

Abstract
Molecules with high nitrogen content are of interest for their potential as high-energy materials. However, many molecules with 100% nitrogen content are unstable and dissociate with low barriers, which limits practical applications. In the present study, cyclic hexamers of the basic unit NCN (70% nitrogen by mass) are studied to determine the structural features and bonding characteristics that lead to more stable molecules. Double- and triple-bonded NCN units are compared to determine which form of NCN contributes the greater stability. Theoretical calculations using density functional theory and couple-cluster theory are carried out on a series of N12C6 molecules to determine trends in stability. Energetic and structural trends, as well as differences between DFT and coupled-cluster theory, are calculated and discussed.
Graphical abstract

Introduction
Molecules consisting predominantly of nitrogen have been the subject of much research because of their potential as high-energy materials. Decomposition reactions of the type Nx → (x/2) N2 can be exothermic by up to 200 kJ/mole per nitrogen atom (approximately 14 kilojoules per gram of material). Experimental synthetic successes in high-energy nitrogen materials include the N5+ and N5− ions1-3 as well as various azido compounds4-7 and even a network polymer of nitrogen8. Additionally, nitrogen-rich salts9 and the N7O+ ion10 have been successfully produced in the laboratory, as well as the CN7− anion11 and a hexamer of diazomethane12. The production of such a diverse group of nitrogen systems demonstrates the potential for such materials as novel high-energy molecules. High-energy nitrogen-based molecules have also been extensively studied by theoretical calculations. Theoretical studies of high-energy nitrogen include cyclic and acyclic compounds13-19, as well as nitrogen cages20-26. Structures and thermodynamics of energetic nitrogen systems have been calculated for both small molecules and larger structures with up to seventy-two atoms.
A study27 of various isomers of N4C2 (70% nitrogen by mass) revealed that the most stable molecule is a dimer of the structural unit NCN. This dimer was the most stable both thermodynamically and with respect to dissociation. A subsequent theoretical study28 on tetramers of NCN included two different forms of the NCN unit, one involving carbon-nitrogen double bonds and one involving carbon-nitrogen triple bonds. A survey of various bonding combinations of the NCN units revealed a stark contrast between the PBE1PBE density functional (DFT) method and coupled-cluster theory. Compared to DFT, coupled-cluster theory greatly favored structures with C-N triple bond, a trend that was shown for both the tetramers and smaller model systems. In the current study, involving hexamers of the basic NCN bonding units, coupled-cluster theory is compared to DFT, but with multiple functionals to determine if the variations are particular to the PBE1PBE functional or more general to DFT.
Computational Methods
Geometries for all molecules in this study were optimized using the PBE1PBE, B3LYP, and B97D density functional methods29-31. Single energy points at the PBE1PBE geometries were carried out using coupled-cluster theory32 (CCSD(T)). Geometries were verified as local minima by PBE1PBE vibrational frequencies. Calculations were carried out using the Dunning cc-pVDZ and cc-pVTZ correlation-consistent basis sets33. Calculations were carried out using the Gaussian09 computational chemistry software34 and its Windows counterpart Gaussian09W.
Results and Discussion
Two different NCN structural units are considered in this study: one with two C=N double bonds and an open valence on each nitrogen atom, and one with a single and triple bond and two open valences on the singly bonded nitrogen. Both are pictured in Figure 1. Fifteen NCN hexamers are considered in this study, with varying composition with respect to the two types of NCN units. The isomer designated A has six double-bonded units bonded into a single eighteen-atom ring. Isomer B has five double-bonded and one triple-bonded unit. Isomers C1, C2, and C3 have four double-bonded and two triple-bonded units. Isomers D1, D2, D3, and D4 have three of each type of NCN units. The isomers E1, E2, and E3 have two double-bonded and four triple-bonded units. Isomer F has one double-bonded and five triple-bonded units, and the isomers G1 and G2 have six triple-bonded units. All fifteen hexamers are shown in Figure 2.
Figure 1.

NCN bonding groups: (a) double-bonded unit, (b) triple-bonded unit. Nitrogen atoms are shown in yellow, carbon atoms in black.
Figure 2.

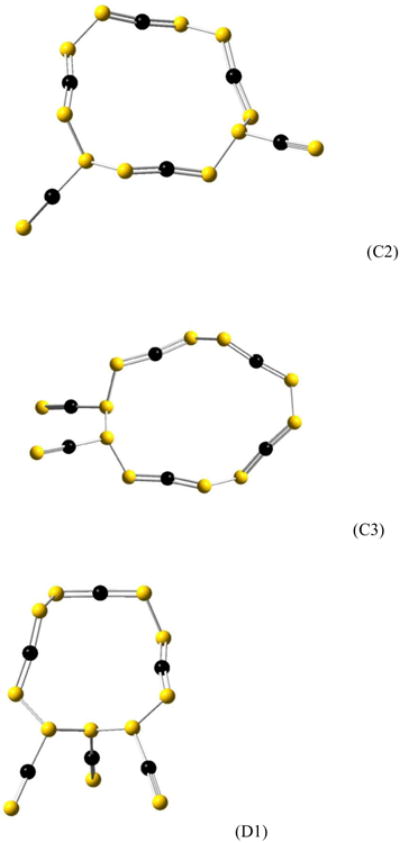



N12C6 molecules (hexamers of NCN): (A) Isomer A, (B) Isomer B, (C1) Isomer C1, (C2) Isomer C2, (C3) Isomer C3, (D1) Isomer D1, (D2) Isomer D2, (D3) Isomer D3, (D4) Isomer D4, (E1) Isomer E1, (E2) Isomer E2, (E3) Isomer E3, (F) Isomer, (G1) Isomer G1, (G2) Isomer G2. Nitrogen atoms are shown in yellow, carbon atoms in black.
B3LYP, PBE1PBE, B97D, CCSD and CCSD(T) relative energies for all fifteen hexamers are shown in Table 1. The clearest trend in the data is that, while B3LYP, PBE1PBE, and B97D disagree by up to 40 kJ/mole for some isomers, the density functional methods agree with each other far more closely than any of them agrees with coupled-cluster theory. All three DFT methods predict isomer C1 as the most stable, and none of the DFT method shows a clear energetic trend in favor of double-bonded or triple-bonded NCN units. This stands in stark contrast to coupled-cluster theory, which shows a strong trend in favor of the triple-bonded NCN units; increasing the number of triple-bonded NCN units is heavily rewarded by CCSD(T). This behavior of CCSD(T) relative to density functional theory is in agreement with similar behavior previously shown28 for tetramers of NCN. Benchmark calculations in the NCN tetramer study28 demonstrated that coupled-cluster theory favors the C-N triple bond more than DFT does. The energetic trends hold for both CCSD and CCSD(T), with the triples correction narrowing the isomer energy differences. Clearly, either DFT or coupled-cluster is failing to calculate accurate isomer energies. For the G isomers, which have the maximum number of triple bonds, the disagreement approaches 200 kJ/mole. T1 diagnostic values for the coupled-cluster calculations on all molecules in this study are approximately 0.02, so it is not likely that multireference character is adversely affecting the coupled-cluster results.
Table 1.
Relative energies of the N12C6 molecules. All calculations carried out using cc-pVDZ atomic orbital basis set. Energies in kJ/mole. Coupled-cluster calculations carried out as single points at PBE1PBE geometries.
| PBE1PBE | B3LYP | B97D | CCSD | CCSD(T) | |
|---|---|---|---|---|---|
| Isomer | |||||
| A | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| B | −5.9 | −4.7 | +0.3 | −44.6 | −38.5 |
| C1 | −46.9 | −41.8 | −27.4 | −110.2 | −99.2 |
| C2 | +38.9 | +32.8 | +54.0 | −64.2 | −49.8 |
| C3 | +15.1 | +13.5 | +31.7 | −74.5 | −60.2 |
| D1 | +11.7 | +13.6 | +34.2 | −116.5 | −96.2 |
| D2 | +10.5 | +12.9 | +27.5 | −97.2 | −83.7 |
| D3 | −26.8 | −19.7 | -11.6 | −128.3 | −115.5 |
| D4 | −11.7 | −6.7 | +10.9 | −108.7 | −94.6 |
| E1 | −12.6 | −4.5 | +17.3 | −150.2 | −126.4 |
| E2 | −4.6 | −2.4 | +23.8 | −153.9 | −129.7 |
| E3 | +20.1 | +19.2 | +44.6 | −149.9 | −125.9 |
| F | +16.3 | +19.0 | a | −176.6 | −147.3 |
| G1 | −24.3 | −19.0 | +17.1 | −252.5 | −212.5 |
| G2 | +122.6 | +113.3 | +152.0 | −111.9 | −86.6 |
The B97D optimization of isomer F was dissociative
DFT and coupled-cluster do, however, agree on a number of points. First, all methods agree that isomer G1 is much more stable than isomer G2. Both isomers have a chair-conformed cyclohexane-like ring of six nitrogen atoms, but isomer G1 has its six C-N triple bonds in axial positions, which minimizes steric repulsion between the electrons in the triple bonds. Conversely, G2 has all the triple bonds in equatorial positions, which causes destabilizing repulsions between the C-N triple bonds. All methods in this study also agree that the energy ordering of the C isomers is C1 < C3 < C2. Isomer C1 has the C-N triple bonds farthest apart and has the most open, torsionally-relaxed structure, which minimizes the interactions between the triple bonds and the neighboring nitrogen lone pairs. All methods also agree that isomer D3 is the most stable of the D isomers.
Basis set effects are shown in Table 2. All three DFT methods were used for isomers A and G1 using both the cc-pVDZ and the larger cc-pVTZ basis set. The effect of the cc-pVTZ is to favor isomer A by approximately 20-25 kJ/mole. While basis set effects are clearly nontrivial for these systems, the effect is not large enough to explain such a large disagreement between DFT and CC theory. Geometry effects are also shown in Table 2. Calculations with the CCSD(T)/cc-pVDZ method have been carried out for isomers A and G1 using optimized geometries from all three DFT methods. The three CCSD(T) calculations all agree with each other within 10 kJ/mole. This close agreement is similar to results35 for MP4 calculations on large nitrogen cages, which also showed that the results of high-level calculations are relatively insensitive to the choice of geometry optimization method.
Table 2.
Basis set effects and geometry effects on the relative energies of isomers A and G1. Energies shown are energies of G1 relative to A. Energies are shown in kJ/mole. Results are shown for the three DFT methods in this study.
| DFT//DFT | DFT//DFT | CCSD(T)//DFT | |
|---|---|---|---|
| cc-pVDZ | cc-pVTZ | cc-pVDZ | |
| PBE1PBE | −24.3 | −0.7 | −212.5 |
| B3LYP | −19.0 | +2.6 | −206.1 |
| B97D | +17.1 | +35.7 | −214.2 |
Table 3 shows significant physical parameters for isomers A and G1. PBE1PBE/cc-pVDZ bond lengths and angles are shown for A and G1, along with the corresponding parameters for small carbon- and nitrogen-bearing small molecules. Both A and G1 show a significant shortening of the N-N single bond relative to hydrazine, but the most significant property unique to isomer G1 is the shortening of the C-N single bond by nearly a tenth of an angstrom relative to methylamine. This implies a high-degree of double-bond character in the C-N bonds of isomer G1, which would indicate electron delocalization that could explain the high degree of stability shown for isomer G1 by all methods in this study. Additionally, the bond angles at the sp3 nitrogen atoms in isomer G1 are 113-115 degrees, which is actually closer to the sp2 ideal of 120 degrees than to the 104.3-degree angles of ammonia with the PBE1PBE/cc-pVDZ method. This high degree of flattening of formally sp3 nitrogen implies significant sp2 character and supports the idea of a stabilizing electron delocalization in isomer G1.
Table 3.
Bond length and angle data for isomers A and G1. All bond lengths are shown in angstroms and compared to similar bonds in reference compounds as indicated. All values are calculated using the cc-pVDZ basis set, and reference values are calculated with PBE1PBE/cc-pVDZ.
| PBE1PBE | B3LYP | B97D | Reference | ||
|---|---|---|---|---|---|
| Isomer | Bond/angle | ||||
| A | N–N | 1.39 | 1.41 | 1.41 | 1.47 (N2H4) |
| C=N | 1.23 | 1.24 | 1.24 | 1.27 (CH2NH) | |
| G1 | N–N | 1.40 | 1.42 | 1.44 | 1.47 (N2H4) |
| C–N | 1.36 | 1.37 | 1.37 | 1.45 (CH3NH2) | |
| C≡N | 1.16 | 1.16 | 1.17 | 1.16 (HCN) | |
| sp3 N angles | 114.2 114.9 114.9 |
114.4 114.7 114.7 |
114.3 113.4 113.4 |
104.3 (NH3) |
Conclusion
Density functional theory appears to treat carbon-nitrogen molecules and carbon-nitrogen bonding very differently from coupled-cluster theory. The DFT functionals have good agreement with each other but not with CCSD(T). Coupled-cluster theory greatly favors the C-N triple bond, and in the absence of strong multireference effects, it is likely that the CCSD(T) treatment is more accurate. While it is possible that the use of additional functionals would change the results, it is unlikely given the sheer magnitude of the disagreement between DFT and CCSD(T).
Supplementary Material
Highlights.
N12C6 molecules are highly energetic
DFT and CCSD(T) disagree on their treatment
3LYP and PBE1PBE agree with each other
CCSD(T) likely gives more accurate results
Acknowledgments
The Alabama Supercomputer Authority is gratefully acknowledged for a grant of computer time on the SGI Ultraviolet in Huntsville, AL. This work was supported by the National Science Foundation (NSF/HBCU-UP grant 0505872). This work was also supported by the National Institutes of Health (NIH/NCMHD 1P20MD000547-01) and the Petroleum Research Fund, administered by the American Chemical Society (PRF 43798-B6). The taxpayers of the state of Alabama in particular and the United States in general are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Christe KO, Wilson WW, Sheehy JA, Boatz JA. Angew Chem Int Ed. 1999;38:2004. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2004::AID-ANIE2004>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 2.Vij A, Pavlovich JG, Wilson WW, Vij V, Christe KO. Angew Chem Int Ed. 2002;41:3051. doi: 10.1002/1521-3773(20020816)41:16<3051::AID-ANIE3051>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; Butler RN, Stephens JC, Burke LA. Chem Commun. 2003;8:1016. doi: 10.1039/b301491f. [DOI] [PubMed] [Google Scholar]
- 3.Dixon DA, Feller D, Christe KO, Wilson WW, Vij A, Vij V, Jenkins HDB, Olson RM, Gordon MS. J Am Chem Soc. 2004;126:834. doi: 10.1021/ja0303182. [DOI] [PubMed] [Google Scholar]
- 4.Knapp C, Passmore J. Angew Chem Int Ed. 2004;43:4834. doi: 10.1002/anie.200401748. [DOI] [PubMed] [Google Scholar]
- 5.Haiges R, Schneider S, Schroer T, Christe KO. Angew Chem Int Ed. 2004;43:4919. doi: 10.1002/anie.200454242. [DOI] [PubMed] [Google Scholar]
- 6.Huynh MV, Hiskey MA, Hartline EL, Montoya DP, Gilardi R. Angew Chem Int Ed. 2004;43:4924. doi: 10.1002/anie.200460366. [DOI] [PubMed] [Google Scholar]
- 7.Klapotke TM, Schulz A, McNamara J. J Chem Soc Dalton Trans. 1996:2985. [Google Scholar]; Klapotke TM, Noth H, Schutt T, Warchhold M. Angew Chem Int Ed. 2000;39:2108. doi: 10.1002/1521-3773(20000616)39:12<2108::aid-anie2108>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]; Klapotke TM, Krumm R, Mayer P, Schwab I. Angew Chem Int Ed. 2003;42:5843. doi: 10.1002/anie.200352656. [DOI] [PubMed] [Google Scholar]
- 8.Eremets MI, Gavriliuk AG, Trojan IA, Dzivenko DA, Boehler R. Nature Materials. 2004;3:558. doi: 10.1038/nmat1146. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Zhang Y, Shreeve JM. Chemistry. 2011;17:1538. doi: 10.1002/chem.201002363. [DOI] [PubMed] [Google Scholar]
- 10.Christe KO, Haiges R, Wilson WW, Boatz JA. Inorg Chem. 2010;49:1245. doi: 10.1021/ic9022213. [DOI] [PubMed] [Google Scholar]
- 11.Klapotke TM, Stierstorfer J. J Am Chem Soc. 2009;131:1122. doi: 10.1021/ja8077522. [DOI] [PubMed] [Google Scholar]
- 12.Klapotke TM, Mayer P, Schulz A, Weigand JJ. Propellants Explosives Pyrotechnics. 2004;29:325. [Google Scholar]
- 13.Chung G, Schmidt MW, Gordon MS. J Phys Chem A. 2000;104:5647. [Google Scholar]
- 14.Strout DL. J Phys Chem A. 2002;106:816. [Google Scholar]
- 15.Thompson MD, Bledson TM, Strout DL. J Phys Chem A. 2002;106:6880. [Google Scholar]
- 16.Li QS, Liu YD. Chem Phys Lett. 2002;353:204. [Google Scholar]; Li QS, Qu H, Zhu HS. Chin Sci Bull. 1996;41:1184. [Google Scholar]
- 17.Li QS, Zhao JF. J Phys Chem A. 2002;106:5367. [Google Scholar]; Qu H, Li QS, Zhu HS. Chin Sci Bull. 1997;42:462. [Google Scholar]
- 18.Gagliardi L, Evangelisti S, Bernhardsson A, Lindh R, Roos BO. Int J Quantum Chem. 2000;77:311. [Google Scholar]
- 19.Law CK, Li WK, Wang X, Tian A, Wong NB. Theochem. 2002;617:121. [Google Scholar]
- 20.Gagliardi L, Evangelisti S, Widmark PO, Roos BO. Theor Chem Acc. 1997;97:136. [Google Scholar]
- 21.Schmidt MW, Gordon MS, Boatz JA. Int J Quantum Chem. 2000;76:434. [Google Scholar]
- 22.Zhou HW, Wong NB, Zhou G, Tian AM. J Phys Chem A. 2006;110:3845. doi: 10.1021/jp056435w. [DOI] [PubMed] [Google Scholar]
- 23.Zhou HW, Wong NB, Zhou G, Tian AM. J Phys Chem A. 2006;110:7441. doi: 10.1021/jp062214u. [DOI] [PubMed] [Google Scholar]
- 24.Bruney LY, Bledson TM, Strout DL. Inorg Chem. 2003;42:8117. doi: 10.1021/ic034696j. [DOI] [PubMed] [Google Scholar]
- 25.Sturdivant SE, Nelson FA, Strout DL. J Phys Chem A. 2004;108:7087. [Google Scholar]
- 26.Strout DL. J Phys Chem A. 2004;108:10911. [Google Scholar]
- 27.Casey K, Thomas J, Fairman K, Strout DL. J Chem Theory Comput. 2008;4:1423. doi: 10.1021/ct8001943. [DOI] [PubMed] [Google Scholar]
- 28.Privott TJ, Casey K, Strout DL. J Und Chem Res. 2014;13:71. [Google Scholar]
- 29.Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]; Adamo C, Barone V. J Chem Phys. 1999;110:6158. [Google Scholar]
- 30.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 31.Grimme S. J Comp Chem. 2006;27:1787. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 32.Purvis GD, Bartlett RJ. J Chem Phys. 1982;76:1910. [Google Scholar]; Scuseria GE, Janssen CL, Schaefer HF., III J Chem Phys. 1988;89:7382. [Google Scholar]
- 33.Dunning TH., Jr J Chem Phys. 1989;90:1007. [Google Scholar]
- 34.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Gaussian Inc.; Wallingford CT: 2010. [Google Scholar]
- 35.Strout DL. J Phys Chem A. 2004;108:2555. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.