John Gibas; Gunnar Hansson (U. Gothenburg) for helpful discussions and advice on mucins; and Michael Delannoy and Tyler Stephens (Johns Hopkins) for their assistance with expansion medium processing.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding This study was funded by National Institutes of Health grants U18-TR000552, T32-DK007632, R24-DK064388, P01-DK072084, and P30-DK089502 and the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Supplementary Material
References
- 1.Karmali M.A., Gannon V., Sargeant J.M. Verocytotoxin-producing Escherichia coli (VTEC) Vet Microbiol. 2010;140:360–370. doi: 10.1016/j.vetmic.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen Y., Sperandio V. Enterohemorrhagic E. coli (EHEC) pathogenesis. Front Cell Infect Microbiol. 2012;2:90. doi: 10.3389/fcimb.2012.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldwater P.N., Bettelheim K.A. Treatment of enterohemorrhagic Escherichia coli (EHEC) infection and hemolytic uremic syndrome (HUS) BMC Med. 2012;10:12. doi: 10.1186/1741-7015-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacheco A.R., Sperandio V. Shiga toxin in enterohemorrhagic E. coli: regulation and novel anti-virulence strategies. Front Cell Infect Microbiol. 2012;2:81. doi: 10.3389/fcimb.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kovbasnjuk O., Mourtazina R., Baibakov B. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc Natl Acad Sci USA. 2005;102:19087–19092. doi: 10.1073/pnas.0506474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sjogren R., Neill R., Rachmilewitz D. Role of shiga-like toxin I in bacterial enteritis: comparison between isogenic Escherichia coli strains induced in rabbits. Gastroenterology. 1994;106:306–317. doi: 10.1016/0016-5085(94)90587-8. [DOI] [PubMed] [Google Scholar]
- 7.Kaper J.B., Nataro J.P., Mobley H.L.T. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 8.Croxen M.A., Finlay B.B. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol. 2010;8:26–38. doi: 10.1038/nrmicro2265. [DOI] [PubMed] [Google Scholar]
- 9.Brockmeyer J., Bielaszewska M., Fruth A. Subtypes of the plasmid-encoded serine protease EspP in shiga toxin-producing Escherichia coli: distribution, secretion, and proteolytic activity. Appl Environ Microbiol. 2007;73:6351–6359. doi: 10.1128/AEM.00920-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Frey E., MacKenzie A.M.R. Human response to Escherichia coli O157:H7 infection: antibodies to secreted virulence factors. Infect Immun. 2000;68:5090–5095. doi: 10.1128/iai.68.9.5090-5095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss A., Brockmeyer J. Prevalence, biogenesis, and functionality of the serine protease autotransporter EspP. Toxins. 2012;5:25–48. doi: 10.3390/toxins5010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.In J., Lukyanenko V., Foulke-Abel J. Serine protease EspP from enterohemorrhagic Escherichia coli is sufficient to induce shiga toxin macropinocytosis in intestinal epithelium. PLoS One. 2013;8:e69196. doi: 10.1371/journal.pone.0069196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato T., Stange D.E., Ferrante M. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 14.Foulke-Abel J., In J., Kovbasnjuk O. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 2014;239:1124–1134. doi: 10.1177/1535370214529398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanDussen K.L., Marinshaw J.M., Shaikh N. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64:911–920. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heijmans J., van Lidth de Jeude J.F., Koo B.K. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013;3:1128–1139. doi: 10.1016/j.celrep.2013.02.031. [DOI] [PubMed] [Google Scholar]
- 17.Donohue-Rolfe A., Kondova I., Oswald S. Escherichia coli O157:H7 strains that express shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J Infect Dis. 2000;181:1825–1829. doi: 10.1086/315421. [DOI] [PubMed] [Google Scholar]
- 18.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabady R.L., Peterson J.H., Skillman K.M. An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc Natl Acad Sci USA. 2004;102:221–226. doi: 10.1073/pnas.0406055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lukyanenko V., Malyukova I., Hubbard A.L. Enterohemorrhagic Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis and transcytosis across intestinal epithelial cells. Am J Physiol Cell Physiol. 2011;301:C1140–C1149. doi: 10.1152/ajpcell.00036.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snippert H.J., van Es J.H., van den Born M. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology. 2009;136:2187–2194.e1. doi: 10.1053/j.gastro.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Jung P., Sato T., Merlos-Suárez A. Isolation and in vitro expansion of human colonic stem cells. Nat Rev. 2011;17:1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 23.Kosinski C., Li V.S.W., Chan A.S.Y. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci USA. 2007;104:15418–15423. doi: 10.1073/pnas.0707210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crawley Scott W., Shifrin David A., Grega-Larson Nathan E. Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell. 2014;157:433–446. doi: 10.1016/j.cell.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johansson M.E.V., Sjövall H., Hansson G.C. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10:352–361. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ermund A., Schutte A., Johansson M.E.V. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer’s patches. Am J Physiol Gastrointest Liver Physiol. 2013;305:G341–G347. doi: 10.1152/ajpgi.00046.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rendon M.A., Saldana Z., Erdem A.L. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc Natl Acad Sci USA. 2007;104:10637–10642. doi: 10.1073/pnas.0704104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grys T.E., Siegel M.B., Lathem W.W. The StcE protease contributes to intimate adherence of enterohemorrhagic Escherichia coli O157:H7 to host cells. Infect Immun. 2005;73:1295–1303. doi: 10.1128/IAI.73.3.1295-1303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grys T.E., Walters L.L., Welch R.A. Characterization of the StcE protease activity of Escherichia coli O157:H7. J Bacteriol. 2006;188:4646–4653. doi: 10.1128/JB.01806-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis S.B., Cook V., Tighe R. Enterohemorrhagic Escherichia coli colonization of human colonic epithelium in vitro and ex vivo. Infect Immun. 2015;83:942–949. doi: 10.1128/IAI.02928-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yashunsky V., Kharilker L., Zlotkin-Rivkin E. Real-time sensing of enteropathogenic E. coli-induced effects on epithelial host cell height, cell-substrate interactions, and endocytic processes by infrared surface plasmon spectroscopy. PLoS One. 2013;8:e78431. doi: 10.1371/journal.pone.0078431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roxas J.L., Koutsouris A., Bellmeyer A. Enterohemorrhagic E. coli alters murine intestinal epithelial tight junction protein expression and barrier function in a Shiga toxin independent manner. Lab Invest. 2010;90:1152–1168. doi: 10.1038/labinvest.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohawk K.L., O’Brien A.D. Mouse models of Escherichia coli O157:H7 infection and shiga toxin injection. J Biomed Biotechnol. 2011;2011:1–17. doi: 10.1155/2011/258185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shifflett D.E., Clayburgh D.R., Koutsouris A. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- 35.Pacheco A.R., Curtis M.M., Ritchie J.M. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492:113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertin Y., Chaucheyras-Durand F., Robbe-Masselot C. Carbohydrate utilization by enterohaemorrhagic Escherichia coli O157:H7 in bovine intestinal content. Environ Microbiol. 2013;15:610–622. doi: 10.1111/1462-2920.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erdem A.L., Avelino F., Xicohtencatl-Cortes J. Host protein binding and adhesive properties of H6 and H7 flagella of attaching and effacing Escherichia coli. J Bacteriol. 2007;189:7426–7435. doi: 10.1128/JB.00464-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasko D.A., Webster D.R., Sahl J.W. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med. 2011;365:709–717. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munera D., Ritchie J.M., Hatzios S.K. Autotransporters but not pAA are critical for rabbit colonization by Shiga toxin-producing Escherichia coli O104:H4. Nat Commun. 2014;5:3080. doi: 10.1038/ncomms4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boisen N., Hansen A.M., Melton-Celsa A.R. The presence of the pAA plasmid in the German O104:H4 shiga toxin type 2a (Stx2a)-producing enteroaggregative Escherichia coli strain promotes the translocation of Stx2a across an epithelial cell monolayer. J Infect Dis. 2014;210:1909–1919. doi: 10.1093/infdis/jiu399. [DOI] [PMC free article] [PubMed] [Google Scholar]