

Abstract
Background
Transgelin is an actin-binding protein that promotes motility in normal cells. Although the role of transgelin in cancer is controversial, a number of studies have shown that elevated levels correlate with aggressive tumor behavior, advanced stage, and poor prognosis. Here we sought to determine the role of transgelin more directly by determining whether experimental manipulation of transgelin levels in colorectal cancer (CRC) cells led to changes in metastatic potential in vivo.
Methods
Isogenic CRC cell lines that differ in transgelin expression were characterized using in vitro assays of growth and invasiveness and a mouse tail vein assay of experimental metastasis. Downstream effects of transgelin overexpression were investigated by gene expression profiling and quantitative PCR.
Results
Stable overexpression of transgelin in RKO cells, which have low endogenous levels, led to increased invasiveness, growth at low density, and growth in soft agar. Overexpression also led to an increase in the number and size of lung metastases in the mouse tail vein injection model. Similarly, attenuation of transgelin expression in HCT116 cells, which have high endogenous levels, decreased metastases in the same model. Investigation of mRNA expression patterns showed that transgelin overexpression altered the levels of approximately 250 other transcripts, with over-representation of genes that affect function of actin or other cytoskeletal proteins. Changes included increases in HOOK1, SDCCAG8, ENAH/Mena, and TNS1 and decreases in EMB, BCL11B, and PTPRD.
Conclusions
Increases or decreases in transgelin levels have reciprocal effects on tumor cell behavior, with higher expression promoting metastasis. Chronic overexpression influences steady-state levels of mRNAs for metastasis-related genes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-016-2105-8) contains supplementary material, which is available to authorized users.
Keywords: Transgelin, Colorectal cancer, Experimental metastasis, Gene regulation, Invasiveness, Biomarker
Background
Colorectal cancer (CRC) is a leading cause of cancer death worldwide. Although early-stage, localized CRC is often curable by surgical resection, some of these patients will experience recurrent, metastatic disease. Currently, the best predictor of risk is lymph node status. There is, however, considerable interest in identifying mechanistically based, molecular markers to improve the ability to forecast individual risk of disease recurrence. In a prior study, we sought to identify such markers by proteomic analysis of samples obtained by laser capture micro-dissection of tumor tissue from node-negative and node-positive patients. Of these, transgelin, a 23 kDa actin binding protein, ranked the highest in a statistical analysis [1].
Transgelin, also known as SM22α, is an abundant protein in normal tissue [2]. It promotes podosome formation [3] and contributes to cell motility in response to injury or inflammation [4, 5]. Transgelin knockout mice are viable and fertile [6], but exhibit reduced smooth muscle contractility [7, 8] and enhanced atherogenesis in a susceptible background [9].
Perhaps because of its abundance, transgelin has been frequently identified in proteomic profiling studies of cancer. Early studies showed that expression is down-regulated in early-stage cancer models [10, 11], leading to the idea that transgelin is a tumor suppressor (reviewed in [12]). However, proteomic studies of transgelin in human cancers often indicate up-regulation in aggressive, late-stage disease [13–16]. Several studies, although not all, have shown a correlation between higher tumor transgelin levels and aggressive behavior, advanced stage, or poor prognosis [1, 17–21]. Consistent with this, various studies have suggested an influence of transgelin on motility or invasiveness in cell-based models [1, 22, 23]. A recent review summarizes current understanding of transgelin in normal tissue and malignancy [24].
One hypothesis that fits with most of the experimental and clinical data is that transgelin is not a marker of cancer per se, but rather a marker of metastatic potential in advanced disease. One way that transgelin may influence metastasis is through direct interaction with cytoplasmic actin. There is also evidence that manipulation of transgelin expression levels affects the expression of other metastasis-related genes [1], suggesting a possible dual mechanism of action.
Here, we adopt an approach based on comparison of isogenic CRC cell populations that differ in transgelin expression but are otherwise identical. We generated new isogenic cell pairs, in which low transgelin-expressing CRC cell lines were transfected with a transgelin cDNA vector to create high-expressing derivatives. We also further characterized a previously created isogenic cell pair, in which high transgelin-expressing cells were stably transfected with miRNA to attenuate transgelin expression. Overexpression in low-expressing cells and attenuation in high-expressing cells had reciprocal effects on cell behavior. In addition, comprehensive gene expression profiling showed that increasing transgelin expression in a low-expressing background led to changes in expression of ~250 other mRNAs. Thus, experimental manipulation of transgelin levels leads to wide-scale transcriptional reprogramming.
Results
Establishment of a new transgelin overexpression cell model
Previously, we described stable transfection of HCT116 and SW480 CRC cells with transgelin miRNA to create two independent cell line pairs that differ in transgelin protein and mRNA levels [1]. In both cases, lower transgelin expression was associated with reduced motility, invasiveness, and resistance to anoikis. This phenotype could furthermore be reversed by transfection with miRNA-resistant transgelin cDNA [1].
To extend and confirm the results of the transgelin knockdown experiments, we tested a complementary approach, creating a new isogenic cell line model in which transgelin was overexpressed in CRC cells that express endogenous transgelin at very low levels. We transfected RKO CRC cells, in which endogenous transgelin is nearly undetectable, with a transgelin cDNA and selected stable transfectants. After selection, virtually all cells expressed a fluorescent transfection marker (Fig. 1a). Immunoblotting showed high levels of transgelin protein in cells that received the transgelin cDNA (RKOTAGLN), whereas transgelin remained undetectable in cells that received an empty control vector (RKOCTRL) (Fig. 1b). Measurement of the relative levels of transgelin mRNA by qPCR showed an increase of about 25-fold (Fig. 1c). The overexpressed transgelin was distributed primarily in the cytoplasm, as indicated by immunofluorescence staining (Fig. 1d).
Fig. 1.
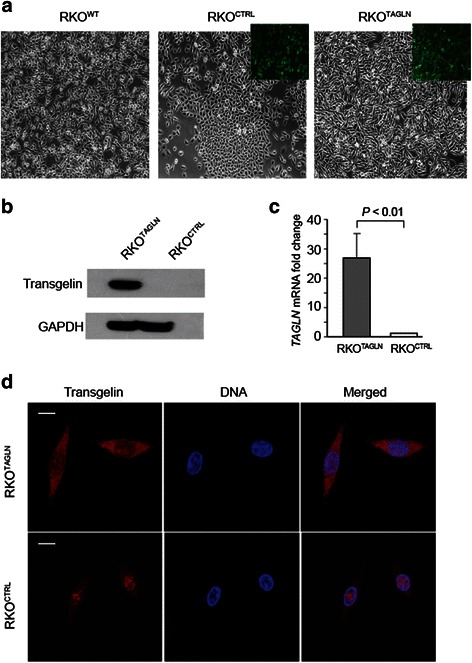
Establishment of transgelin-overexpressing RKO cell lines. a. Phase contrast images of unmodified RKO cells (RKOWT) and derivatives stably transfected with empty vector (RKOCTRL) or with transgelin cDNA (RKOTAGLN). Inset, confocal fluorescence imaging showing the transfection marker, EmGFP. b. Immunoblot analysis of total cell extracts from RKOCTRL or RKOTAGLN cells. c. Fold change in transgelin mRNA expression as determined by real-time PCR analysis, normalized to GAPDH. Mean of three experiments, error bars denote standard deviation. P value determined by Student’s t-test. d. Anti-transgelin immunostaining of RKOCTRL or RKOTAGLN cells. Cells were counterstained for DNA with DAPI; merged image is indicated. Scale bar, 5 μm
Effects of transgelin on invasiveness, clonogenicity, and anchorage-independent growth
We investigated the phenotype of the newly created RKO cell pair using in vitro assays. Transgelin overexpression led to a 2 to 3-fold increase in invasiveness in a Transwell assay (Fig. 2a). There was also an increase in the ability to form colonies when plated at low density (Fig. 2b), and in the number and size of colonies in a soft-agar growth assay (Fig. 2c). Differences were highly significant in all three assays (P < 0.01). Interestingly, transgelin expression had essentially no effect on growth rate or cell cycle distribution under standard cell culture conditions (Fig. 2d, e), suggesting that transgelin expression primarily affects behaviors relevant to metastasis (such as invasiveness, clonogenicity, and anchorage-independent growth) rather than growth per se.
Fig. 2.
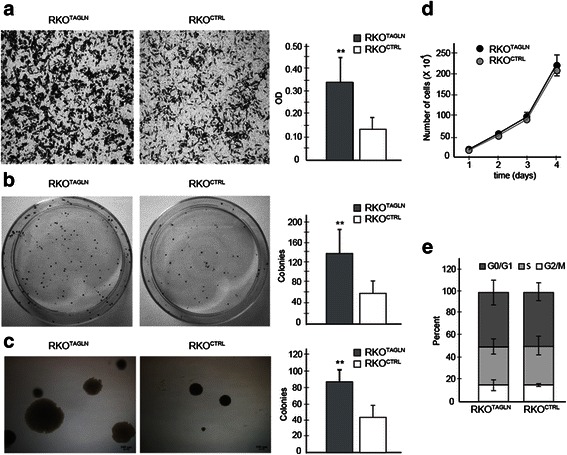
Effects of TAGLN overexpression on in vitro cell behavior. a. Invasiveness. Left, representative images showing invasion of RKOTAGLN and RKOCTRL cells through Matrigel-coated Transwell filters, right, quantification of filter staining. b. Clonogenicity. Left, representative images of plates seeded with RKOTAGLN or RKOCTRL cells, right, quantification of colony formation after 12 days. c. Growth in soft agar. Left, representative images of colonies formed by RKOTAGLN and RKOCTRL cells, right, quantification colony formation after 17 days. d. Cell proliferation. Graph shows cell count in replicate cultures of RKOTAGLN and RKOCTRL, counted daily for four days. Graphs in panels a-d show mean of three experiments. Error bars denote standard deviation. e. Cell cycle distribution. Graph shows the percentage of RKOTAGLN and RKOCTRL cells in G0/G1, S, and G2/M phases of the cell cycle. Data are mean of technical replicates from a single representative experiment. Error bars denote standard deviation. ** P < 0.01 by Student’s t-test
Effect of transgelin on experimental metastasis
We next tested the behavior of isogenic cell pairs in a model of experimental metastasis. RKOTAGLN or RKOCTRL cells were injected via the tail vein into scid mice. We also tested the behavior of previously described HCT 116 cells stably transfected with a transgelin miRNA knockdown vector (HCT116 TAGLN-KD) or with empty control vector (HCT116CTRL) [1] in the same assay.
Tumor burden in each experimental group was measured by quantitative histologic analysis. Mice receiving RKOTAGLN cells had more tumors than those receiving RKOCTRL cells, and the tumors occupied a greater fraction of the lung area (Fig. 3A). Similar results were seen with HCT116CTRL and HCT116TAGLN-KD cells (Fig 3B). In both instances, the member of the isogenic pair that had higher transgelin levels also had a greater tumor burden.
Fig. 3.
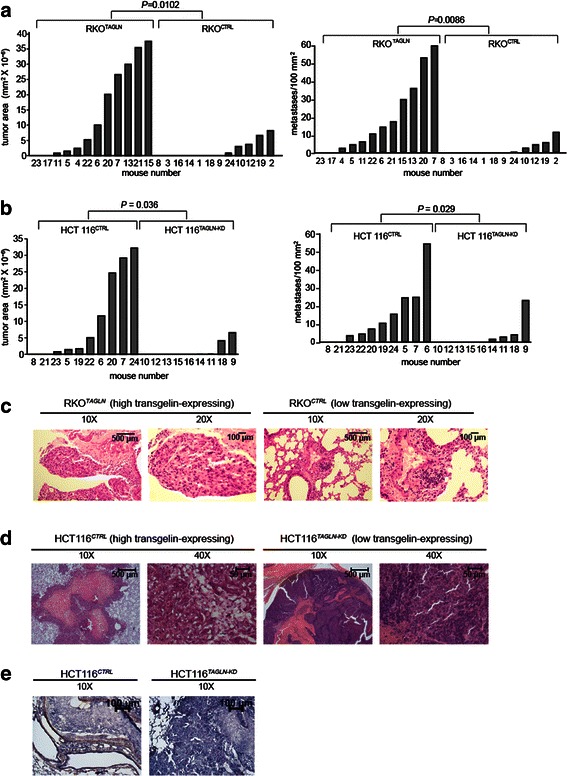
Experimental metastasis assay. Mice were injected with test cells via the tail vein as described in Materials and Methods. a. Aperio Precision image analysis on representative lung sections from animals injected with RKO cell derivatives. Twelve mice were used in each group. Left, total tumor area per lung section; right, number of metastases per unit area of lung tissue. P value reflects results of a non-parametric Wilcoxon rank sum test. b. Same analysis for HCT 116 cell derivatives. Ten mice were used in the HCT116CTRL group and 9 were used in the HCT116TAGLN-KD group. Statistical analysis as in Panel a. c. Histology of representative tumor sections from mice injected with RKO cell derivatives. d. Same for mice injected with HCT116CTRL derivatives. HCT116CTRL-derived tumor is a lung metastasis, HCT116TAGLN-KD-derived tumor arose near the injection site. e. HCT116-derived tumors stained with anti-transgelin
Although transgelin levels affected the number and size of metastases, there were no consistent differences in tumor histology (Fig. 3c, d). Immunostaining of HCT116-derived tumors with anti-transgelin antibody showed that tumors derived from the two cell populations retained their respective transgelin phenotypes in vivo, with no evidence of reversion (Fig. 3e). We did note that injection with HCT116 TAGLN-KD cells resulted in an unexpected incidence tumors near the injection site, instead of or in addition to the lung metastases (6/10 with HCT116 TAGLN-KD versus 1/10 with HCT116CTRL). Tumors near the injection site were not grossly evident in mice injected with the RKO cells.
Gene expression profiling to identify downstream genes
Prior studies suggest that transgelin is capable of both activating and repressing genes involved in tumor progression [1, 25, 26], although only a small number of individual genes have been examined to date. To define the full scope of transgelin-mediated gene regulation, we performed comprehensive gene expression profiling on RKOCTRL and RKOTAGLN cells using Affymetrix microarray technology.
Based on criteria of adjusted P value <0.05 and a minimum 2-fold change, 256 transcripts were significantly affected, with approximately equal numbers of transcripts increased and decreased (Fig. 4a). The most significantly affected categories of genes were those involved in cytoskeletal and actin binding (Fig. 4b). Other categories that were significantly affected included GTPase regulatory activities, other enzyme regulatory activities and identical protein binding (the ability to form homodimers or higher-order multimers). Table 1 shows the ten most highly up-regulated and ten most highly down-regulated genes. Interestingly, prior studies implicate a number of these in metastasis or cell motility (see Discussion).
Fig. 4.
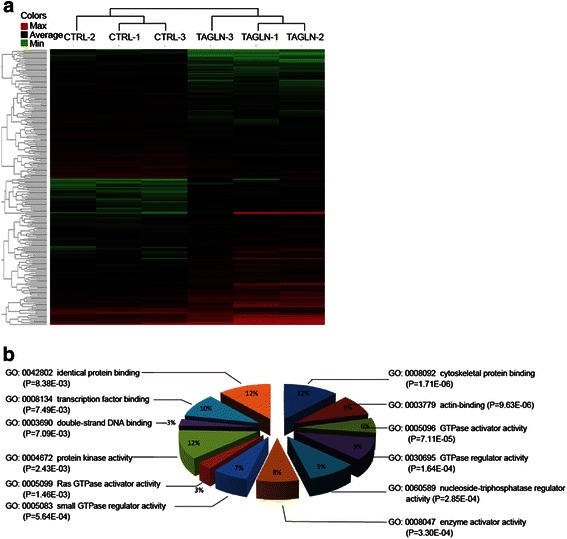
Effects of TAGLN overexpression on other genes. Gene expression profiling was performed using the Affymetrix platform. a. Hierarchical clustering analysis of 256 genes that show significant expression differences (P < 0.05) and a minimum 2-fold change between RKOTAGLN and RKOCTRL cells. Color represents relative gene expression on a log-transformed scale. b. Gene Ontology analysis for genes in Panel a
Table 1.
Transcripts up- or down-regulated in transgelin cDNA-transfected RKO cells
| Rank | Symbol | Gene name | Fold up | P value | Function |
| 1 | HOOK1 | Hook homolog 1 | 95.7 | 0.011 | actin binding |
| 2 | SDCCAG8 | serologically defined colon cancer antigen 8 | 85.8 | 0.023 | microtubule organizing center |
| 3 | ENAH | enabled homolog | 17.7 | 0.045 | actin binding |
| 4 | TNS1 | Tensin 1 | 14.3 | 0.014 | actin binding |
| 5 | ST8SIA4 | ST8 alpha-N-acetyl-neuraminide alpha-2, 8-sialyltransferase 4 | 13.2 | 0.047 | sialyltransferase activity |
| 6 | ARHGAP29 | Rho GTPase activating protein 29 | 13.0 | 0.046 | GTPase activator activity |
| 7 | FBXL14 | F-box and leucine-rich repeat protein 14 | 11.3 | 0.005 | protein binding |
| 8 | ZNF704 | zinc finger protein 704 | 10.5 | 0.020 | zinc ion binding |
| 9 | CCDC141 | coiled-coil domain containing 141 | 8.7 | 0.036 | protein binding |
| 10 | SPECC1 | sperm antigen with calponin homology and coiled-coil domains 1 | 8.2 | 0.020 | cell adhesion |
| Rank | Symbol | Gene name | Fold down | P value | Function |
| 1 | EMB | embigin | 15.9 | 0.005 | cell adhesion |
| 2 | WIPF1 | WAS/WASL interacting protein family, member 1 | 13.0 | 0.006 | actin binding |
| 3 | BCL11B | B-cell CLL/lymphoma 11B (zinc finger protein) | 12.0 | 0.0004 | nucleic acid binding |
| 4 | IL15 | interleukin 15 | 11.3 | 0.010 | signal transducer activity |
| 5 | PTPRD | protein tyrosine phosphatase, receptor type, D | 11.0 | 0.021 | phosphoprotein phosphatase |
| 6 | FRRS1 | ferric-chelate reductase 1 | 10.4 | 0.004 | ferric-chelate reductase |
| 7 | INSM1 | insulinoma-associated 1 | 10.1 | 0.045 | nucleic acid binding |
| 8 | PTGIS | prostaglandin I2 (prostacyclin) synthase | 9.8 | 0.006 | monooxygenase |
| 9 | PDGFB | platelet-derived growth factor beta polypeptide (simian sarcoma viral (v-sis) oncogene homolog) | 9.8 | 0.004 | PDGF receptor binding |
| 10 | POU3F1 | POU class 3 home box 1 | 8.7 | 0.005 | DNA binding |
Gene expression profiling was performed on RKO cell populations that were stably transfected with empty control vector or with transgelin cDNA expression vector. Experiment was performed using Affymetrix microarray technology as described in Methods. P values have been adjusted for multiple testing
Confirmation of gene expression changes by qPCR
To confirm the microarray results and investigate their generality, we performed qPCR analysis to independently determine mRNA levels for seven genes, chosen from Table 1 based on cancer relevance (see Discussion for details on the function of individual genes). We performed the analysis using RNA from the RKO cells, which were used for microarray, and also from an isogenic cell pair derived from DLD-1 cells. Like RKO, DLD-1 is a widely studied human CRC line with low endogenous transgelin levels. Transgelin protein expression was undetectable by immunoblotting in cells stably transfected with empty control vector (DLD-1CTRL) and was greatly increased following transfection with the transgelin expression vector (DLD-1TAGLN) (Fig. 5a).
Fig. 5.
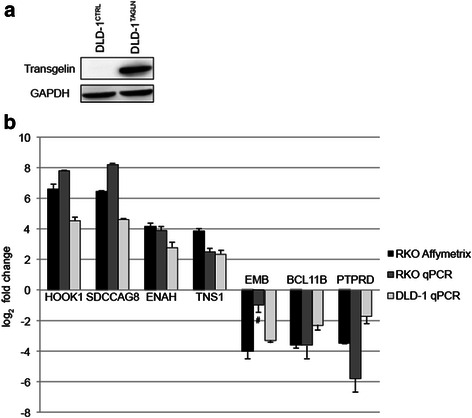
Expression of selected mRNAs determined by qPCR in RKO and DLD-1 cell derivatives. a. Immunoblot analysis of total cell extracts from DLD-1 derivatives stably transfected with empty vector (DLD-1CTRL) or with transgelin cDNA (DLD-1TAGLN). b. Real-time RT-PCR analysis for the levels of mRNA in RKO and DLD-1 cells of selected top-ranked differentially expressed genes identified by the cDNA microarray. Gene expression was normalized to GAPDH. Error bars denote standard deviation. All changes were significant (P < 0.05 by Student’s t-test) except for the one result indicated (#)
The effect of chronic transgelin expression in RKO and DLD-1 cells was strikingly similar and correlated well with the microarray data (Fig. 5b and Additional file 1: Table S1). HOOK1, SDCCAG8, ENAH, and TNS1 were up-regulated in the presence of transgelin, with effect sizes ranging from 5-fold to more than one hundred-fold. EMB, BCL11B, and PTPRD were down-regulated 3- to 60-fold. The significance of these changes in gene expression will be taken up in the Discussion; all of the genes are related to the cytoskeleton or cancer, and concordant regulation by transgelin in two independent CRC lines suggests a general effect with a common mechanism.
Discussion
Up-regulation of transgelin in low-expressing CRC cells and down-regulation in high-expressing cells have reciprocal effects
We characterized a new isogenic pair of high and low transgelin-expressing cell populations. In contrast to our previous study [1], where we knocked down expression in two high expressing CRC lines, here we increased transgelin expression in a low-expressing line. The overexpressing variant showed gain of metastasis-related behaviors in cell-based assays. Thus, overexpressing cells showed increased ability to invade Matrigel, an increased ability to form colonies when plated at low density, and more and larger colonies in soft agar. The reciprocal effects of up-regulation and down-regulation further support the idea that transgelin levels are causally related to metastatic behavior.
Transgelin levels affect metastatic potential
Here, we show that experimental manipulation of transgelin expression levels affects the metastatic potential of CRC cells in vivo. In the experimental metastasis assay, control HCT116 cells and RKO cells with stable overexpression of transgelin produced more and larger tumors in the lung than their isogenic, low transgelin-expressing counterparts. The widely used tail vein assay provides a composite measure of the efficiency of sequential events required for metastasis, including survival of isolated cells in the bloodstream, extravasation into the lung, and subsequent colonization and proliferation to form observable tumors [27]. Each of these steps has a parallel counterpart that was previously measured in cell culture [1], including resistance to anoikis, invasiveness in a Transwell assay, and colony formation at low density, respectively. Although a number of proteomic studies (but not all) have shown a correlation between transgelin levels and advanced stage disease and poor prognosis, data here imply a direct causal relationship between transgelin levels and metastatic potential in vivo.
Mechanism of the transgelin effect
In normal cells and tissue, transgelin interacts directly with the actin, influencing cell motility [3–5]. This mechanism may apply in cancer cells as well. In addition to direct interaction with actin, transgelin may also influence cell phenotype by affecting gene regulation. This idea is based on transgelin’s partial nuclear localization, its homology to transgelin 3, which has transcriptional regulatory activity [28], and other evidence [1, 25, 26]. To address the influence of transgelin on gene regulation in greater depth, we performed comprehensive gene expression profiling comparing low- and high-expressing RKO cells. Altering transgelin levels significantly affected the expression of a set of ~250 other genes, with statistical overrepresentation of cytoskeletal and actin-binding proteins, together with various other regulatory genes.
The five most-highly upregulated genes in Table 1 are connected with cancer, and in some cases specifically with tumor metastasis. HOOK1 (up-regulated 96-fold) interacts with microtubules and is up-regulated in breast cancer [29]. SDCCAG8 (up-regulated 86-fold) organizes the centrosome and is related to a colon cancer autoantigen [30]. ENAH (up-regulated 18-fold), which is the mammalian enabled homology and is also known as Mena, is a transcriptional target of the wnt/beta-catenin pathway [31]. It facilitates cell invasion via phosphatidylinositol 3-kinase-dependent local accumulation of actin filaments [32]. Increased ENAH/Mena expression levels correlate with invasiveness of breast and salivary gland tumors[33, 34], and are also seen in colorectal cancer and in polyps with high grade dysplasia [35]. TNS1 (up-regulated 14-fold), which is also known as Tensin 1, has actin cross-linking activity and localizes to focal adhesions. Prior studies show that increased expression of Tensin 1 correlates with tumor cell migration [36, 37]. A sialyltransferase (ST8SIA4) has been shown to promote metastatic dissemination in pancreatic cancer by interfering with E-cadherin dependent cell adhesion [38].
Of the most highly down-regulated genes, EMB (down-regulated 16-fold) is thought to mediate adhesion to the extracellular matrix [39]. BCL11B (down regulated 12-fold) is a tumor suppressor that is widely mutated in acute lymphocytic leukemia [40] and has recently been shown to be methylated, and thus likely down-regulated, in prostate cancers [41]. PTPRD (down-regulated 11-fold), a tumor suppressor that is frequently mutated in glioblastoma [42, 43], Ewing’s sarcoma [44], lung cancer [45], cutaneous squamous cell carcinoma [46], and laryngeal squamous cell carcinoma [47]. PTPRD has previously been shown to suppress colon cancer cell migration in cooperation with β-catenin/TCF signaling [48].
Based on an analysis of a more limited set of genes, we had previously hypothesized that transgelin regulates the epithelial-to-mesenchymal transition (EMT). In one cell line, fibronectin and vimentin (mesenchymal markers) and occludin (an epithelial marker) were affected. In another, only fibronectin was affected. In the RKO model, however, neither these nor other markers of EMT were altered. Thus the effect of transgelin on EMT, if any, appears to be cell-line specific.
It remains unclear whether the effect of transgelin on gene regulation occurs by a direct or indirect mechanism. Transgelin localized partially in the nucleus of the CRC cells; it shares 85 % similarity to TAGLN3, which is believed to be a transcriptional regulator [28]; Bioinformatic analysis using the DP-bind Web server (http://lcg.rit.albany.edu/dp-bind) identified several segments of transgelin as having high DNA-binding potential (data not shown); studies also suggests that nuclear actin-binding proteins participate widely in the transcription processes (reviewed in [49]). We were, however, unable, to detect direct association of transgelin with RNA polymerase II or chromatin (HMZ, YYF, and YL, unpublished results), which suggests that transgelin most likely works indirectly, through interaction with other transcriptional regulatory proteins, rather than by direct interaction with the polymerase or template.
Transgelin in the context of human CRC
The original impetus for the studies reported here was to identify biomarkers that could be quantified at the time of surgical resection and used to predict metastatic risk. Transgelin is abundant in normal tissue and is not a marker of cancer per se. In our work, however, it discriminated well between node-positive and node-negative CRC specimens [1].
Evaluation of potential biomarkers should ideally include evidence that they influence the biological process of interest; that is, adding or subtracting transgelin in a given context influences phenotypic behaviors associated with metastatic risk. Combining the results presented with those of a previous study [1], we have created four isogenic pairs (HCT116, SW480, RKO, and DLD-1), three have been characterized with respect to in vitro phenotypes (HCT116, SW480, and RKO) and two (HCT116 and RKO) have been characterized in an animal model of experimental metastasis. The combined results are consistent with a contribution of transgelin to metastatic potential.
Human CRC is characterized by genomic instability and variability, and caution is warranted in generalizing from results with individual cell lines. However, laboratory findings provide a rationale for continuing to explore the potential of transgelin, perhaps in conjunction with other biomarkers, as a predictor of individual risk. As noted earlier, proteomic studies of human cancer are not in universal agreement with respect to the role of transgelin, and it will be important to identify the root cause of the discrepancies, which could relate either to different patient populations or to technical factors. Separately, the finding that a number of gene products are co-regulated with transgelin suggests that they may be attractive targets for drug therapy in advanced CRC.
Conclusions
The results support a hypothesis that transgelin expression levels influence metastatic potential in CRC cell lines, and this may occur in part due to altered expression of downstream target genes that affect cell motility.
Methods
Isogenic cell line pairs
RKO and DLD-1 colon carcinoma cells were obtained from ATCC (Manassas, VA; CRL-2577 or CRL-221 respectively). Cells were transfected with pcDNA6.2/EmGFP-Bsd/V5-DEST or pcDNA6.2/EmGFP-Bsd/V5-TAGLN-mut vectors; the latter was generated using pDONR221-TAGLN-mut from a previous study[1] by an LR recombination). Stable transfectants were selected in medium containing 25 μg/ml blasticidin (Invitrogen, Carlsbad, CA). Immunoblot analysis was performed using anti-transgelin IgG or anti-GAPDH IgG1 (Abcam, Cambridge, MA) with ECL substrate for detection (Thermo Fisher Scientific, Rockford, IL). Immunofluorescence was performed on paraformaldehyde-fixed cells as described [1]. For qPCR, total RNA was extracted using Trizol (Invitrogen), cDNA was prepared, and qPCR was performed using primers in Additional file 2: Table S2. Reactions were performed using a SYBR Green PCR kit (Qiagen, Germantown, MD) and a LightCycler 480 instrument (Roche Applied Science, Indianapolis, IN).
Creation and characterization of HCT116CTRL and HCT116TAGLN-KD populations has been described [1]. In brief, cells were transfected with plasmid expressing an artificial microRNA (Invitrogen, Hmi416875) or with control vector and drug selection was applied to obtain cell populations that differed by about five-fold in transgelin expression levels.
Transwell invasion assay
Cell invasion assays were performed as described [1, 50] using a Transwell permeable support (8.0 μm pore size in 6-well format, Corning Life Sciences, Corning, NY). Filters were coated with 650 μg/ml Matrigel (BD Biosciences, Sparks, MD) according to the manufacturer’s protocol. Cells were harvested and resuspended in serum-free medium, and 2 × 105 cells were applied onto the upper chamber of the Transwell apparatus. The bottom chamber contained 0.6 ml of medium supplemented with 10 % fetal bovine serum. Cells were incubated for 48 hours. Cells that did not migrate were removed by cotton swabbing. Remaining cells were fixed with 4 % formaldehyde for 20 minutes at room temperature and stained with 0.1 % crystal violet for 15 minutes, also at room temperature. The stained cells were extracted with 10 % acetic acid, and the absorbance at 595 nm was measured.
Clonogenic survival assay
Cells were plated at 2 × 102 per 60 mm dish and incubated with complete growth medium for 12 days. Colonies were fixed and stained with staining buffer (0.1 % crystal violet, 4 % formaldehyde) for 30 minutes at room temperature.
Soft agar colony formation assay
Eagle’s MEM supplemented with 20 % fetal bovine serum, was adjusted to 0.6 % agar and distributed in a 6-well plate to form a base layer. Cells (2.5 × 102 cells per well) were suspended in the same medium containing 0.35 % agar and the suspension was dispensed to each well. Cultures were fed twice weekly with an overlay of complete medium. After 17 days colonies of 100 or more cells were counted.
Cell proliferation assay
Replicate cultures were seeded in 6-well dishes (1 × 105 cells/well). Triplicate samples of each cell population were harvested and counted daily for four days.
Cell cycle analysis
For cell cycle analysis, 5 × 105 cells were fixed with 70 % ethanol overnight at 4 °C, washed twice with PBS, and incubated with 50 μg RNase (Sigma-Aldrich, USA) and 9 μg propidium iodide (Invitrogen, USA) for 30 minutes at 4 °C in the dark. DNA content was analyzed using a FACSCalibur flow cytometer (Becton Dickinson, USA).
Tail vein assay of experimental metastasis
For the tail vein assay [51], 2 × 106 cells were suspended in 0.15 ml of Hank’s solution, filtered to obtain a single-cell suspension, and injected using a 28-gauge needle into a tail vein of six-week-old CB.17 scid mice. Mice were sacrificed after 6–8 weeks or when they exhibited signs of disease (weight loss and decreased grooming). Animals were necropsied and lungs and any other organs that showed gross tumors were fixed, paraffin embedded, and stained with hematoxylin and eosin for examination by a pathologist. The number of tumors in lung, heart, kidney, rib, back (near the injection site), leg, and foot was scored. Representative lung tissue sections were imaged using an Aperio Scanscope eSlide capture device and analyzed using Aperio Precision image analysis software.
cDNA microarray analysis
Gene expression profiling was performed using an Affymetrix Gene Chip Human Genome U133 Plus 2.0 Array, which contains 54,000 probe sets representing 38,500 well-characterized human genes (Affymetrix, Santa Clara, CA). Total RNA was isolated from triplicate cultures using the TRIzol method. After passing a quality control measurement, RNA was amplified and labeled using the Affymetrix 3’ IVT Express kit and hybridized to the array for 16 hours. This was followed by washing and staining using the Affymetrix fluidics station 450. Arrays were scanned using an Affymetrix 3000 7G plus instrument. AGCC software was used to generate .cel files. CHP files were generated with the mas5 method using the Affymetrix Expression Console. Data were imported into the Agilent Gene Spring GX software for further analysis. Statistical significance of inter-group differences was evaluated by a t-test with Bonferroni correction. Unsupervised hierarchical clustering analysis was performed using Spotfire 5.0 software (TIBCO, Somerville, MA). The row dendrogram was generated using Ward’s clustering method with a half square Euclidean distance measure. The column dendrogram was generated using the single linkage clustering method and a Euclidean distance measure. Gene ontology analysis was based on David 6.7 [52, 53] (http://david.abcc.ncifcrf.gov).
Ethics statement
Ethics approvals were obtained from the Institutional Animal Care and Use Committee of Georgia Regents University (Protocol Number 2008–0155) or by the Experimental Animal Care and Use Committee of Sun Yat-Sen University (Protocol Number 201306). All experiments were conducted in accordance with the institutional guidelines of Sun Yat-Sen University and Georgia Regents University for the care and use of experimental animals.
Acknowledgements
The National Natural Science Foundation of China (No. 30901782, YL), the Fundamental Research Funds for the Central Universities (No. 11ykpy32, YL), and the Yat-Sen Scholarship for Young Scientist (YL) supported this work. Grant [2013] 163 from Key Laboratory of Malignant Tumor Molecular Mechanism and Translational Medicine of Guangzhou Bureau of Science and Information Technology, and Grant KLB09001 from the Key Laboratory of Malignant Tumor Gene Regulation and Target Therapy of Guangdong Higher Education Institutes also supported this work. A National Institutes of Health/National Cancer Institute grant CA95941 (WSD) provided additional support. The data discussed in this manuscript have been deposited in NCBI's Gene Expression Omnibus (Zhou et al., 2013) and are accessible through GEO Series accession number GSE48998 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48998).
Additional files
- Comparison of the genes altered by transgelin overexpression in RKO and DLD-1 cells. Table shows gene symbols, gene names, expression fold changes obtained by cDNA microarray and qPCR in RKO and DLD-1 cells. (DOCX 16 kb)
- Primers for qPCR. Table shows gene symbols and primer sequences. All primers are written in 5’ to 3’ direction. (DOC 78 kb)
Footnotes
Competing interests
The authors declare no competing interests.
Authors’ contributions
HMZ, YYF, QKC, and HS developed and characterized the RKO and DLD-1 model; YL, LLD, FZH, JKC, and MQR designed and performed the tail vein assay experiments; PMW conceived, performed and analyzed quantitative histology experiments; JRL performed all other pathology; WSD supervised work at Georgia Regents University, analyzed data, and significantly contributed to writing and editing the manuscript; YL initiated tail vein experiments at Georgia Regents University, performed, supervised, and analyzed work at Sun Yat-Sen Memorial Hospital, and drafted the manuscript. All authors read and approved the final version of the manuscript.
Contributor Information
Hui-min Zhou, Email: erikoapril@163.com.
Yuan-yuan Fang, Email: fangyy2@sina.cn.
Paul M. Weinberger, Email: pweinberger@gru.edu
Ling-ling Ding, Email: llding7538@hotmail.com.
John K. Cowell, Email: jcowell@gru.edu
Farlyn Z. Hudson, Email: fhudson@gru.edu
Mingqiang Ren, Email: mren@gru.edu.
Jeffrey R. Lee, Email: jlee@gru.edu
Qi-kui Chen, Email: qkchen@21cn.com.
Hong Su, Email: sophiasucn@hotmail.com.
William S. Dynan, Email: wdynan@emory.edu
Ying Lin, Email: linwy@mail.sysu.edu.cn.
References
- 1.Lin Y, Buckhaults PJ, Lee JR, Xiong H, Farrell C, Podolsky RH, et al. Association of the actin-binding protein transgelin with lymph node metastasis in human colorectal cancer. Neoplasia. 2009;11:864–873. doi: 10.1593/neo.09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shapland C, Hsuan JJ, Totty NF, Lawson D. Purification and properties of transgelin: a transformation and shape change sensitive actin-gelling protein. J Cell Biol. 1993;121:1065–1073. doi: 10.1083/jcb.121.5.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gimona M, Kaverina I, Resch GP, Vignal E, Burgstaller G. Calponin repeats regulate actin filament stability and formation of podosomes in smooth muscle cells. Mol Biol Cell. 2003;14:2482–2491. doi: 10.1091/mbc.E02-11-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu H, Konigshoff M, Jayachandran A, Handley D, Seeger W, Kaminski N, et al. Transgelin is a direct target of TGF-beta/Smad3-dependent epithelial cell migration in lung fibrosis. FASEB J. 2008;22:1778–1789. doi: 10.1096/fj.07-083857. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa A, Sakatsume M, Wang X, Sakamaki Y, Tsubata Y, Alchi B, et al. SM22alpha: the novel phenotype marker of injured glomerular epithelial cells in anti-glomerular basement membrane nephritis. Nephron Exp Nephrol. 2007;106:e77–87. doi: 10.1159/000103020. [DOI] [PubMed] [Google Scholar]
- 6.Zhang JC, Kim S, Helmke BP, Yu WW, Du KL, Lu MM, et al. Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol Cell Biol. 2001;21:1336–1344. doi: 10.1128/MCB.2001.21.4.1336-1344.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeidan A, Sward K, Nordstrom I, Ekblad E, Zhang JC, Parmacek MS, et al. Ablation of SM22alpha decreases contractility and actin contents of mouse vascular smooth muscle. FEBS Lett. 2004;562:141–146. doi: 10.1016/S0014-5793(04)00220-0. [DOI] [PubMed] [Google Scholar]
- 8.Je HD, Sohn UD. SM22alpha is required for agonist-induced regulation of contractility: evidence from SM22alpha knockout mice. Mol Cells. 2007;23:175–181. [PubMed] [Google Scholar]
- 9.Feil S, Hofmann F, Feil R. SM22alpha modulates vascular smooth muscle cell phenotype during atherogenesis. Circ Res. 2004;94:863–865. doi: 10.1161/01.RES.0000126417.38728.F6. [DOI] [PubMed] [Google Scholar]
- 10.Shields JM, Rogers-Graham K, Der CJ. Loss of transgelin in breast and colon tumors and in RIE-1 cells by Ras deregulation of gene expression through Raf-independent pathways. J Biol Chem. 2002;277:9790–9799. doi: 10.1074/jbc.M110086200. [DOI] [PubMed] [Google Scholar]
- 11.Sitek B, Luttges J, Marcus K, Kloppel G, Schmiegel W, Meyer HE, et al. Application of fluorescence difference gel electrophoresis saturation labelling for the analysis of microdissected precursor lesions of pancreatic ductal adenocarcinoma. Proteomics. 2005;5:2665–2679. doi: 10.1002/pmic.200401298. [DOI] [PubMed] [Google Scholar]
- 12.Assinder SJ, Stanton JA, Prasad PD. Transgelin: an actin-binding protein and tumour suppressor. Int J Biochem Cell Biol. 2009;41:482–486. doi: 10.1016/j.biocel.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Ryu JW, Kim HJ, Lee YS, Myong NH, Hwang CH, Lee GS, et al. The proteomics approach to find biomarkers in gastric cancer. J Korean Med Sci. 2003;18:505–509. doi: 10.3346/jkms.2003.18.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi Y, Chiu JF, Wang L, Kwong DL, He QY. Comparative proteomic analysis of esophageal squamous cell carcinoma. Proteomics. 2005;5:2960–2971. doi: 10.1002/pmic.200401175. [DOI] [PubMed] [Google Scholar]
- 15.Mikuriya K, Kuramitsu Y, Ryozawa S, Fujimoto M, Mori S, Oka M, et al. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int J Oncol. 2007;30:849–855. [PubMed] [Google Scholar]
- 16.Huang Q, Chen W, Wang L, Lin W, Lin J, Lin X. Identification of transgelin as a potential novel biomarker for gastric adenocarcinoma based on proteomics technology. J Cancer Res Clin Oncol. 2008;134:1219–1227. doi: 10.1007/s00432-008-0398-y. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Song MQ, Zhu JS, Zhou Z, Xu ZP, Chen WX, et al. Identification of differentially-expressed proteins between early submucosal non-invasive and invasive colorectal cancer using 2D-DIGE and mass spectrometry. Int J Immunopathol Pharmacol. 2011;24:849–859. doi: 10.1177/039463201102400404. [DOI] [PubMed] [Google Scholar]
- 18.Kim HJ, Kang UB, Lee H, Jung JH, Lee ST, Yu MH, et al. Profiling of differentially expressed proteins in stage IV colorectal cancers with good and poor outcomes. J Proteomics. 2012;75:2983–2997. doi: 10.1016/j.jprot.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Zhou L, Zhang R, Zhang L, Sun Y, Yao W, Zhao A, et al. Upregulation of transgelin is an independent factor predictive of poor prognosis in patients with advanced pancreatic cancer. Cancer Sci. 2013;104:423–430. doi: 10.1111/cas.12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao L, Wang H, Deng YJ, Wang S, Liu C, Jin H, et al. Transgelin as a suppressor is associated with poor prognosis in colorectal carcinoma patients. Mod Pathol. 2009;22:786–796. doi: 10.1038/modpathol.2009.29. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Shi R, Wang Y, Niu X. TAGLN suppresses proliferation and invasion, and induces apoptosis of colorectal carcinoma cells. Tumour Biol. 2013;34:505–513. doi: 10.1007/s13277-012-0575-0. [DOI] [PubMed] [Google Scholar]
- 22.Lee EK, Han GY, Park HW, Song YJ, Kim CW. Transgelin promotes migration and invasion of cancer stem cells. J Proteome Res. 2010;9:5108–5117. doi: 10.1021/pr100378z. [DOI] [PubMed] [Google Scholar]
- 23.Tsai ST, Tsou CC, Mao WY, Chang WC, Han HY, Hsu WL, et al. Label-free quantitative proteomics of CD133-positive liver cancer stem cells. Proteome Sci. 2012;10:69. doi: 10.1186/1477-5956-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dvorakova M, Nenutil R, Bouchal P. Transgelins, cytoskeletal proteins implicated in different aspects of cancer development. Expert Rev Proteomics. 2014;11:149–165. doi: 10.1586/14789450.2014.860358. [DOI] [PubMed] [Google Scholar]
- 25.Nair RR, Solway J, Boyd DD. Expression cloning identifies transgelin (SM22) as a novel repressor of 92-kDa type IV collagenase (MMP-9) expression. J Biol Chem. 2006;281:26424–26436. doi: 10.1074/jbc.M602703200. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Chang YJ, Miyamoto H, Ni J, Niu Y, Chen Z, et al. Transgelin functions as a suppressor via inhibition of ARA54-enhanced androgen receptor transactivation and prostate cancer cell growth. Mol Endocrinol. 2007;21:343–358. doi: 10.1210/me.2006-0104. [DOI] [PubMed] [Google Scholar]
- 27.Elkin M, Vlodavsky I. Tail vein assay of cancer metastasis. Curr Protoc Cell Biol. 2001;Chapter 19(Unit 19):12. doi: 10.1002/0471143030.cb1902s12. [DOI] [PubMed] [Google Scholar]
- 28.Feng J, Lawson MA, Melamed P. A proteomic comparison of immature and mature mouse gonadotrophs reveals novel differentially expressed nuclear proteins that regulate gonadotropin gene transcription and RNA splicing. Biol Reprod. 2008;79:546–561. doi: 10.1095/biolreprod.108.068106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bubnov V, Moskalev E, Petrovskiy Y, Bauer A, Hoheisel J, Zaporozhan V. Hypermethylation of TUSC5 genes in breast cancer tissue. Exp Oncol. 2012;34:370–372. [PubMed] [Google Scholar]
- 30.Kenedy AA, Cohen KJ, Loveys DA, Kato GJ, Dang CV. Identification and characterization of the novel centrosome-associated protein CCCAP. Gene. 2003;303:35–46. doi: 10.1016/S0378-1119(02)01141-1. [DOI] [PubMed] [Google Scholar]
- 31.Najafov A, Seker T, Even I, Hoxhaj G, Selvi O, Ozel DE, et al. MENA is a transcriptional target of the Wnt/beta-catenin pathway. PLoS One. 2012;7:e37013. doi: 10.1371/journal.pone.0037013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi K, Suzuki K. WAVE2, N-WASP, and Mena facilitate cell invasion via phosphatidylinositol 3-kinase-dependent local accumulation of actin filaments. J Cell Biochem. 2011;112:3421–3429. doi: 10.1002/jcb.23276. [DOI] [PubMed] [Google Scholar]
- 33.Gurzu S, Krause M, Ember I, Azamfirei L, Gobel G, Feher K, et al. Mena, a new available marker in tumors of salivary glands? Eur J Histochem. 2012;56:e8. doi: 10.4081/ejh.2012.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- 35.Gurzu S, Jung I, Prantner I, Ember I, Pavai Z, Mezei T. The expression of cytoskeleton regulatory protein Mena in colorectal lesions. Rom J Morphol Embryol. 2008;49:345–349. [PubMed] [Google Scholar]
- 36.Chen H, Duncan IC, Bozorgchami H, Lo SH. Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc Natl Acad Sci U S A. 2002;99:733–738. doi: 10.1073/pnas.022518699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Lo SH. Regulation of tensin-promoted cell migration by its focal adhesion binding and Src homology domain 2. Biochem J. 2003;370:1039–1045. doi: 10.1042/bj20021308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schreiber SC, Giehl K, Kastilan C, Hasel C, Muhlenhoff M, Adler G, et al. Polysialylated NCAM represses E-cadherin-mediated cell-cell adhesion in pancreatic tumor cells. Gastroenterology. 2008;134:1555–1566. doi: 10.1053/j.gastro.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 39.Guenette RS, Sridhar S, Herley M, Mooibroek M, Wong P, Tenniswood M. Embigin, a developmentally expressed member of the immunoglobulin super family, is also expressed during regression of prostate and mammary gland. Dev Genet. 1997;21:268–278. doi: 10.1002/(SICI)1520-6408(1997)21:4<268::AID-DVG4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118:4169–4173. doi: 10.1182/blood-2010-11-318873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahapatra S, Klee EW, Young CY, Sun Z, Jimenez RE, Klee GG, et al. Global methylation profiling for risk prediction of prostate cancer. Clin Cancer Res. 2012;18:2882–2895. doi: 10.1158/1078-0432.CCR-11-2090. [DOI] [PubMed] [Google Scholar]
- 42.Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009;106:9435–9440. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stallings RL, Nair P, Maris JM, Catchpoole D, McDermott M, O'Meara A, et al. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006;66:3673–3680. doi: 10.1158/0008-5472.CAN-05-4154. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, Janku F, Subbiah V, Angelo LS, Naing A, Anderson PM, et al. Germline PTPRD Mutations in Ewing Sarcoma: Biologic and Clinical Implications. Oncotarget. 2013;4:884–889. doi: 10.18632/oncotarget.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohno T, Otsuka A, Girard L, Sato M, Iwakawa R, Ogiwara H, et al. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer. 2010;49:342–352. doi: 10.1002/gcc.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambert SR, Harwood CA, Purdie KJ, Gulati A, Matin RN, Romanowska M, et al. Metastatic cutaneous squamous cell carcinoma shows frequent deletion in the protein tyrosine phosphatase receptor Type D gene. Int J Cancer. 2012;131:E216–226. doi: 10.1002/ijc.27333. [DOI] [PubMed] [Google Scholar]
- 47.Giefing M, Zemke N, Brauze D, Kostrzewska-Poczekaj M, Luczak M, Szaumkessel M, et al. High resolution ArrayCGH and expression profiling identifies PTPRD and PCDH17/PCH68 as tumor suppressor gene candidates in laryngeal squamous cell carcinoma. Genes Chromosomes Cancer. 2011;50:154–166. doi: 10.1002/gcc.20840. [DOI] [PubMed] [Google Scholar]
- 48.Funato K, Yamazumi Y, Oda T, Akiyama T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp Ther Med. 2011;2:457–463. doi: 10.3892/etm.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng B, Han M, Bernier M, Wen JK. Nuclear actin and actin-binding proteins in the regulation of transcription and gene expression. FEBS J. 2009;276:2669–2685. doi: 10.1111/j.1742-4658.2009.06986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen Y, Zhang YW, Zhang ZX, Miao ZH, Ding J. hTERT-targeted RNA interference inhibits tumorigenicity and motility of HCT116 cells. Cancer Biol Ther. 2008;7:228–236. doi: 10.4161/cbt.7.2.5259. [DOI] [PubMed] [Google Scholar]
- 51.Sossey-Alaoui K, Safina A, Li X, Vaughan MM, Hicks DG, Bakin AV, et al. Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am J Pathol. 2007;170:2112–2121. doi: 10.2353/ajpath.2007.060975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]