

Abstract
Previously we have shown that prenatal moderate arsenic exposure (50 ppb) disrupts glucocorticoid receptor (GR) programming and that these changes continue into adolescence in males. However, it was not clear what the molecular mechanisms were promoting these GR programming changes or if these changes occurred in arsenic-exposed females. In the present studies, we assessed the effects of arsenic on protein and mRNA of the glucocorticoid receptor (GR) and 11β-hydroxysteroid dehydrogenase (Hsd) isozymes and compared the levels of methylation within the promoters of the Nr3c1 and Hsd11b1 genes in female fetal brain at embryonic days (E) 14 and 18. Prenatal arsenate exposure produced sex specific effects on the glucocorticoid system. Compared to males, females were resistant to arsenic induced changes in GR, 11β-Hsd-1 and 11β-Hsd-2 protein levels despite observed elevations in Nr3c1 and Hsd11b2 mRNA. This sex-specific effect was not due to differences in the methylation of the GR promoter as methylation of the Nr3c1 gene was either unchanged (region containing the egr-1 binding site) or similarly reduced (region containing the SP-1 transcription factor binding site) in both males and females exposed to arsenic. Arsenic did produce sex and age-specific changes in the methylation of Hsd11b1 gene, producing increased methylation in females at E14 and decreased methylation at E18.These changes were not attributed to changes in DNMT levels. Since arsenate metabolism could interfere with the generation of methyl donor groups, we assessed glutathione (GSH), S-adenosylmethionine (SAM) and As 3 methyltransferase (As3MT). Exposed males and females had similar levels of As3MT and SAM; however, females had higher levels of GSH/GSSH. It is possible that this greater anti-oxidative capacity within the females provides protection against low to moderate arsenate. Our data suggest that the GR signaling system in female offspring was not as affected by prenatal arsenic and predicts that female arsenic-exposed mice should have normal GR feedback regulation.
Keywords: Arsenate, Prenatal, Glucocorticoid, 11B-Hydroxysteroid, Dehydrogenase-1, DNA methylation, glutathione, S-adenosylmethionine
1. Introduction
It is estimated that over 100 million people are exposed to arsenic via drinking water. The World Health Organization (WHO) recommends that 0.010 mg/l (10 ppb) arsenic in water is safe for ingestion. While the WHO standard is in the parts-per-billion range, several millions of people worldwide either consume unregulated well-water or live in regions where the standard amount of arsenic is in the parts-per-million range. Arsenic is a common, pervasive environmental contaminant that affects almost every major organ system in the body including the brain. Inorganic arsenic, found in drinking water, exists in two oxidation states as As3+ (arsenite) and As5+ (arsenate), both of which readily accumulate in the body. While arsenite is considered to be more toxic than arsenate because of its thiol reactivity, both inorganic forms are considerably more toxic than their organic counterparts [19] and arsenate is converted to arsenite in vivo through the activity of a purine nucleoside phosphorylase, arsenate reductase. Arsenic biotransformation after ingestion involves several redox reactions and associated methylation steps to form arsenical species that are excreted from the body; these include the trivalent and pentavalent forms of inorganic arsenic and the mono- and dimethylated metabolites of each inorganic form. Several studies have demonstrated that the speciation of arsenic, not only the total exposure, impacts type and severity of molecular dysfunction [9], [47]. Metabolism of arsenic after exposure actually produces more toxic species (conversion of arsenate to arsenite) using glutathione (GSH) and other thiol groups as reducing agents. Arsenite is then distributed throughout tissues into cytosol where it undergoes methylation, which requires S-adenosyl methionine (SAM) as the methyl donor. This has led several investigators to suggest that arsenic may deplete the levels of SAM in the body, thereby reducing the levels of methyl donors available for DNA methylation, leading to aberrant DNA methylation and other aberrant epigenetic processes including histone posttranslational modifications that require methyl groups [32], [33], [8], [1], [24], [26], [30], [43]. However, the mechanisms by which arsenic impedes epigenetic processes are still under investigation.
There are several components of epigenetic gene regulation including DNA methylation, histone posttranslational modifications (PTM), and noncoding RNAs. DNA methylation, located at CpG dinucleotides, is the most widely studied component of epigenetics. Three highly conserved enzymes, the DNA methyltransferases (DNMT1, DNMT3A, DNMT3B), impart single methyl groups (CH3) from the universal methyl donor S-adenosyl methionine (SAM). These methylated CpG sites can occur in heterochromatin, or regions of the DNA not actively transcribed. CpG regions are present in 5⿲ regulatory regions of genes including transcriptional start sites and are typically unmethylated, with only 10% containing DNA methylation [13]. CpG dinucleotides within promoters without CpG islands are usually methylated based on the tissue specificity and are indicative of the transcriptional status of the gene. In essence, methylation of a CpG is indicative of repression of gene expression, while demethylation of a CpG can be indicative of active transcription [10]. Genes that contain CpG islands, short (0.5⿿4 kb) stretches of CG rich DNA, include most constitutively expressed genes and genes important for developmental regulation [38]. CpG islands are often unmethylated, where methylation then is able to permit dynamic and temporal regulation of gene expression. DNA methylation is critical to development of an organism, and in the context of developmental toxins, is the most widely studied epigenetic mechanism for toxicity [40]. If arsenic exposure reduces methylation capacity or alters the machinery necessary for methylation, then this could result in aberrant DNA methylation and gene expression patterns.
Recent work in our lab has shown that prenatal arsenic exposure alters glucorticoid programming [22], [20], [21], [11], [42], [4], [43]. This altered programming occurs in utero and can be detected as early as embryonic day 14 [4]. The changes to the glucocorticoid system are perpetuated into adulthood animal [20], [21], [11]. However, our previous work focused only on males and the impact of arsenic on female offspring was not assessed.The goal of the studies presented here are to compare the impact of arsenic on male and female offspring.
In an effort to identify the molecular mechanism for arsenic-induced changes to the glucocorticoid receptor system, we have evaluated the levels of DNA methylation within specific regions of the promoter regions of the glucocorticoid receptor gene (Nr3c1) and 11βHSD1 gene (Hsd11B1), a critical hydroxysteroid dehydrogenase linked to glucocorticoid sensitivity. 11βHSD1 is an oxoreductase which converts inactive cortisone to active corticosterone and increases in expression during fetal development. In addition, we assessed levels of DNMTs, the DNA methyl binding protein (MeCP2), and transcription factors which regulate the expression of GR. Finally, we assessed the metabolic enzymes involved in arsenic metabolism in embryos from male and female exposed mice.
2. Materials and methods
2.1. Chemical hazards
Arsenic is classified as a human co-carcinogen; all arsenicals were handled with caution in accordance with MSDS standards.
2.2. Prenatal arsenic exposure (PAE) paradigm
The Institutional Animal Care and Use Committee at the University of New Mexico (UNM) approved the animal protocols, including the arsenic exposure paradigm, used in this study. C57BL/6 mice obtained from Jackson Labs were maintained on a 12 h reverse light/dark cycle (lights off at 08:00 and lights on at 20:00 h) with ad libitum access to food and water in the Animal Resource Facility at UNM. Arsenic exposure was performed as previously described [44]. Briefly, singly-housed female mice aged 55 days were acclimated to drinking 50 parts-per-billion (ppb) arsenic water (sodium arsenate, Sigma⿿Aldrich) for 10 days prior to mating.The 50 ppb level was chosen due to the fact that water standard prior to 2006 in the United States was 50 ppb. Moreover, the detoxification capacity of the mouse liver is significantly greater than that of humans [17], [2] making the 50 ppb concentration more relevant to much lower human exposures. Arsenic water was prepared weekly using standard tap water. Control mice were administered tap water which contains approximately 2⿿5 ppb arsenic. Tap water was chosen as the control as there are key minerals which would be missing from Milli-Q water. It is likely that the local tap water contains pesticides. These were not measured or controlled for in this study. Female mice were paired with breeding males for a single day. Pregnant females (dams) continued to drink arsenic-laced water throughout pregnancy until fetuses were removed at E14 or E18. Dams were decapitated and embryos removed. Fetal telencephalon and liver were dissected and frozen in liquid nitrogen. Male and female embryos were identified using qPCR of Sry levels in DNA extracted from the embryo body as described previously [4]. For each experiment, at least 7 different litters from different dams were used; e.g., n = 7 represents the number of different litters used with one animal per litter to avoid litter effects.
2.3. DNA methylation analysis of Nr3c1 and Hsd11b1
Evaluation of methylation status of was completed with a Methylated DNA Immunoprecipitation (MeDIP)-qPCR assay. Whole frozen (⿿80 °C) embryonic mouse brains were homogenized and gDNA was extracted using the Qiagen DNeasy Blood & Tissue Kit (cat #: 69504; Valencia, CA) according to the manufacturer⿿s methods. gDNA samples were treated with Life Technologies PureLink RNase A (cat #: 12091-021; Grand Island, NY) to remove any contaminating RNA. gDNA was quantified with the Qubit® dsDNA HS Assay Kit (cat #: Q32851; Life Technologies) on a Qubit® 2.0 Fluorometer. MeDIP was conducted using the Epigentek Methylamp Methylated DNA Capture (MeDIP) Kit (cat #: P-1015-24; Farmingdale, NY). 2 μg of gDNA from each sample was fragmented with a Kontes Micro Ultrasonic Cell Disrupter (model: KT50) by 3 ÿ 12 s pulses at 20% separated by 40 s incubations on ice. gDNA fragment sizes ranged from 200 to 1000 bp with an average of about 600 bp and were verified on a 3% agarose gel. The 2 μg of fragmented gDNA were then divided into two equal reactions and melted at 95 °C for 10 min then placed immediately on ice. 5 μl of each melt reaction (10 μl total per sample) was set aside as Input DNA. The remaining pair of IP samples was then processed with the MeDIP kit per the user manual only to be merged into one final elution volume of 20 μl. Input and IP ssDNA was quantified with the Qubit® ssDNA Assay Kit (cat #: Q10212). ssDNA Input and IP samples were run on a LightCycler 96 qPCR instrument (Roche Diagnostics, Indianapolis, IN) with Roche FastStart Essential DNA Green Master (cat #: 6402712001). qPCR primers (Supplemental Figs. 1 and 2) were designed against gene promoter CpG island regions with putative or functional transcription factor binding sites using the NCBI Primer-BLAST online software [52]. Primer efficiencies were found to be between 90 and 110%. qPCR dissociation curves and gel electrophoresis were used to determine primer specificity. qPCR was conducted under standard profile conditions. Data were analyzed using the relative quantification method with the Input DNA as an endogenous control. MeDIP reaction efficiencies were determined and οCq values were adjusted to account for efficiency variances. Statistical significance was determined by Student⿿s t-test using GraphPad Prism 6 software.
2.4. Nr3c1, Hsd11b1, and Hsd11b2 mRNA assessment
mRNA levels for Nr3c1, Hsd11b1, and Hsd11b2 were determined using previously published methods [4].
2.5. Tissue preparation for protein quantification
Frozen (⿿80 °C) mouse brain tissue, post nuclear lysate (PNL) and nuclear (NUC) fractions were prepared from embryonic day 14 (E14) and E18 brain. The PNL contains both the membrane and cytosolic fractions. Tissue fractions were homogenized and prepared following established protocols [4].
2.6. Western assessment
Immunoblotting for DNA methylation related proteins (MeCp2, DNMT1, DNMT3a, DNMT3b), transcription factors (Egr-1, SP-1, and cJun/Ap-1), and arsenic 3 methyltransferase (As3MT) was conducted essentially as previously described using our established protocols [11], [44]. Brain tissue lysates from one animal were prepared and the nuclear fraction (NUC) and post nuclear lysate (PNL, containing cytosolic and membrane proteins) isolated for protein expression analysis. Total protein concentration was determined by Bradford assay. Antibody and protein optimization was performed for each target protein in each type and fraction of tissue; Coomassie staining was demonstrated to be linear for all protein concentrations used. Membranes were incubated overnight at 4 °C in primary antibodies diluted in PBS-T (see Table 1). Membranes were washed, then incubated for 45 min in their respective secondary antibodies solutions ([1:15,000], 0.01% SDS, 0.1% Tween, in PBS pH 7.4). Secondary antibodies were from Li-COR: rabbit IRDye 680RD 926-68073 and mouse IRDye 800CW 926-32212. Quantification of protein expression was performed using Image Studio version 4.1. Blots were normalized by Coomassie staining using IRDye® Blue Protein Stain (LI-COR; #3343C056) or with traditional Coomassie staining solution (0.1% (w/v) Coomassie Brilliant Blue R-250, 50% methanol, 7% Glacial Acetic Acid, in ultra-pure H2O) [27]. Values are presented as protein expression normalized to Coomassie staining and renormalized to controls.
Table 1.
Antibody source concentration and product information.
| Age/fraction/tissue | Total protein | Antibody concentration | Target protein | Product code | Company |
|---|---|---|---|---|---|
| E14 Nuclear male whole brain |
12 μg | [1:1000] | C-Jun | cs#9185 | Cell signaling |
| 12 μg | [1:1000] | SP 1 | sc-14027 | Santa Cruz biotechnology | |
| 18 μg | [1:1000] | Egr-1 | cs#4153 | Cell signaling | |
| 12 μg | [1:500] | Dnmt1 | sc-20701 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3b | sc-20704 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3a | M0229S | New England biolabs | |
| 15 μg | [1:500] | MeCP 2 | sc-5755 | Santa Cruz biotechnology | |
| E14 Nuclear female whole brain |
12 μg | [1:1000] | C-Jun | cs#9185 | Cell signaling |
| 12 μg | [1:1000] | SP1 | sc-14027 | Santa Cruz biotechnology | |
| 18 μg | [1:1000] | Egr-1 | cs#4153 | Cell signaling | |
| 12 μg | [1:500] | Dnmt1 | sc-20701 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3b | sc-20704 | New England biolabs | |
| 15 μg | [1:1000] | Dnmt3a | M0229S | Santa Cruz biotechnology | |
| 15 μg | [1:500] | MeCP 2 | sc-5755 | Santa Cruz Biotechnology | |
| 13.5 μg | [1:500] | As3MT | sc-376537 | Santa Cruz Biotechnology | |
| E18 Nuclear male whole brain |
13.5 μg | [1:1000] | c-Jun | cs#9185 | Cell signaling |
| 13 μg | [1:1000] | SP1 | sc-14027 | Santa Cruz biotechnology | |
| 18 μg | [1:1000] | Egr-1 | cs#4153 | Cell signaling | |
| 12 μg | [1:500] | Dnmt1 | sc-20701 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3b | sc-20704 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3a | M0229S | New England biolabs | |
| 15 μg | [1:500] | MeCP 2 | sc-5755 | Santa Cruz biotechnology | |
| 13.5 μg | [1:500] | As3MT | sc-376537 | Santa Cruz biotechnology | |
| 13 μg | [1:1000] | SP1 | sc-14027 | Santa Cruz biotechnology | |
| E18 Nuclear female whole brain |
14 μg | [1:1000] | c-Jun | cs#9185 | Cell signaling |
| 14 μg | [1:1000] | SP1 | sc-14027 | Santa Cruz biotechnology | |
| 18 μg | [1:1000] | Egr-1 | cs#4153 | Cell signaling | |
| 12 μg | [1:500] | Dnmt1 | sc-20701 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3b | sc-20704 | Santa Cruz biotechnology | |
| 15 μg | [1:1000] | Dnmt3a | M0229S | New England biolabs | |
| 15 μg | [1:500] | MeCP 2 | sc-5755 | Santa Cruz biotechnology | |
| 13.5 μg | [1:500] | As3MT | sc-376537 | Santa Cruz Biotechnology | |
| 20 μg | [1:500] | GR | sc-1004 | Santa Cruz biotechnology | |
| 20 μg | [1:500] | HSD11b2 | sc-20176 | Santa Cruz biotechnology | |
| 20 μg | [1:500] | HSD11b1 | sc-19259 | Santa Cruz biotechnology | |
2.7. Evaluation of glutathione levels
Glutathione levels were determined usinga colorimetric assay (Detect X, Arbor Assays #K006-H1, Ann Arbor, MI) as described by the manufacturer. Levels of glutathione disulfide (GSSG or GSSH) and glutathione (GSH) at both endpoint (4 min) and kinetic (minute-by-minute analysis) were determined using a TECAN plate reader (Infinite M200, analyzed using Magellan software).
2.8. Speciation of arsenic
Speciation analysis of arsenite was performed as previously reported [4] using HPLC-ICP-MS male and female E18 brain tissue. Quantification was conducted using calibration standards for Arsenite (AsIII), Arsenate (AsV), Monomethyl Arsenic (MMA), and Dimethyl Arsenic (DMA) which were prepared similar to the tissue samples using the buffered mobile phase (2 mM octanesulfonic acid and 2 mM malonic acid, pH 7). The detection limit was 0.010 μg/L for iAsIII DMA, MMA and iAsV. The system was calibrated using a blank and four point calibration standards. Samples were analyzed in automated mode. Chromatogram retention times were adjusted, peaks were identified, and the calibration curves linearity was verified for each species. The analytical data were reprocessed and validated.
2.9. SAM/SAH level analysis
Levels of SAM and SAH were determined by HPLC using the method described by She et al. [37]. SAM (iodide salt), SAH, HPLC grade methanol, and 1-heptanesulfonic acid were obtained by Sigma⿿Aldrich (St. Louis, MO). Ammonium dihydrogen phosphate (99.9% pure) was obtained from Acros Organics (New Jersey, USA). SAM and SAH standards were dissolved in sample buffer (4% perchlorate, 1% sodium metabisulfite, and 1% EDTA) and then diluted to generate a standard range of 25, 50,100, 200, 400 pmol. Tissues were weighted, diluted in 4ÿ volume of the sample buffer, and sonicated. They were centrifuged at 750 ÿ g for 5 min and filtered using cellulose acetate spin columns (0.45 μm) and then placed in autosampler vials for HPLC detection. The HPLC system was a PerkinElmer Flexar system equipped with an injector, column oven, and a UV detector set at 254 nm and a Flexar 225/275 autosampler. A TSKgel ODS-80Tm column (25 cm ÿ 4.6 mm, 5 μm, Tosoh Co., Tokyo, Japan) was used. The column was maintained at 35 °C while samples in the autosampler were held at 20 °C. The mobile phase was 40 mM NH4H2PO4, 8 mM 1-heptanesulfonic acid sodium salt, and 18% (v/v) methanol, pH 3.0 by HCl and filtered through a 0.45 μm filter. The mobile phase was isocratically applied with a flow rate of 1 ml/min.Under these conditions, the SAH peak was seen at approximately 28 min and the SAM peak at 32 min. Data were analyzed using the Chromera software.
2.10. Statistical analysis
Protein expression was normalized to the average of total protein expression evaluated by Coomassie staining. All data are presented as mean ± SEM normalized to control values, and a p value of <0.05 was set for statistical significance. Data were analyzed by Student⿿s t-test using GraphPad software (GraphPad Software, v. 6.0; San Diego, CA) or, for the GSH/GSSH by a two-way ANOVA and the SAM and SAH data by three way ANOVA (SPSS, IBM v.22). The number of litters is reported for each experiment, with only one animal per litter used to avoid litter confounds.
3. Results
3.1. Impact of prenatal arsenic on GR, hydroxysteroid dehydrogenase isozyme in female embryonic brain
To assess the impact of prenatal arsenate exposure on the expression of glucocorticoid receptor system in female brain tissue, protein levels of the glucocorticoid receptor, 11β-Hsd isozymes were determined by western blot in both the post nuclear lysate (PNL) and the nuclear (Nuc) fraction from embryonic brain at E14 and E18 (Fig.1A⿿D) Unlike our findings with male embryonic brain where we observed significant effects of arsenic on the levels of GR and 11β-Hsd isozymes, the females exposed to arsenate during development displayed no differences in protein levels for GR or the 11β-Hsd isozymes (Fig. 1 A⿿D). Since the female embryos tested here were the siblings to those in our earlier study on the male embryos [4], it indicates that the identical prenatal environment produced a sexually dimorphic effect. However, while the protein expression was unaltered (Fig. 1A⿿D), the mRNA for Nr3c1 (GR gene) was significantly elevated in the female E14 brain (Fig. 2A, t(11) = 2.3, p = 0.04), but not altered at E18 (Fig. 2D). Similarly, 11β-Hsd-2 mRNA expression was significantly increased in the arsenic exposed females (Fig. 2C, t(13) = 3.37, p = 0.005) at E14 but was not different from controls at E18 (Fig. 2F). 11β-Hsd-1 mRNA expression levels were not altered by arsenic at either developmental time point (Fig. 2B and E)
Fig. 1.

The effect of prenatal arsenic (50 ppb) in female post nuclear (PNL) and nuclear (Nuc) fractions from E14 and E18 brain. Protein expression of Glucocorticoid receptor (GR), 11B-HSD1 and 2 in the arsenic exposed group (filled bars) compared to control (open bars), did not differ. Data are mean ± SEM, n = 7⿿9. The intensity of the specific antibody was corrected to within blot Coomassie stain as described in (Caldwell et al., 2015).
Fig. 2.

Prenatal arsenic (50 ppb) significantly increase the level of mRNA expression of Nr3c1 (GR) and Hsd11b2 (11b-hydroxysteroid dehydrogenase 2) in E14 female brain, but there was no effect of arsenic at E18.No effect on the levels of Hsd11b1 (11b-hydroxysteroid dehydrogenase 1) was observed at either developmental time-point. Fold expression of the target genes were acquired by the οοCt (Comparative CT) method. Data presented are mean ± SEM n = 7⿿11, *p = .04, **p = 0.005. Arsenic are presented in the filled columns and controls presented in the unfilled columns.
3.2. Prenatal arsenic exposure (PAE) reduced methylation in Nr3c1 promoter in the region encompassing SP-1 transcription factor binding site, but not the Egr-1 region
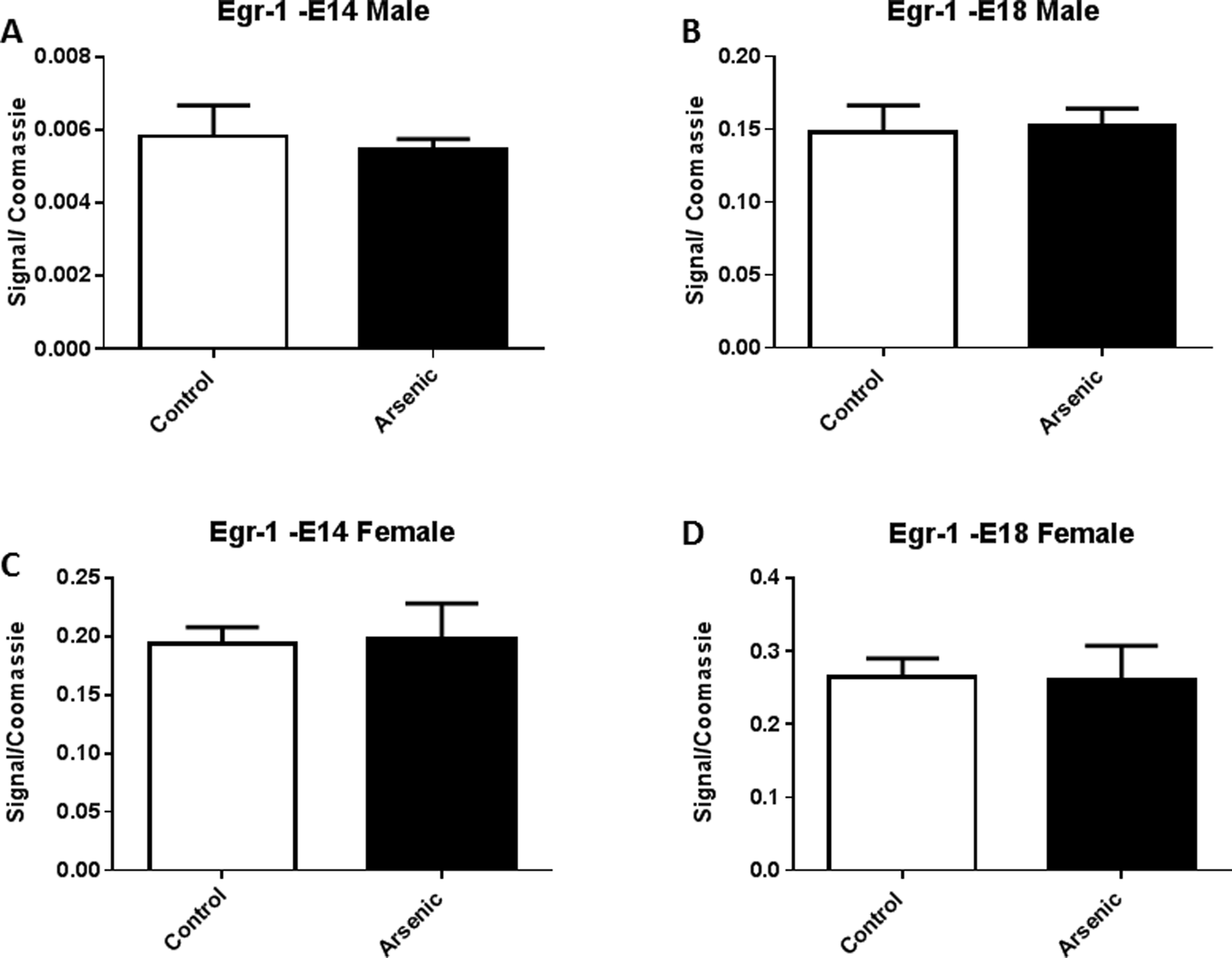
In both male and female embryonic day 14 brain, arsenic reduced the level of methylation in the SP-1 transcription factor binding region of the Nr3c1 promoter (Fig. 3A and C, t(10) = 2.6, p = .03) and t(10) = 2.2, p = .05 respectively). This effect was not present just 4 gestational days later on E18 (Fig. 3B and D) in either the male or female brain, suggesting a possible compensatory mechanism to counter the effect. Methylation in the egr-1 binding region of Nr3c1 was not different in either male of female offspring from the arsenic exposed dams at either gestational time point (Fig. 4A⿿D).
Fig. 3.

Prenatal arsenic (50 ppb) significantly decreased the level of methylation within the SP-1 binding site region of Nr3c1 (see Supplemental Fig. 1) in E14 female and male brain. No effect of prenatal arsenic was seen for either sex on the levels of Nr3c1 methylation at either developmental time-point. The levels of methylation within region 1 of Nr3c1 (see Supplemental Fig. 1) was assessed by MeDIP qPCR (see methods). Region 1 primers were developed to encompass the SP-1 transcription factor binding site.Data are expressed as mean fold methylation +/⿿ SEM, n = 5⿿7 different litters. Data from male (A and B) and female (C and D) are presented separately for E14 (A and C) and E18 (B and D) brains. Arsenic are presented in the filled columns and controls presented in the unfilled columns. E14 male t(10) = 2.6, p = 0.03, and E14 female t(10) = 2.2, p = 0.05.
Fig. 4.

There were no differences in the levels of methylation within the Egr-1 binding site (Region 6) in Nr3c1 between male and female at either developmental time-point. The levels of methylation within region 6 of Nr3c1 (see Supplemental Fig. 1) were assessed by MeDIP qPCR see methods). Region 1 primers were developed to encompass the Egr-1 transcription factor binding site. Data are expressed as mean fold methylation +/⿿ SEM, n = 5⿿7 different litters. Data from male (A and B) and female (C and D) are presented separately for E14 (A and C) and E18 (B and D) brains. Arsenic exposed animals are presented in the filled columns and controls presented in the unfilled columns.
3.3. Prenatal arsenic exposure produced sex and age specific changes in the levels of methylation within the promoter region of Hsd11b 1
Levels of methylation within the promoter region (Supplemental Fig. 3) of Hsd11b1 gene were significantly lower in the female E14 brain (Fig. 5C [t(10) = 2.47, p = 0.003] and elevated at E18 [t(10) = 3.07, p = 0.012] exposed to prenatal arsenic (Fig. 5D). Methylation levels were not altered by arsenic in male E14 (Fig. 5A) or E18 (Fig. 5B) brain.
Fig. 5.

Prenatal arsenic (50 ppb) significantly decreased the level of methylation of Hsd11b1 (see Supplemental Fig. 2) in E14 female brain, while increasing methylation in arsenic exposed females at E18. No effect was seen on the levels of Hsd11b1 methylation in male brain following arsenic exposure at either developmental time-point. Data are expressed as mean fold methylation +/⿿ SEM, n = 6⿿7 different litters. Data from male (A and B) and female (C and D) are presented separately for E14 (A and C) and E18 (B and D) brains. Arsenic are presented in the filled columns and controls presented in the unfilled columns. t(10) = 2.47, p = 0.033 for E14 and t(10) = 3.07, p = 0.012.
3.4. Assessment of DNA methyltransferases (DNMTs) in response to prenatal arsenic exposure
Given the observation that arsenic altered the level of methylation within specific promoter regions of Nr3c1, we assessed the levels of DNA methyltransferase. DNMT1 levels were not significantly altered by prenatal arsenic in males (Fig. 6A and B) at either E14 or E18. Similarly, female arsenic exposed mice did not show a change in the levels of DNMT1 at either time point (Fig. 6C and D). Further, DNMT3a and DNMT3b were not altered by prenatal arsenic in either sex at either developmental time point (Fig. 7, Fig. 8A⿿D). Although we should note that DNMT3b levels did approach significance (p = 0.064) in the male E18 mice.
Fig. 6.

The effect of prenatal arsenic (50 ppb) in male (top row) and female (bottom row) nuclear (Nuc) fractions from E14 (left column) and E18 (right column) brain on DNMT1 protein expression. Arsenic exposed group (filled bars) compared to control (open bars), did not differ. Data are mean ± SEM, n = 7⿿9. The intensity of the specific antibody was corrected to within blot Coomassie stain as described in (Caldwell et al., 2015).
Fig. 7.

The effect of prenatal arsenic (50 ppb) in male (top row) and female (bottom row) nuclear (Nuc) fractions from E14 (left column) and E18 (right column) brain on DNMT3a protein expression. Arsenic exposed group (filled bars) compared to control (unfilled bars), did not differ. Data are mean ± SEM, n = 7⿿9. The intensity of the specific antibody was corrected to within blot Coomassie stain as described in (Caldwell et al., 2015).
Fig. 8.

The effect of prenatal arsenic (50 ppb) in male (top row) and female (bottom row) nuclear fractions from E14 (left column) and E18 (right column) brain on DNMT3b protein expression. Arsenic exposed group (filled bars) compared to control (unfilled bars), did not differ. Data are mean ± SEM, n = 7⿿9. The intensity of the specific antibody was corrected to within blot Coomassie stain as described in (Caldwell et al., 2015).
3.5. Assessment of MeCP2 levels in response to prenatal arsenic exposure
MeCP2 is a part of a family of proteins which binds specifically to methylated CpG domains on DNA but can also interact with SIN3A to recruit histone deacetylases. Given our observation of changes in histone acetylation [43] and the indication of altered methylated levels in the Nr3c1 promoter region, we evaluated the levels of MeCP2 in E14 and E18 brain from male and female PAE.No differences in MeCP2 protein levels were found at either developmental time point in either sex (Fig. 9).
Fig. 9.

The effect of prenatal arsenic (50 ppb) in male (top row) and female (bottom row) nuclear (Nuc) fractions from E14 (left column) and E18 (right column) brain on MeCP2 protein expression. Arsenic exposed group (filled bars) compared to control (unfilled bars), did not differ. Data are mean ± SEM, n = 7⿿9. The intensity of the specific antibody was corrected to within blot Coomassie stain as described in (Caldwell et al., 2015).
3.6. Assessment of transcription factor levels in response to prenatal arsenic exposure
Three primary transcription factors regulating Nr3c1 are Egr-1, SP-1 and cJun. We assessed the levels of these transcription factors in the nuclear fraction of the E14 and E18 male and female brain. There were no significant differences in any of the transcription factor levels between control and arsenic at either developmental time points for either sex (Supplemental Figs. 3⿿5).
3.7. Assessment of the levels of GSH and GSSH in liver and brain at E18 in male and females following prenatal arsenic exposure
Reduced glutathione (γ-glutamylcysteinylglycine, GSH) is a major tissue antioxidant. The formation of a disulfide bond between two GSH molecules gives rise to glutathione disulfide (GSSH or GSSG). During increased oxidative stress the levels of GSSH will rise and the ratio of GSH to GSSH will decrease. GSH plays a key role in arsenic metabolism converting arsenate to arsenite. Levels of glutathione (GSH) and the reduced form (GSSH) were assessed both at end point, or assay completion, and as a kinetic assessment. This enzymatic reduction reaction also plays a critical role in the metabolic processing of arsenate. The levels of GSH/GSSH were measured in male and female liver and brain at E18 both at a pseudo-end point (Fig. 10A and C) and at multiple times during the reaction to provide an assessment of the kinetic rate (Fig. 10 B and D). Using end point data, there was a significant prenatal effect [F(1,16) = 26.2, p = 0.0001] and a significant sex effect [F(1,16) = 16.08, p = 0.001], but no interaction present (Fig. 10A). At end point, arsenic increased the GSH/GSSH ratio in both male and female brain similarly, though female control tissue had higher GSH/GSSH levels than male controls (Fig. 10A, p = .003). In brain, there was a prenatal treatment main effect [F(1,16) = 108.4, p = 0.0001] and a sex main effect [F(1,16) = 175.8, p = 0.0001] and a prenatal ÿ sex interaction [F(1,16) = 66.5, p = 0.0001] when the data were measured as a kinetic reaction (Fig. 10B). There was a clear increase in GSH/GSSH ratio due to arsenic in the female brain tissue (Fig. 10B, p = 0.005), indicating a greater oxidative capacity and a more rapid enzymatic rate in female brain tissue. In the liver, neither kinetic nor end point data revealed any significant findings (Fig. 10C and D).
Fig. 10.

The effect of prenatal arsenic (50 ppb) in male (left) and female (right) brain (top row) and liver (bottom row) homogenates from E18 on glutathione enzymatic activity expressed as the ratio GSH/GSSH. Data are taken at the single 4 min end point (left column of figures) or as a kinetic reaction (right column of figures) integrated every minute for 4 min. Data are mean ± SEM, n = 5⿿8 with arsenic exposed group indicated in the filled bars compared to control unfilled bars.
3.8. Assessment of arsenic 3 methyltransferase and SAM levels
Arsenate is converted to arsenite by GSH then it is methylated to monomethyl, dimethyl, and trimethyl forms by the combined action of As3MT and SAM. We evaluated the levels of As3MT in the PNL fraction brain from male and female at E14 and E18 (Fig. 11A⿿D). There were no significant differences in the levels between the exposure groups at either developmental time-points for ether sex (Fig. 11A⿿D).
Fig. 11.

The effect of prenatal arsenic (50 ppb) in male (top row) and female (bottom row) post nuclear lysate (PNL) fractions from E14 (left column) and E18 (right column) brain on As3MT protein expression. Arsenic exposed group (filled bars) compared to control (unfilled bars), did not differ. Data are mean ± SEM, n = 7⿿9. The intensity of the specific antibody was corrected to within blot Coomassie stain as described in (Caldwell et al., 2015).
S-adenosylmethionine (SAM) and its reduced form S-adenosyl homocysteine (SAH) levels were determined in E14 and E18 from arsenic and control male and female brain and liver (Table 2). SAM/SAH ratio have been used to estimate the capacity of biological methylation [7]. In brain, there was in significant effect of age for SAM [F(1,40) = 21.5, p = 0.0001] and SAH [F(1,40) = 5.6, p = 0.023] levels. With age, brain levels of SAM increased overall and the levels of SAH reduced. There were no effects of prenatal exposure or sex nor interaction between any of the independent variables on brain levels of SAM or SAH. Similarly in liver there was a significant effect of age on SAM [F(1,35) = 130.0, p = 0.0001] and SAH [F(1,35) = 59.1, p = 0.0001] but unlike the brain the levels of both SAM and SAH decreased with age. In liver, there was a significant interaction between age and sex for SAH levels [F(1,35) = 10.5, p = 0.003]. The nature of the interaction was due to a sharper decrease in the females due to age than in the males. The very low SAH levels in the control males at E14 is the likely source. No other main effects or interactions were significant.
Table 2.
Levels of SAM and SAH determined in brain (top table) and liver (bottom table) in arsenic and control E14 and E18 male and female tissues. Data are presented as average (+/⿿ SEM) in nmol/g tissue wet weight from n = 5⿿6.
| Brain | E14 male |
E14 female |
E18 male |
E18 female |
||||
|---|---|---|---|---|---|---|---|---|
| Control | Arsenic | Control | Arsenic | Control | Arsenic | Control | Arsenic | |
| SAM | 2.3 (±0.5) | 2.3 (±0.6) | 2.6 (±0.5) | 2.5 (±0.4) | 4.0 (±0.4) | 3.5 (±0.7) | 4.1 (±0.5) | 4.7 (±0.2) |
| SAH | 0.12 (±0.02) | 0.07 (±0.02) | 0.10 (±0.03) | 0.13 (±0.02) | 0.08 (±0.001) | 0.08 (±0.002) | 0.06 (±0.001) | 0.08 (±0.001) |
| Liver | E14 male |
E14 female |
E18 male |
E18 female |
||||
|---|---|---|---|---|---|---|---|---|
| Control | Arsenic | Control | Arsenic | Control | Arsenic | Control | Arsenic | |
| SAM | 2.1 (±0.2) | 2.4 (±0.2) | 2.5 (±0.2) | 2.1 (±0.2) | 6.3 (±0.5) | 5.1 (±0.5) | 6.5 (±0.4) | 6.6 (±0.4) |
| SAH | 0.15 (±0.03) | 0.23 (±0.03) | 0.29 (±0.03) | 0.27 (±0.03) | 4.0 (±0.4) | 6.8 (±0.7) | 4.5 (±0.3) | 5.1 (±0.05) |
3.9. Levels of arsenate, arsenite, monomethylarsenicals (MMA III) and dimethylated arsenicals (DMA V) in male and female E18 brain
A possible sex dependent difference in the bioaccumulation of arsenicals in the fetal brain was assessed at E18 using our previously published ICP-MS procedures [4]. There were no significant sex differences in the levels of any of the arsenicals measured.There was a greater concentration of arsenate compared with arsenite and higher levels of DMA relative to MMA (Table 3).
Table 3.
Levels of arsenicals in brain from male and female E18 arsenic exposed mice. Data are presented as mean ppb ± SEM, n = 6⿿7.
| In ppb | AsV | AsIII | MMA | DMA |
|---|---|---|---|---|
| E18 male | 1.6 (±0.2) | 0.28 (±0.02) | 0.02 (±0.001) | 0.13 (±0.04) |
| E18 female | 1.3 (±0.2) | 0.34 (±0.03) | 0.03 (±0.001) | 0.14 (±0.03) |
4. Discussion
During development, methylation and demethylation of DNA and posttranslational modifications of histones are dynamically involved in regulating gene and protein levels in a temporal and cell specific manner. Dysregulation of the extent and timing of these epigenetic controls by environmental toxins can alter the developmental programming of key biological systems. Determining the impact of fetal toxic exposures on the status of factors associated with epigenetic control of genetic programs could provide insight into how fetal programming leads to disease susceptibility in adulthood. While the impact of arsenic exposure on the epigenome has been extensively studied in the context of cancer research, the influence of this toxicant in the developing brain, particularly as it relates to epigenetics, is not well understood. Arsenic has been shown to alter the levels of histone methylation [8] and DNA methylation [35], but only a few have explored potential sex-dependent effects of prenatal arsenic on epigenetic modifications [29], [3], [43]. A study of mother newborn pairs from Bangladesh found DNA methylation was increased in male, but not female, newborn cord blood [29]. In our own work, we found developmental exposure in mice to 50 ppb arsenic affected histone3 lysine9 trimethylation patterns in a sex and brain region dependent manner [43]. The study presented here extends these sex dependent effect of prenatal arsenic.
In our previous studies, we found that in male embryos arsenic exposure significantly decreased expression of both protein and mRNA in brain of GR and the 11β-HSD1 enzyme [4]. Expression of 11βHSD1 plays a key role in glucorticoid synthesis and in the expression and set point of the GR negative feedback regulation. This feedback regulation mechanism is established early in development (about E16.5). In adult arsenic exposed males we observed a continuing failure of GR negative feedback regulation along with a persistent decrease in GR and 11β-HSD1 [11], [4].
In the present study examining arsenic exposed female-embryos, we found no changes in the protein levels of GR, 11β-HSD1, and 11β-HSD2. However, there was an increase in Nr3c1 (GR) and Hsd11b2 message at E14 in the brain from female arsenic exposed condition.These findings were quite different from those we observed in the male fetus [4]. In the males, prenatal arsenic produced a decrease in mRNA for Nr3c1, Hsd11b1, and Hsd11b2 at E14 along with a decrease in protein expression of GR and 11-βHsd1 but an increase in 11-βHsd2.This increase in 11-βHsd2 along with the decrease in 11-βHsd1 could indicate a developmental delay in the males. Normally, there should be an increase in 11-βHsd1 expression with a corresponding decrease in11-βHsd2 at E18. In the present data looking at females, the only significant changes in mRNA due to arsenic exposure was an increase in Nr3c1 and Hsd11B2 at E14 with no corresponding changes in these proteins. The findings suggest that there is a translational regulation which is occurring within the females in response to arsenic that is not engaged in the male exposed fetal brains. To our knowledge, these studies are the first to demonstrate that gestational exposure to moderate levels of arsenic results in sexually dimorphic changes to the glucocorticoid system.
Levels of methylation within the CpG islands surveyed in our study found that arsenic exposure at E14 reduced methylation in both male and female for the SP-1 transcription factor binding area of the promoter (region-1, corresponding to the promoter region of exon 1f) of Nr3c1 but not the Egr-1 binding region (region 6). In general, the levels of methylation in these regions of Nr3c1 inversely correlate with the levels of glucocorticoid mRNA and often the levels of protein as well [23], [28]. In E14 males, prenatal arsenic decreased the levels of methylation in region 1 (Fig. 3), but decreased Nr3c1 mRNA and decreased GR protein levels [4]. In females, while we did observe an increase in the levels of Nr3c1 mRNA at E14, we did not see a corresponding increase in GR protein, suggesting a decreased translation rate or a post translational regulation.
11βHsd-1 is commonly found in tissues co-localized with GR [39]. Levels of Hsd11b1 mRNA can be increased by corticosterone and activity at the GR suggesting that there is a feed-forward system between GR and Hsd11b1 [51]. It is believed that this relationship between GR and 11Hsdb1 is also key to establishing GR negative feedback regulation [5]. The 11Hsdb1 gene has two promoter sites; promoter site 1 contains a glucocorticoid response element and promoter site 2 contains a CCAAT/enhancer-binding protein (C/EBP) transcription factor site which is required for GR regulation of Hsd11b1 [49], [36]. Since we had already demonstrated that the GR levels were altered in male brain in response to arsenic [4], the primers we used were designed to target promoter 2. For Hsd11b1, fold methylation was decreased in the arsenic exposed females at E14, but then increased relative to controls at E18. This differential regulation between the two time points is a bit surprising as normally expression of Hsd11b1 should be increasing by E18. However, while there were significant methylation changes in the females, there were no corresponding mRNA or protein changes in Hsd11b1 due to arsenic exposure.Arsenic has been shown to down regulate C/EBP in mesenchymal stem cells [50] and reduce the binding of C/EBP to DNA in adipocytes [12]. It is possible that, while there was altered methylation at this site in the female arsenic exposed mice, arsenic mediated changes in the levels of C/EBP could have occurred. Interestingly, there were no methylation changes seen in the Hsd11b1 promoter in the male arsenic exposed mice. We evaluated the levels of several transcription factors including Egr-1, cJun and SP-1 (Supplemental Figs. 3⿿5) and found no effect of sex or arsenic exposure, but we have not assessed C/EBP. While it is intriguing to speculate that there are changes to c/EBP in our arsenic exposure model, future studies would be needed to test this hypothesis.
These age and sex specific methylation changes indicate that it is unlikely arsenic is producing a global methylation change and suggests there is a dynamically regulated epigenetic process. In support of this conclusion, we did not observe any significant changes in the levels of DNMT1, DNMT3a or 3b, which would be expected if there were global methylation differences due to arsenic in our embryos [6].
Finally, because most mammals enzymatically convert inorganic arsenic (iAs) into methylated metabolites, predominately mono- and dimethylated arsenicals we examined the levels and activities of the key enzymes and methyl donor compounds in the male and female brain and liver. To confirm that male and female fetuses were being exposed to the same levels of arsenic and were generating the same levels of speciated forms, we assessed E18 brain levels of arsenite, arsenate, MMA, and DMA levels. In our studies, we observed the largest portion of iAs as arsenate with 15% as arsenite in males and about 20% as arsenite in the female brain at E18.There were no statistically significant differences between the sexes in the arsenical species, suggesting that arsenate metabolism is not significantly impacted by the sex of the fetus. A study by Lu et al. [18] indicates when pregnant mice are treated with either 10 ppm arsenate or arsenite in their water throughout gestation that As(III) levels accumulate in the liver when measured on postnatal day (PND) 1⿿35. It is thought that the arsenite could possibly produce a greater interference of methylation potential in the offspring than As (V), because the levels of DMA were greater at PND1 in the As(III) treated group. However, they do report that the levels of iAs were similar in liver at PND 1 and PND10 and that the DMA levels were not different at PND10 and PND15. Additionally, the authors note that in the brain exposure with either arsenate or arsenite produces similar levels of iAs and DMA accumulation from PND10⿿21, likely due to an immature blood brain barrier in the early stages of gestation.Although the Lu et al. [18] study did not separately assess male and female offspring at PND 1 and their work was conducted on postnatal time points, our findings are in general agreement.
In vivo, arsenate is converted to arsenite via glutathione [45], [25], [48] followed by the conversion to MMA and DMA with the combined action of S-adenosylmethionine (SAM) and As3MT. Given the critical role of glutathione and SAM in the generation of methyl donor groups it has been suggested that arsenic exposures can affect the levels or activity of these enzymes and thereby alter the ability to methylate DNA and histones. We assessed the levels of glutathione, As3MT, and SAM/SAH in the arsenic exposed male and female embryos. We did not observe a difference in the levels of As3MT between the male and female arsenic exposed brains at either E14 or E18. This is in contrast to a study using frogs which showed 0.5⿿100 ppm arsenate first decreased As3MT at the lower level of exposure but then produced an increase in As3MT levels in the 100 ppm concentration condition [14], thus it is possible that at higher arsenate exposures As3MT levels could be affected.
In our study, there was a significant difference between male and female fetal brain in the ratio of GSH/GSSH. Female brain tissue from arsenic exposed embryos had an elevated GSH/GSSH ratio; which was particularly noted when the glutathione assay was conducted using a kinetic analysis. This relationship was not seen in liver, however, as there was no sex difference nor an effect of arsenic exposure. Brain GSH levels are produced locally and are limited by the available levels of l-cysteine [46]. The sex differences in brain GSH/GSSH ratios are interesting, but not entirely surprising. There is evidence that females may have a more rapid development of the enzymes within this anti- oxidative pathway. Levels of glutathione peroxidase mRNA were higher in female mice early in the postnatal period [41]. Cysteine uptake, a key component of glutathione, is greater in female premature infants than male counterparts [16]. Glutathione-S-transferase levels have also been reported to be higher in females than males [15]. These findings are in agreement with the findings of Ramos-Chavez et al. [31], where they observed higher levels of GSH in postnatal day 15 hippocampus from female but not male offspring of arsenic exposed dams. This suggests that there may be a greater redox rate in females exposed to prenatal arsenic. However, despite this difference, the levels of arsenicals were not different between the male and female fetal brain tissue, suggesting that at these low levels of arsenate (50 ppb) the glutathione rate difference had no substantive effect on arsenic metabolism. Interestingly, like the present study, Ramos-Chavez et al. [31] also did not find a difference between the sexes in the brain levels of arsenic species. Thus, while it is assumed that arsenic metabolism will utilize and perhaps deplete the enzymes needed for epigenetic methylation, our study using a 50 ppb arsenate exposure during gestation did not produce significant decreases in these enzymes. There is evidence that arsenate is being metabolized and converted to arsenite and the MMA and DMA forms but this metabolic activity was not demanding enough to produce a substantive change in the levels of these enzymes.
While the levels of SAM and SAH changed with age, we did not observe any impact of prenatal arsenic or sex on the levels measured in either brain or liver. The fact that arsenic exposure did not alter brain levels of SAM or SAH is not surprising since Rios et al. [34] reported 3 ppm arsenic exposures did not affect the levels of brain SAM in adult female mice. They did, however, observe a decrease in the liver from these mice. The total levels of brain and liver SAM in the Rios et al. [34] study are substantially greater (μmol/g tissue) than were observed (nmol/g tissue) in our study using embryonic tissue. These findings along with the lack of an arsenic effect on As3MT and a lack of an effect overall on GSH/GSSH levels, suggests that the changes in methylation seen in the Hsd11b1 and Nr3c1 promoter regions are not likely the result of the depletion of methyl donor groups due to arsenic metabolism. However, we should note that enzyme activity rates were not measured for As3MT and, while the steady state assessment did not change due to arsenic exposure, it is possible that the rates of the reactions were altered by the exposures which could have altered the dynamic levels of methyl donor groups. Moreover, the levels of arsenic tested in this study are significantly lower than others where changes in SAM and glutathione were reported.
5. Conclusion
In summary, prenatal arsenate exposure (50 ppb) produced significant sex dependent effects on the glucocorticoid system in embryonic brain.Compared to the impact on males, females were resistant to arsenic induced changes in GR, 11β-Hsd-1, and 11β-Hsd-2 proteins despite elevations in Nr3c1 and Hsd11b2 mRNA.This sexual dimorphic effect on GR could not be accounted for by altered promoter methylation since both male and female exposed fetal brain showed a decrease in methylation at E14. Unlike the males, females exposed to arsenate at E14 showed a decrease in methylation of Hsd11b1 gene, but no corresponding mRNA increase. There were no changes in the key transcription factors, DNMTs, to explain this differential effect. With respect to arsenate metabolism, males and females exposed to 50 ppb arsenate had similar levels of As3MT and SAM, however, there was an indication that the GSH/GSSH production rate was faster in the females. It is possible that this greater anti-oxidative capacity within the females provides protection against low to moderate arsenate. Finally, our data provides evidence that prenatal arsenic exposure impacts levels of methylation of targeted regions within the promoter of the glucocorticoid receptor gene (Nr3c1) and in females, alters methylation status of the hydroxysteroid dehydrogenase 1 gene (Hsd11b1) in an age specific manner.
Funding
This work was supported by grants from the National Institute of Environmental Health Sciences [RO1ES019583 to AMA].
Acknowledgments
We thank Kevin Caldwell for helpful discussions and critical reading of the manuscript.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.toxrep.2015.10.003.
Appendix A. Supplementary data
The following are Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Argos M. Arsenic exposure and epigenetic alterations: recent findings based on the illumina 450K DNA methylation Array. Curr. Environ. Health Rep. 2015;2:137–144. doi: 10.1007/s40572-015-0052-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boogaard P.J., Sumner S.C., Bond J.A. Glutathione conjugation of 1,2:3,4-diepoxybutane in human liver and rat and mouse liver and lung in vitro. Toxicol. Appl. Pharmacol. 1996;136:307–316. doi: 10.1006/taap.1996.0037. [DOI] [PubMed] [Google Scholar]
- 3.Broberg K., Ahmed S., Engstrom K., Hossain M.B., Jurkovic Mlakar S., Bottai M., Grander M., Raqib R., Vahter M. Arsenic exposure in early pregnancy alters genome-wide DNA methylation in cord blood, particularly in boys. J. Dev. Orig. Health Dis. 2014;5:288–298. doi: 10.1017/S2040174414000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caldwell K.E., Labrecque M.T., Solomon B.R., Ali A., Allan A.M. Prenatal arsenic exposure alters the programming of the glucocorticoid signaling system during embryonic development. Neurotoxicol. Teratol. 2015;47:66–79. doi: 10.1016/j.ntt.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman K., Holmes M., Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013;93:1139–1206. doi: 10.1152/physrev.00020.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen T., Ueda Y., Dodge J.E., Wang Z., Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang P.K., Gordon R.K., Tal J., Zeng G.C., Doctor B.P., Pardhasaradhi K., McCann P.P. S-Adenosylmethionine and methylation. FASEB J. 1996;10:471–480. [PubMed] [Google Scholar]
- 8.Cronican A.A., Fitz N.F., Carter A., Saleem M., Shiva S., Barchowsky A., Koldamova R., Schug J., Lefterov I. Genome-wide alteration of histone H3K9 acetylation pattern in mouse offspring prenatally exposed to arsenic. PLoS One. 2013;8:e53478. doi: 10.1371/journal.pone.0053478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dodmane P.R., Arnold L.L., Kakiuchi-Kiyota S., Qiu F., Liu X., Rennard S.I., Cohen S.M. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology. 2013;312:18–29. doi: 10.1016/j.tox.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh R.P., Nikitina T., Horowitz-Scherer R.A., Gierasch L.M., Uversky V.N., Hite K., Hansen J.C., Woodcock C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry. 2010;49:4395–4410. doi: 10.1021/bi9019753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goggin S.L., Labrecque M.T., Allan A.M. Perinatal exposure to 50 ppb sodium arsenate induces hypothalamic⿿pituitary⿿adrenal axis dysregulation in male C57BL/6 mice. Neurotoxicology. 2012;33:1338–1345. doi: 10.1016/j.neuro.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou Y., Xue P., Woods C.G., Wang X., Fu J., Yarborough K., Qu W., Zhang Q., Andersen M.E., Pi J. Association between arsenic suppression of adipogenesis and induction of CHOP10 via the endoplasmic reticulum stress response. Environ. Health Perspect. 2013;121:237–243. doi: 10.1289/ehp.1205731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 14.Koch I., Zhang J., Button M., Gibson L.A., Caumette G., Langlois V.S., Reimer K.J., Cullen W.R. Arsenic(+3) and DNA methyltransferases, and arsenic speciation in tadpole and frog life stages of western clawed frogs (Silurana tropicalis) exposed to arsenate. Metallomics. 2015;7:1274–1284. doi: 10.1039/c5mt00078e. [DOI] [PubMed] [Google Scholar]
- 15.Krishna N.S., Getchell T.V., Dhooper N., Awasthi Y.C., Getchell M.L. Age- and gender-related trends in the expression of glutathione S-transferases in human nasal mucosa. Ann. Otol. Rhinol. Laryngol. 1995;104:812–822. doi: 10.1177/000348949510401012. [DOI] [PubMed] [Google Scholar]
- 16.Lavoie J.C., Rouleau T., Truttmann A.C., Chessex P. Postnatal gender-dependent maturation of cellular cysteine uptake. Free Radic. Res. 2002;36:811–817. doi: 10.1080/1071576021000005230. [DOI] [PubMed] [Google Scholar]
- 17.Lim C.K., Yuan Z.X., Lamb J.H., White I.N., De Matteis F., Smith L.L. A comparative study of tamoxifen metabolism in female rat, mouse and human liver microsomes. Carcinogenesis. 1994;15:589–593. doi: 10.1093/carcin/15.4.589. [DOI] [PubMed] [Google Scholar]
- 18.Lu C., Zhao F., Sun D., Zhong Y., Yu X., Li G., Lv X., Sun G., Jin Y. Comparison of speciated arsenic levels in the liver and brain of mice between arsenate and arsenite exposure at the early life. Environ. Toxicol. 2014;29:797–803. doi: 10.1002/tox.21808. [DOI] [PubMed] [Google Scholar]
- 19.Marchiset-Ferlay N., Savanovitch C., Sauvant-Rochat M.P. What is the best biomarker to assess arsenic exposure via drinking water? Environ. Int. 2012;39:150–171. doi: 10.1016/j.envint.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Finley E.J., Ali A.M., Allan A.M. Learning deficits in C57BL/6J mice following perinatal arsenic exposure: consequence of lower corticosterone receptor levels? Pharmacol. Biochem. Behav. 2009;94:271–277. doi: 10.1016/j.pbb.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Finley E.J., Goggin S.L., Labrecque M.T., Allan A.M. Reduced expression of MAPK/ERK genes in perinatal arsenic-exposed offspring induced by glucocorticoid receptor deficits. Neurotoxicol. Teratol. 2011;33:530–537. doi: 10.1016/j.ntt.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez E.J., Kolb B.L., Bell A., Savage D.D., Allan A.M. Moderate perinatal arsenic exposure alters neuroendocrine markers associated with depression and increases depressive-like behaviors in adult mouse offspring. Neurotoxicology. 2008;29:647–655. doi: 10.1016/j.neuro.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGowan P.O., Sasaki A., D⿿Alessio A.C., Dymov S., Labonte B., Szyf M., Turecki G., Meaney M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao Z., Wu L., Lu M., Meng X., Gao B., Qiao X., Zhang W., Xue D. Analysis of the transcriptional regulation of cancer-related genes by aberrant DNA methylation of the cis-regulation sites in the promoter region during hepatocyte carcinogenesis caused by arsenic. Oncotarget. 2015;6(25):21493–21506. doi: 10.18632/oncotarget.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller W.H., Jr. Molecular targets of arsenic trioxide in malignant cells. Oncologist. 2002;7(Suppl. 1):14–19. doi: 10.1634/theoncologist.7-suppl_1-14. [DOI] [PubMed] [Google Scholar]
- 26.Pelch K.E., Tokar E.J., Merrick B.A., Waalkes M.P. Differential DNA methylation profile of key genes in malignant prostate epithelial cells transformed by inorganic arsenic or cadmium. Toxicol. Appl. Pharmacol. 2015;286:159–167. doi: 10.1016/j.taap.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrone-Bizzozero N.I., Sower A.C., Bird E.D., Benowitz L.I., Ivins K.J., Neve R.L. Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14182–14187. doi: 10.1073/pnas.93.24.14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perroud N., Dayer A., Piguet C., Nallet A., Favre S., Malafosse A., Aubry J.M. Childhood maltreatment and methylation of the glucocorticoid receptor gene NR3C1 in bipolar disorder. Br. J. Psychiatry. 2014;204:30–35. doi: 10.1192/bjp.bp.112.120055. [DOI] [PubMed] [Google Scholar]
- 29.Pilsner J.R., Hall M.N., Liu X., Ilievski V., Slavkovich V., Levy D., Factor-Litvak P., Yunus M., Rahman M., Graziano J.H., Gamble M.V. Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS One. 2012;7:e37147. doi: 10.1371/journal.pone.0037147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rager J.E., Tilley S.K., Tulenko S.E., Smeester L., Ray P.D., Yosim A., Currier J.M., Ishida M.C., Gonzalez-Horta M., del C., Sanchez-Ramirez B., Ballinas-Casarrubias L., Gutierrez-Torres D.S., Drobna Z., Del R., azo L.M., Garcia-Vargas G.G., Kim W.Y., Zhou Y.H., Wright F.A., Styblo M., Fry R.C. Identification of novel gene targets and putative regulators of arsenic-associated DNA methylation in human urothelial cells and bladder cancer. Chem. Res. Toxicol. 2015;28:1144–1155. doi: 10.1021/tx500393y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos-Chavez L.A., Rendon-Lopez C.R., Zepeda A., Silva-Adaya D., Del R., azo L.M., Gonsebatt M.E. Neurological effects of inorganic arsenic exposure: altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front. Cell. Neurosci. 2015;9(21) doi: 10.3389/fncel.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reichard J.F., Puga A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics. 2010;2:87–104. doi: 10.2217/epi.09.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren X., McHale C.M., Skibola C.F., Smith A.H., Smith M.T., Zhang L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect. 2011;119:11–19. doi: 10.1289/ehp.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rios R., Santoyo M.E., Cruz D., Delgado J.M., Zarazua S., Jimenez-Capdeville M.E. Methyl group balance in brain and liver: role of choline on increased S-adenosyl methionine (SAM) demand by chronic arsenic exposure. Toxicol. Lett. 2012;215:110–118. doi: 10.1016/j.toxlet.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Rojas D., Rager J.E., Smeester L., Bailey K.A., Drobna Z., Rubio-Andrade M., Styblo M., Garcia-Vargas G., Fry R.C. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol. Sci. 2015;143:97–106. doi: 10.1093/toxsci/kfu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sai S., Esteves C.L., Kelly V., Michailidou Z., Anderson K., Coll A.P., Nakagawa Y., Ohzeki T., Seckl J.R., Chapman K.E. Glucocorticoid regulation of the promoter of 11beta-hydroxysteroid dehydrogenase type 1 is indirect and requires CCAAT/enhancer-binding protein-beta. Mol. Endocrinol. 2008;22:2049–2060. doi: 10.1210/me.2007-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.She Q.B., Nagao I., Hayakawa T., Tsuge H. A simple HPLC method for the determination of S-adenosylmethionine and S-adenosylhomocysteine in rat tissues: the effect of vitamin B6 deficiency on these concentrations in rat liver. Biochem. Biophys. Res. Commun. 1994;205:1748–1754. doi: 10.1006/bbrc.1994.2871. [DOI] [PubMed] [Google Scholar]
- 38.Straussman R., Nejman D., Roberts D., Steinfeld I., Blum B., Benvenisty N., Simon I., Yakhini Z., Cedar H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009;16:564–571. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 39.Sun K., He P., Yang K. Intracrine induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by glucocorticoid potentiates prostaglandin production in the human chorionic trophoblast. Biol. Reprod. 2002;67:1450–1455. doi: 10.1095/biolreprod.102.005892. [DOI] [PubMed] [Google Scholar]
- 40.Szyf M. The implications of DNA methylation for toxicology: toward toxicomethylomics, the toxicology of DNA methylation. Toxicol. Sci. 2011;120:235–255. doi: 10.1093/toxsci/kfr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tondreau M.Y., Boucher E., Simard M., Tremblay Y., Bilodeau J.F. Sex-specific perinatal expression of glutathione peroxidases during mouse lung development. Mol. Cell. Endocrinol. 2012;355:87–95. doi: 10.1016/j.mce.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 42.Tyler C.R., Allan A.M. Adult hippocampal neurogenesis and mRNA expression are altered by perinatal arsenic exposure in mice and restored by brief exposure to enrichment. PLoS One. 2013;8:e73720. doi: 10.1371/journal.pone.0073720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyler C.R., Hafez A.K., Solomon E.R., Allan A.M. Developmental exposure to 50 parts-per-billion arsenic influences histone modifications and associated epigenetic machinery in a region- and sex-specific manner in the adult mouse brain. Toxicol. Appl. Pharmacol. 2015;288:40–51. doi: 10.1016/j.taap.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tyler C.R., Solomon B.R., Ulibarri A.L., Allan A.M. Fluoxetine treatment ameliorates depression induced by perinatal arsenic exposure via a neurogenic mechanism. Neurotoxicology. 2014;44:98–109. doi: 10.1016/j.neuro.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vahter M., Envall J. In vivo reduction of arsenate in mice and rabbits. Environ. Res. 1983;32:14–24. doi: 10.1016/0013-9351(83)90187-1. [DOI] [PubMed] [Google Scholar]
- 46.Valdovinos-Flores C., Gonsebatt M.E. The role of amino acid transporters in GSH synthesis in the blood⿿brain barrier and central nervous system. Neurochem. Int. 2012;61:405–414. doi: 10.1016/j.neuint.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q.Q., Lan Y.F., Rehman K., Jiang Y.H., Maimaitiyiming Y., Zhu D., Narenmandula H. Effect of arsenic compounds on the mouse embryonic stem cells differentiation into cardiomyocytes in vitro. Chem. Res. Toxicol. 2014;28(3):351–353. doi: 10.1021/tx500286t. [DOI] [PubMed] [Google Scholar]
- 48.Waters S.B., Devesa V., Fricke M.W., Creed J.T., Styblo M., Thomas D.J. Glutathione modulates recombinant rat arsenic (+3 oxidation state) methyltransferase-catalyzed formation of trimethylarsine oxide and trimethylarsine. Chem. Res. Toxicol. 2004;17:1621–1629. doi: 10.1021/tx0497853. [DOI] [PubMed] [Google Scholar]
- 49.Williams L.J., Lyons V., MacLeod I., Rajan V., Darlington G.J., Poli V., Seckl J.R., Chapman K.E. C/EBP regulates hepatic transcription of 11beta-hydroxysteroid dehydrogenase type 1. A novel mechanism for cross-talk between the C/EBP and glucocorticoid signaling pathways. J. Biol. Chem. 2000;275:30232–30239. doi: 10.1074/jbc.M001286200. [DOI] [PubMed] [Google Scholar]
- 50.Yadav S., Anbalagan M., Shi Y., Wang F., Wang H. Arsenic inhibits the adipogenic differentiation of mesenchymal stem cells by down-regulating peroxisome proliferator-activated receptor gamma and CCAAT enhancer-binding proteins. Toxicol. In Vitro. 2013;27:211–219. doi: 10.1016/j.tiv.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z., Guo C., Zhu P., Li W., Myatt L., Sun K. Role of glucocorticoid receptor and CCAAT/enhancer-binding protein alpha in the feed-forward induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts. J. Endocrinol. 2007;195:241–253. doi: 10.1677/JOE-07-0303. [DOI] [PubMed] [Google Scholar]
- 52.Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.