

Abstract
Epigenetic studies increasingly require analysis of a small number of cells that are of one specific type and derived from patients or animals. In this report, we demonstrate a simple microfluidic device that integrates sonication and immunoprecipitation (IP) for epigenetic assays, such as chromatin immunoprecipitation (ChIP) and methylated DNA immunoprecipitation (MeDIP). By incorporating an ultrasonic transducer with a microfluidic chamber, we implemented microscale sonication for both shearing chromatin/DNA and mixing/washing of IP beads. Such integration allowed highly sensitive tests starting with 100 cross-linked cells for ChIP or 500 pg of genomic DNA for MeDIP (compared to 106–107 cells for ChIP and 1–10 μg of DNA for MeDIP in conventional assays). The entire on-chip process of sonication and IP took only 1 h. Our tool will be useful for highly sensitive epigenetic studies based on a small quantity of sample.
Epigenetics focuses on molecular mechanisms that change gene activity and expression without alteration in DNA sequence.1 Epigenetic mechanisms include histone modifications, DNA methylation, nucleosome positions, and expression of noncoding RNA. Histones are subject to a variety of post-translational modifications (e.g., methylation and acetylation) within their amino-terminal tails. DNA methylation leads to the addition of a methyl group at the carbon-5 position of cytosine residues within CpG dinucleotides, forming 5-methylcytosine (5mC). These epigenetic modifications have profound influences on gene expression by changing chromatin structures and creating binding sites for effector proteins.
Immunoprecipitation (IP) has been the most important approach for identifying and studying epigenetic changes, such as histone modification and DNA methylation, both at specific loci and at the genome-wide level. Chromatin immunoprecipitation (ChIP) assay is the technique of choice for examining histone modifications in living cells or tissues.2−5 Typical ChIP assays involve gathering of a large number of cells (∼106–107 cells), fixation by cross-linking reagents (e.g., formaldehyde) to covalently attach histone to DNA sequence, cell lysis, immunoprecipitation of the modified histone of interest, reversal of cross-linking, digestion of the histone, and finally qPCR amplification/identification of the enriched and purified DNA. A similar process has been used for DNA methylation analysis. Methylated DNA immunoprecipitation (MeDIP) uses a monoclonal antibody specific for 5mC to target single-stranded methylated DNA fragments during immunoprecipitation and identifies these fragments using qPCR.6−9 Both ChIP and MeDIP can be coupled with next generation sequencing to generate profiles of genome-wide epigenetic changes.
Despite their broad applications, conventional ChIP or MeDIP assays require a large number of cells (106∼107 cells for ChIP, 1–10 μg of DNA for MeDIP) as the starting material. Such low sensitivity was largely due to low efficiency in collecting immunoprecipitated DNA and loss of materials during various steps of the complicated and manual procedures. The sample size limitation largely prevents applying these assays to a broad range of primary samples from patients and animals that come in very small quantities despite their direct biomedical relevance. There have been numerous works in recent years that address these issues to improve ChIP and MeDIP sensitivity.10−14
Microfluidics has been explored by other groups and us for conducting epigenetic assays with high sensitivity via drastic reduction in the volume and a high level of integration and automation.15−18 Ultrasensitive 50-cell ChIP-qPCR and 100-cell ChIP-seq were demonstrated in our earlier works.16,18 Despite the success, continued improvement will require further integration of additional steps in the microfluidic platform. Chromatin/DNA fragmentation is a critical step involved in ChIP and MeDIP. These assays require a size range of 100–600 bp for the fragments. The fragment size critically affects the results: small fragments lead to low signal due to breaks within amplicons, whereas large fragment sizes lead to low resolution and high background signal. Both enzymatic digestion (using micrococcal nuclease) and sonication have been used for shearing chromatin/DNA. Enzymatic fragmentation suffers from biases toward certain chromatin regions or sequence-specific cleavage.19 Thus, sonication is much more widely used in epigenetic assays. Microchip sonication was previously reported for shearing λ-DNA and cross-linked chromatin with the integrity of protein epitopes verified by immunoblotting after the procedure.20 However, there has been no report of microscale sonication of cross-linked cells or integrated immunoprecipitation assays. None of the previous microfluidic ChIP assays incorporated sonication for chromatin/DNA shearing. Either enzymatic fragmentation15,16,21 or off-chip sonication18 that handles >100 μL volumes was implemented.
Here, we describe a novel microfluidic device that integrates a composite piezoelectric transducer for both on-chip sonication and acoustofluidic IP/washing. The piezoelectric transducer was actuated at its resonant frequency and generated lamb wave (a type of transverse wave) in a neighboring microfluidic chamber for either shearing chromatin/DNA (at high intensity Vpeak > 20 V) or enhancing microscale mixing/washing (at low intensity Vpeak ∼ 10 V). The microfluidic chamber contained microscale crescent-shaped structures for enhancing cavitation and acoustic streaming. We demonstrated microfluidic epigenetic assays starting from as few as 100 cross-linked cells (for ChIP) or 500 pg of genomic DNA (for MeDIP). Histone modification (H3K4me3) and DNA methylation (5-mC) in GM 12878 cells (a lymphoblastoid cell line) at specific loci were examined by qPCR in these experiments. The on-chip processes (including sonication, IP, and washing) required around 40 min. This integrated device presents high potential for epigenetic assays starting from small quantities of samples.
Materials and Methods
Fabrication of the Microfluidic Device
The microfluidic device was fabricated out of polydimethylsiloxane (PDMS) using soft lithography.22 Briefly, a photomask was generated with microscale patterns designed by computer-aided design software FreeHand MX (Macromedia) and printed with high resolution (5,080 dpi) on a transparency. The master was made of a negative photoresist SU-8 2075 (95 μm thick, Microchem) spin-coated on a 3 in. silicon wafer (University Wafer). PDMS at a mass ratio of 10:1 RTV615 A/RTV615 B (General Electric silicone RTV 615, MG chemicals) was poured onto the master in a Petri dish to generate an ∼5 mm thick PDMS layer. The PDMS layer was solidified by baking at 80 °C for 2 h, peeled off from the master, and punched to produce inlet and outlet holes. Glass slides were treated in a basic solution (5:1:1 H2O/27% NH4OH/30% H2O2 volumetric ratio) at 75 °C for 2 h, rinsed with ultrapure water, and then thoroughly blown dry. The PDMS layer and the precleaned glass slide were treated with oxygen plasma and immediately brought into contact against each other to form a sealed device. Finally, the assembled device was baked at 80 °C for 1 h to improve bonding between PDMS and glass.
Cell Sample Preparation
GM12878 cells were obtained from Coriel Institute for Medical Research and used in our previous research.18 The cell line was tested for mycoplasma contamination using ABI MycoSEQ mycoplasma detection assay (Applied Biosystems). Cells were propagated in RPMI 1640 (11875-093, Gibco) plus 15% fetal bovine serum (26140-079, Gibco), 100 U penicillin (15140-122, Gibco), and 100 mg/mL of streptomycin (15140-122, Gibco) at 37 °C in a humidified incubator containing 5% CO2. Cells were subcultured every 2 days to maintain them in the exponential growth phase.
ChIP: Harvested cells were centrifuged at 300 g for 5 min and resuspended in culture medium. The concentration of cells in the medium was measured using a hemocytometer. A specific number of cells (ranging from 100–10,000) were cross-linked in 150 μL of culture medium by adding 10 μL of 16% formaldehyde (28908, Thermo Scientific) for 5 min, and the cross-linking was then terminated by adding 8.4 μL of 2.5 M freshly made glycine and incubating for 5 min at room temperature. The cross-linked cells were then pelleted and resuspended in 8 μL of ChIP IP buffer (20 mM Tris-HCl, pH 8.0, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% (w/v) sodium doxycholate, 0.1% SDS, 1% (v/v) Triton-100X with freshly added 1 mM PMSF (78830-1G, Sigma-Aldrich) and 1% protease inhibitor cocktail (PIC) (P8340, Sigma-Aldrich)) and loaded into the microfluidic device.
MeDIP: Genomic DNA (gDNA) from 106 GM 12878 cells was extracted and purified using QIAamp DNA blood mini kit (51104, Qiagen). Extracted gDNA was dissolved in 200 μL of ultrapure water before use. gDNA aliquots of various sizes (ranging from 500 pg to 50 ng) generated from the stock were dissolved in 8 μL of MeDIP buffer (10 mM monobasic sodium phosphate dihydrate, 10 mM dibasic sodium phosphate, 140 mM NaCl, 0.05% (v/v) Triton-100X) and denatured at 97 °C for 15 min. Denatured DNA solution was then loaded into the microfluidic device. DNA concentrations were measured using a Qubit 2.0 fluorometer with ssDNA HS Assay kit (Q10212, Life Technologies).
Setup of the Microfluidic Device for ChIP/MeDIP
A schematic and an optical image of the microfluidic device are shown in Figure 1. A Langevin-type transducer (MPI-2525D-60H, purchased from UltrasonicsWorld, manufactured by M.P. Interconsulting) was mounted to the glass slide of the microfluidic chip using two-component epoxy glue (2-Ton Epoxy, Devcon) and cured overnight at room temperature. The distance between the center of the microfluidic chamber and that of the transducer was ∼2.5 cm. A function generator (4011A, BK Precision) was used to generate an AC signal that was amplified by a high-power ultrasonic driver/amplifier (PDUS200, Micromechatronics). The amplified AC signal could be switched on/off by a LabVIEW programmed relay (5501, Coto Technology) via a data requisition card (NI SCB-68, National Instruments). The signal drove the transducer at its resonant frequency. Sine-wave AC was used unless otherwise stated. The working voltage (set by the user) and current of the amplifier were monitored using a digital oscilloscope (2530, BK precision) via a built-in monitor port on the ultrasonic amplifier. The resonant frequency of the device was slightly shifted from the fundamental resonant frequency of the transducer (60 kHz) due to bonding to the microfluidic device. The working resonant frequency (in the range of 59–62 kHz) was pinpointed by tracking the frequency that yielded the maximum output current. To monitor the temperature of the microfluidic chamber, a cement-on surface thermocouple (CO1-T, Omega Engineering) was attached to the glass substrate of the microfluidic chamber (i.e., the surface area that was separated from the microfluidic chamber only by the slide thickness of ∼1 mm). The detected temperature was displayed and recorded by a temperature controller (CN8202, Omega Engineering). The microfluidic device was placed on a frozen ice pack during sonication for cooling. The LabVIEW-programmed relay automatically switched on/off the transducer to make it work in 10 s cycles with sonication on for a fraction of each cycle (3 s sonication time per cycle unless otherwise stated) to avoid overheating.
Figure 1.
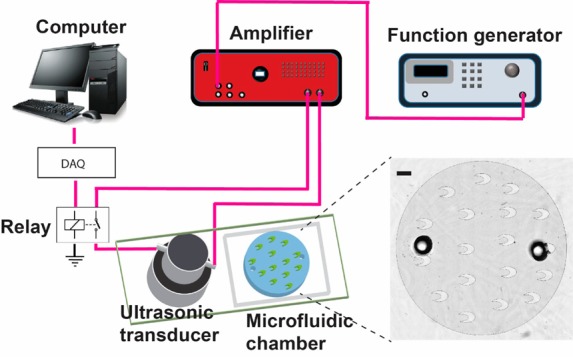
A schematic of the integrated system for on-chip sonication and IP. The ultrasonic transducer was bonded to the glass substrate of the microfluidic chip using epoxy glue. The microscale crescent shapes in the chamber are shown in the schematic as well as in the inset microscopic image. Scale bar is 1 mm.
Preparation of Immunoprecipitation (IP) Beads
Superparamagnetic Dynabeads Protein A (2.8 μm, 30 mg/mL, 10001D, Invitrogen) were used for immunoprecipitation; 150 μg (5 μL of the original suspension) of beads were washed twice with freshly prepared ChIP/MeDIP buffer and resuspended in 150 μL of ChIP/MeDIP buffer, which contained an antibody (against H3K4me3, 07-473, Millipore or 5-mC, 61255, Active Motif) at 6 μg/mL. Beads were gently mixed with the antibody at 4 °C on a rotator mixer at 24 rpm for 1 h. Antibody-coated beads were washed twice with the ChIP/MeDIP buffer and resuspended in 2 μL of the same buffer.
Microfluidic ChIP/MeDIP
In all steps involving sonication, sine-wave AC and 3 s sonication time per 10 s cycle were used. First, cross-linked cells or gDNA suspended in 8 μL of ChIP buffer (PMSF and PIC freshly added) or MeDIP buffer was loaded into the microfluidic chamber using a pipet via the inlet. After cell/gDNA loading, the inlet and outlet were tightly sealed using an adhesive and transparent sealing tape (Microseal “B” adhesive seals, Bio-Rad) to avoid solution loss during sonication. Then, the transducer was driven at its resonant frequency with high acoustic intensity (ChIP: Vpeak = 25 V and 30 cycles; MeDIP: Vpeak = 20 V and 18 cycles) to perform on-chip chromatin/DNA shearing.
After shearing, precoated magnetic IP beads were loaded into the microfluidic chamber under magnetic force generated by a cylindrical permanent magnet (NdFeB, D48-N52, 0.25 in. diameter and 0.5 in. thickness, K&J Magnetics). After bead loading, the transducer was actuated with low acoustic intensity (Vpeak = 10 V; 180 cycles) to perform acoustic streaming-enhanced IP. The IP process was finished in 30 min.
After IP, the magnetic IP beads were retained inside the chamber using the magnet, and the solution was replaced by a washing buffer. For ChIP experiments, a low-salt washing buffer (20 mM Tris-HCl, pH 8.0,150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% (v/v) Triton-100X) was used. For MeDIP experiments, MeDIP buffer was used. After filling of the washing buffer, the transducer was actuated with low acoustic intensity (Vpeak = 10 V, 12 cycles) for 2 min to remove nonspecific adsorption. The acoustic-streaming-enhanced washing step was then repeated once using a high-salt washing buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% (v/v) Triton-100X) for ChIP experiments and the MeDIP buffer for MeDIP experiments. Finally, the IP beads were collected out of the microfluidic chamber into a 0.6 mL LoBind Eppendorf tube containing 100 μL of either ChIP buffer or MeDIP buffer using a pipet and under the direction of the magnet. To generate input DNA, we conducted the same processes without undergoing the IP step. The microfluidic chip was cooled by placing it on a frozen ice pack during sonication.
Extraction of ChIP/MeDIP DNA
ChIP/MeDIP samples (either IP sample or input sample) were purified using Ipure kit (C03010012, Diagenode) following the manufacturer protocol. Purified ChIP/MeDIP DNA was dissolved in 10 μL of DNase-free water and used directly for ChIP-qPCR.
Chromatin/DNA Fragment Size Analysis Using Gel Electrophoresis
Chromatin/gDNA fragments were processed by IPure kit to extract DNA. Purified DNA was dissolved in 10 μL of water and analyzed on GelRed (41003, Biotium) stained 1% agarose gel.23−25 The size of the bands was determined using 100–3000 bp DNA ladders (170–8206, Bio-Rad).
Real-Time PCR
Real-time PCR was conducted using iQ SYBR Green Supermix (1708882, Bio-Rad) on a CFX96 real-time PCR machine (Bio-Rad) with C1000Tm thermal cycler base. All PCR assays were performed using the following thermal cycling profile: 95 °C for 10 min followed by 40 cycles of (95 °C for 15 s, 58 °C for 40 s, 72 °C for 30s). Primer concentrations were 400 nM. All primers were ordered from Integrated DNA Technologies. ChIP-qPCR/MeDIP-qPCR results were represented as relative fold enrichment, which is the ratio of percent input between a positive locus and a selected negative locus. Percent input was computed using the equation
where Ctinput and CtIP are the Ct values (a cycle threshold (Ct) value is determined by the intersection of an amplification curve and a threshold line) of input and ChIP/MeDIP DNA, respectively; dilution factor (DF) is defined as (sample volume of input + sample volume of IP)/(sample volume of input).18
Results
Design and Operation of the Microfluidic Sonication Device
The microfluidic sonication system, as shown in Figure 1, consists of several major components: (1) A function generator driving an alternating current (AC) signal that was amplified by a signal amplifier. The amplified signal actuated a Langevin transducer that was bound on the glass surface of a PDMS/glass microfluidic chip. (2) The Langevin transducer has a structure consisting of piezoelectric elements “sandwiched” between two pieces of metal and generates much higher acoustic power than regular piezo transducers.26 Such high acoustic energy transferred into the microfluidic chamber via the glass substrate in the form of a transverse acoustic wave.20,27 (3) A circular microfluidic chamber had a diameter of 12 mm, a depth of 95 μm, and a volume of 7.7 μL. One inlet and one outlet were created to have reagents and materials in and out of the chamber.
With actuation of the transducer, solution in the microfluidic chamber was subjected to a rapid change of pressure. If the amplitude of pressure change is beyond a certain threshold, small air bubbles that are originally dissolved in the liquid can rapidly collapse. This process is referred to as inertial cavitation and is the main cause of chromatin/DNA fragmentation by sonication.28,29 Because cavitation initiates at the gas–liquid interface, we increased intensity of cavitation by adding microscale crescent-shaped structures, as shown in the inset image of Figure 1. These microscale crescent PDMS structures enhanced cavitation in several ways: First, these crescent structures entrapped air (as solution flowed in from the inlet), thus increasing the air–liquid interface.30 Second, hydrophobicity of PDMS lowered the nucleation energy barrier compared to that with a hydrophilic surface31 and promoted cavitation by creating nanobubbles of sizes close to critical radii for bubble nucleation.31−33 Third, the sharp tip of the crescent structures significantly promoted cavitation and acoustic streaming (required by subsequent steps of mixing and washing). We used an angle of 12° for the tip based on optimization in previous works.34,35
Figure 2 shows cavitation inside devices of different designs and under various sonication conditions. Cavitation was only present close to the edge of the chamber when there were no crescent structures (Figure 2a and Video S1). In comparison, the microscale crescent structures generated significantly more cavitation (Figure 2b–f). As we increased sine AC voltage from 17 to 25 V, there was increased cavitation, and at 25 V, air bubbles originated from various connected crescent shapes and covered a large area of the chamber due to complex interplay of primary and secondary Bjerknes forces (Figure 2b, c and Video S2).36 We also tested the performance under different waveforms (i.e., square, sine, and triangle waves) generated by the function generator. As shown in Figure 2d–f, at the same frequency and voltage, square wave (Figure 2d) generated the strongest acoustic cavitation, and triangle wave generated the least cavitation (Figure 2f). This trend can be explained by the relative magnitude of time-averaged power for various waveforms, when VPeak is the same (i.e., square > sine > triangle for Ptime averaged). We chose to use sine wave for subsequent experiments due to the fact that it offered the best consistency and stability for transducer operation.
Figure 2.

Acoustic cavitation inside the microfluidic chamber under various Vpeak and waveforms. The resonant frequency was 61 kHz in all cases. Scale bar is 1 mm. (a) Cavitation at the edge of the chamber (without microscale crescent structures) under sine-wave AC with Vpeak of 20 V. (b) Cavitation inside the chamber with multiple crescent structures under sine-wave AC with Vpeak of 17 V. (c) Cavitation inside the chamber with multiple crescent structures under sine-wave AC with Vpeak of 25 V. (d–f) Cavitation around a crescent shape under a VPeak of 20 V and different waveforms of square (d), sine (e), and triangle (f).
On-Chip Sonication of Cross-Linked Cells and Genomic DNA
In ChIP, cells are typically cross-linked using formaldehyde to capture protein–DNA interactions. Cells after cross-linking are hard to lyse chemically.37 Thus, sonication in a buffer containing an ionic detergent such as SDS is typically required for complete release and shearing of chromatin.37,38
We demonstrated the performance of our device for sonication-based chromatin shearing starting from cross-linked cells. The on-chip sonication was performed in the ChIP buffer that contained 0.1% SDS (compared to 1% SDS in common sonication buffers) to avoid adverse effects on the downstream ChIP assay. Cells maintained their physical shape before sonication (Figure 3a). After on-chip sonication for 6 cycles (each 10 s cycle has 3 s sonication and 7 s rest period) (Figure 3b), cells rapidly disintegrated with debris and nuclei remaining. After on-chip sonication for 12 cycles (Figure 3c), there were no sizable pieces left, and this suggested complete chromatin release from nuclei. We also examined chromatin shearing under various sonication conditions by resolving DNA fragment size using gel electrophoresis (Figure 3d–f). Both sonication intensity and cycle number critically affected fragment size and distribution of sheared chromatin. We first confirmed that the crescent microstructures substantially enhanced chromatin fragmentation (Figure 3d). Furthermore, in the devices with the crescent shapes, we observed a steady decrease in the fragment size from 100 to 3000 bp to 100–500 bp when we increased Vpeak (that supported the transducer) from 15 to 25 V (Figure 3e). With fixed sonication intensity (Vpeak), fragmentation of chromatin also increased with larger cycle number (i.e., longer overall duration) (Figure 3f). Finally, we also demonstrated that the on-chip sonication shearing was effective for shearing purified genomic DNA that is typically the starting material in MeDIP experiments (Figure 3g). Compared with sonication of cross-linked cells, shearing genomic DNA required much less acoustic energy to generate a similar fragment size; 100–500 bp was the optimal fragment size range for DNA and was produced under 20 V for 18 cycles (compared to 25 V for 30 cycles when cross-linked cells were the starting material). Our on-chip sonication showed good reproducibility (Figure 3g shows repeats for 20 V/18 cycles).
Figure 3.

Chromatin/gDNA fragmentation by on-chip sonication. Sonication was conducted under sine-wave 61 kHz AC with a sonication time of 3 s in each 10 s cycle. (a–c) Microscopic images of cross-linked GM12878 cells before and after sonication. Scale bar is 100 μm. (a) Before sonication. (b) Sonication for 6 cycles with Vpeak of 20 V. (c) Sonication for 12 cycles with Vpeak of 20 V. (d–f) Gel electrophoresis of DNA fragments after chromatin fragmentation (starting from 50,000 cross-linked cells in each experiment). (d) Chromatin fragmentation is stronger with crescent structures than without them in the microfluidic chamber under the same sonication conditions (30 cycles, Vpeak = 20 V). (e) Chromatin fragmentation increases with higher Vpeak. Thirty cycles were applied for each sample. (f) Chromatin fragmentation increases with higher number of sonication cycles (12–48 cycles). Vpeak was 20 V for all the samples. (g) gDNA fragmentation under various sonication conditions measured by gel electrophoresis (Vpeak of 15–25 V, 12–24 cycles); 200 ng of genomic DNA was used in each experiment.
Integrated Sonication and ChIP/MeDIP Assay
Our simple device permitted integration of sonication-based shearing and immunoprecipitation. Thus, we were able to conduct ChIP or MeDIP directly from cross-linked cells or purified genomic DNA, respectively, using the device. Figure 4 shows the process for ChIP, and the MeDIP process is very similar with the exception of starting with single-stranded gDNA. Briefly, cross-linked cells or single-stranded genomic DNA were loaded into the microfluidic chamber for on-chip sonication-based shearing (Vpeak > 20 V) first (Figure 4a, b). After generation of chromatin or DNA fragments, beads that were coated with a specific antibody (H3K4me3 antibody in the case of ChIP or 5-mC antibody in the case of MeDIP) were loaded into the microfluidic chamber under magnetic force generated by a permanent magnet (Figure 4c). Then, low-intensity acoustic wave (that does not induce cavitation) was applied (with Vpeak = 10 V) to generate acoustic streaming (Video S3 and S4) to enhance mixing of beads and solution while keeping nonspecific adsorption minimal (Figure 4d). During this period (30 min), immunoprecipitation (either ChIP or MeDIP) was finished. After immunoprecipitation, magnetic beads were retained inside the chamber by the magnet, whereas a washing buffer was used to flush untargeted chromatin/DNA and other impurities out of the chamber (Figure 4e). A wash step was then conducted under acoustic streaming to further remove nonspecific adsorption from the bead surface (Figure 4f, g). After washing, the magnetic beads were collected out of the chamber for downstream purification and analysis (Figure 4h). The entire on-chip processing time was approximately 40 min, and the IP step took only 30 min.
Figure 4.
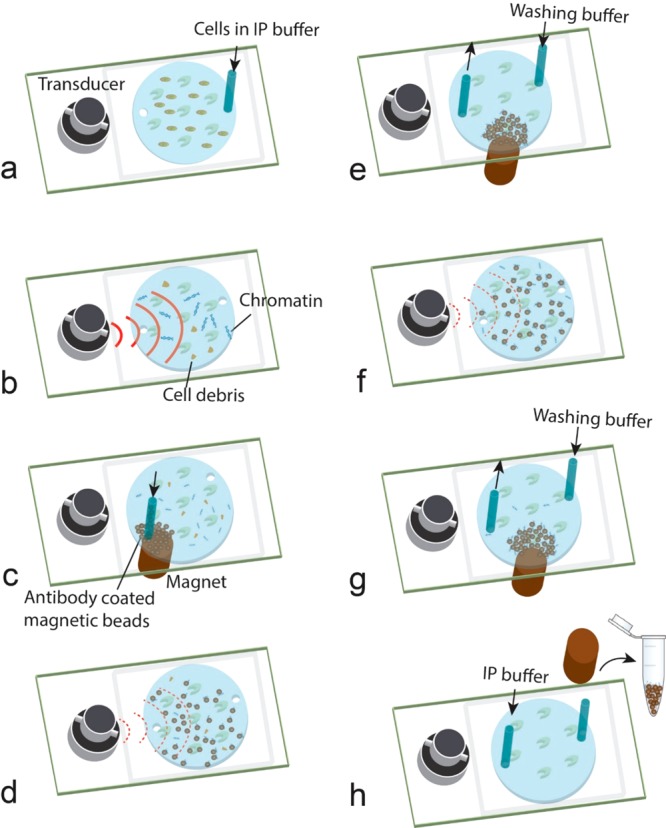
Procedure for integrated sonication and ChIP on a microfluidic platform starting from cross-linked cells. (a) Cross-linked cells are loaded into the chamber. (b) High-intensity sonication for chromatin fragmentation. (c) Antibody-coated IP beads are loaded into the chamber. (d) Acoustic streaming-enhanced immunoprecipitation (30 min). (e) A washing buffer is flowed in while keeping the beads in the chamber. (f) Acoustic streaming-enhanced washing (2 min). (g) Chamber flushing by the washing buffer. (h) Collection of IP beads out of the microfluidic chip. The microfluidic chip was placed on a frozen ice pack during steps b–f. In all steps involving sonication (or acoustic streaming), sine-wave AC and 3 s sonication time per 10 s cycle were used.
Heat management was an important consideration in our device operation. On-chip sonication generated a substantial amount of heat and the rise in the temperature particularly affected IP results by damaging protein/antibody. We placed the microfluidic chip on top of a frozen ice pack during sonication. We also conducted sonication in short intervals with periods of sonication off to facilitate cooling. We obtained temperature profiles of the microfluidic chamber over time under various sonication conditions (ranging from 1 to 9 s sonication time out of each 10 s cycle) (Figure 5a). There was no substantial temperature increase when there was sonication for 1s during a 10 s cycle. However, when the sonication time in each cycle increased to 3, 5, and 9 s, there was an increasingly sharp temperature rise at the beginning of the process. In all cases, the temperature eventually reached a plateau (17.2, 38.1, and 66.7 °C for 3, 5, and 9 s, respectively).
Figure 5.

Effect of temperature rise on ChIP-qPCR results. (a) Variation over time in the substrate temperature at the location of the microfluidic chamber under various sonication conditions. The measurement was conducted during 30 cycle sonication with a Vpeak of 25 V. The sonication time in each 10 s cycle was varied from 1 to 9 s; 10,000 cross-linked GM12878 cells were used in each experiment. The data points were the average of two replicates. The data points are connected to guide the eye. (b–e) ChIP-qPCR results corresponding to various temperature profiles in (a).
We tested how the temperature rise affected ChIP results. We used ChIP-qPCR to measure the percent input at known positive and negative loci for H3K4me3 (with the primer sequences listed in Table S1). We found that the enrichment of ChIP DNA was critically affected by our sonication protocol. When a 1 s sonication time was used in a 10 s cycle, there was high percent input at all loci (Figure 5b). This result suggests incomplete chromatin fragmentation that created false positives. By increasing the sonication time to 3 s in a 10 s cycle, the temperature of the glass substrate at the location of the microfluidic chamber increased to around 17.2 °C (Figure 5a). The enrichment of ChIP DNA was significantly improved as percent inputs at two positive loci (UNKL and C9orf3) were above 25%, whereas the ones at two negative loci (N1 and N2) were around 1% (i.e., a relative fold enrichment of 25) (Figure 5c). However, further increasing the sonication time to 5s led to decreased percent input in one of the positive loci (C9orf3) from 25 to 7.5% and an increase at the two negative loci (to 4 and 2.5%) (Figure 5d), likely due to increased temperature (38.1 °C). Elevated temperature degrades proteins (both histone and antibody) and potentially de-cross-links the protein and DNA.38,39 This is confirmed by loss of most signal in the positive loci when the sonication time and temperature were further increased to 9 s and 66.7 °C, respectively (Figure 5e). Thus, our results indicated that 3 s sonication in a 10 s cycle (for 30 cycles) was optimal for ChIP starting from cross-linked cells.
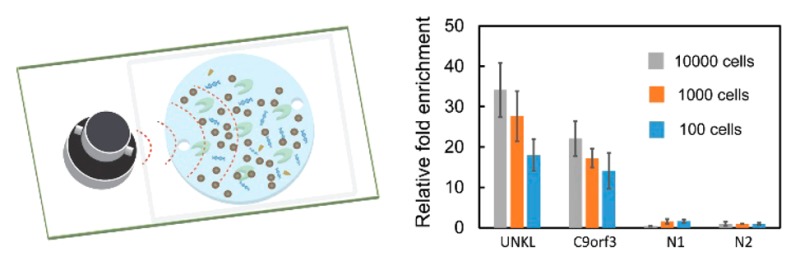
We demonstrated that our device permitted sensitive and integrated ChIP and MeDIP tests (with primer sequences listed in Table S1 and S2) based on a small quantity of cells/DNA (Figure 6). Using the optimized protocol, we tested with various samples ranging from 10,000 down to 100 cross-linked cells on the microfluidic device. The identification of H3k4me3 at specific loci was conducted off-chip using qPCR. With decreasing cell sample size, the fold enrichment at positive loci (UNKL and C9orf3, in reference to the negative locus N2) varied from 34 and 22 to 18 and 14, respectively (Figure 6a). Such a decrease in the enrichment was expected due to the decreased amount of targeted chromatin with smaller sample sizes and increased nonspecific adsorption of untargeted chromatin. Nevertheless, the enrichment of 18 and 14 with only 100 cells was more than sufficient for differentiating positive from negative loci. The enrichment of 27 and 17 at UNKL and C9orf3 with 1000 cells were slightly lower than the optimized results obtained using MOWChIP (i.e., 37 and 19).18 Thus, the two approaches appeared to generate results of similar quality. Using the same device, we also started with various amounts of single-stranded genomic DNA (50 ng, 5 ng, and 500 pg) to conduct an on-chip MeDIP assay with the same procedure except with a lower sonication intensity for shearing. We tested two known positive loci (SNRPN and MAGEA1 promoters) and two negative loci (GABRB3 and GAPDH promoters) with MeDIP followed by qPCR.40 The relative fold enrichment values at the two positive loci were 58 and 34 even with 500 pg DNA, confirming the high data quality.
Figure 6.

Microfluidic ChIP-qPCR and MeDIP-qPCR conducted using our device. (a) qPCR analysis of ChIP DNA at two known positive loci and two negative loci using cross-linked cells from 10,000 to 100 cells. (b) qPCR analysis of MeDIP DNA at two known positive loci and two negative loci using single-stranded gDNA ranging from 50 ng down to 500 pg.
Discussion
Several features of our device permit its use for sensitive and rapid epigenetic assays. First, we integrate sonication of cells/DNA with the rest of the procedures so that we can start with tiny amounts of cross-linked cells or genomic DNA. In this case, we fully take advantage of the small volume of a microfluidic chamber to avoid dilution in tubes and material loss during transfer. Second, acoustic streaming is used to facilitate both mixing (of chromatin/DNA and beads) and washing (to remove nonspecific adsorption) at the microscale. Acoustic streaming facilitates transport of molecules and promotes their interaction with the bead surface by reducing the thickness of diffusion boundary layers. A low-amplitude acoustic wave also allows removal of nonspecific binding without compromising specific binding.41,42 These steps are critical for generating ChIP or MeDIP DNA of high quality. Third, our assays are rapid. The on-chip procedures can be finished in less than 1 h, and the entire ChIP/MeDIP assay takes less than 3 h including off-chip protein digestion/DNA purification (0.5 h) and qPCR (1.5 h). In particular, the immunoprecipitation step is shortened to 30 min. Taken together, our device offers a new platform for performing immunoprecipitation-based epigenetic assays with high level of integration and sensitivity. Our device will greatly facilitate studies of scarce samples derived from patients and small lab animals.
Acknowledgments
This work was supported by National Institutes of Health Grants CA174577, EB017235, EB017855, and HG008623 to C.L.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.5b04707.
Primer sequences used in ChIP-qPCR and MeDIP-qPCR and in-depth video descriptions (PDF)
Video showing cavitation at the edge of a microfluidic chamber without crescent structures (AVI)
Video showing cavitation in a microfluidic chamber containing multiple crescent shapes (AVI)
Video showing acoustic streaming around a crescent shape (AVI)
Video showing a close look of acoustic streaming around the tip of a crescent shape (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Goldberg A. D.; Allis C. D.; Bernstein E. Cell 2007, 128, 635. 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Massie C. E.; Mills I. G. EMBO Rep. 2008, 9, 337. 10.1038/embor.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P. J. Nat. Rev. Genet. 2009, 10, 669. 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham P. J. Nat. Rev. Genet. 2009, 10, 605. 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P. Mol. Biotechnol. 2010, 45, 87. 10.1007/s12033-009-9239-8. [DOI] [PubMed] [Google Scholar]
- Weber M.; Davies J. J.; Wittig D.; Oakeley E. J.; Haase M.; Lam W. L.; Schubeler D. Nat. Genet. 2005, 37, 853. 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- Chavez L.; Jozefczuk J.; Grimm C.; Dietrich J.; Timmermann B.; Lehrach H.; Herwig R.; Adjaye J. Genome Res. 2010, 20, 1441. 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C.; Chavez L.; Vilardell M.; Farrall A. L.; Tierling S.; Bohm J. W.; Grote P.; Lienhard M.; Dietrich J.; Timmermann B.; Walter J.; Schweiger M. R.; Lehrach H.; Herwig R.; Herrmann B. G.; Morkel M. PLoS Genet. 2013, 9, e1003250. 10.1371/journal.pgen.1003250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down T. A.; Rakyan V. K.; Turner D. J.; Flicek P.; Li H.; Kulesha E.; Graf S.; Johnson N.; Herrero J.; Tomazou E. M.; Thorne N. P.; Backdahl L.; Herberth M.; Howe K. L.; Jackson D. K.; Miretti M. M.; Marioni J. C.; Birney E.; Hubbard T. J. P.; Durbin R.; Tavare S.; Beck S. Nat. Biotechnol. 2008, 26, 779. 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M.; Zhu J.; Bernstein B. E. Nat. Methods 2010, 7, 615. 10.1038/nmeth.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaranarayanan P.; Mendoza-Parra M. A.; Walia M.; Wang L.; Li N.; Trindade L. M.; Gronemeyer H. Nat. Methods 2011, 8, 565. 10.1038/nmeth.1626. [DOI] [PubMed] [Google Scholar]
- Taiwo O.; Wilson G. A.; Morris T.; Seisenberger S.; Reik W.; Pearce D.; Beck S.; Butcher L. M. Nat. Protoc. 2012, 7, 617. 10.1038/nprot.2012.012. [DOI] [PubMed] [Google Scholar]
- Jakobsen J. S.; Bagger F. O.; Hasemann M. S.; Schuster M. B.; Frank A. K.; Waage J.; Vitting-Seerup K.; Porse B. T. BMC Genomics 2015, 16, 46. 10.1186/s12864-014-1195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D.; Weiner A.; Lorenzo-Vivas E.; Zaretsky I.; Jaitin D. A.; David E.; Keren-Shaul H.; Mildner A.; Winter D.; Jung S.; Friedman N.; Amit I. Science 2014, 345, 943. 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A. R.; Hiatt J. B.; Lu R.; Attema J. L.; Lobo N. A.; Weissman I. L.; Clarke M. F.; Quake S. R. Lab Chip 2009, 9, 1365. 10.1039/b819648f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng T.; Bao N.; Litt M. D.; Glaros T. G.; Li L.; Lu C. Lab Chip 2011, 11, 2842. 10.1039/c1lc20253g. [DOI] [PubMed] [Google Scholar]
- Wu A. R.; Kawahara T. L.; Rapicavoli N. A.; van Riggelen J.; Shroff E. H.; Xu L.; Felsher D. W.; Chang H. Y.; Quake S. R. Lab Chip 2012, 12, 2190. 10.1039/c2lc21290k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Chen C.; He B.; Tan K.; Lu C. Nat. Methods 2015, 12, 959. 10.1038/nmeth.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavin M.; Cappabianca L.; Kress C.; Thomassin H.; Grange T. Mol. Cell. Biol. 2004, 24, 7891. 10.1128/MCB.24.18.7891-7901.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng Q.; Lomonosov A. M.; Furlong E. E.; Merten C. A. Lab Chip 2012, 12, 4677. 10.1039/c2lc40595d. [DOI] [PubMed] [Google Scholar]
- Rotem A.; Ram O.; Shoresh N.; Sperling R. A.; Goren A.; Weitz D. A.; Bernstein B. E. Nat. Biotechnol. 2015, 33, 1165. 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Chen F.; Bao N.; He H.; Xu P.; Jana S.; Jung S.; Lian H.; Lu C. Lab Chip 2013, 13, 171. 10.1039/C2LC40950J. [DOI] [PubMed] [Google Scholar]
- Dahl J. A.; Collas P. Nat. Protoc. 2008, 3, 1032. 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- Fanelli M.; Amatori S.; Barozzi I.; Minucci S. Nat. Protoc. 2011, 6, 1905. 10.1038/nprot.2011.406. [DOI] [PubMed] [Google Scholar]
- Adli M.; Bernstein B. E. Nat. Protoc. 2011, 6, 1656. 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelle N.; Guyomar D.; Richard C.; Gonnard P.; Eyraud L. Ultrasonics 1996, 34, 187. 10.1016/0041-624X(95)00077-G. [DOI] [Google Scholar]
- Ahmed D.; Mao X.; Shi J.; Juluri B. K.; Huang T. J. Lab Chip 2009, 9, 2738. 10.1039/b903687c. [DOI] [PubMed] [Google Scholar]
- Elsner H. I.; Lindblad E. B. DNA 1989, 8, 697. 10.1089/dna.1989.8.697. [DOI] [PubMed] [Google Scholar]
- Hawley S. A.; Macleod R.; Dunn F. J. Acoust. Soc. Am. 1963, 35, 1285. 10.1121/1.1918685. [DOI] [Google Scholar]
- Tandiono; Ohl S.-W.; Ow D. S.-W.; Klaseboer E.; Wong V. V.; Camattari A.; Ohl C.-D. Lab Chip 2010, 10, 1848. 10.1039/c002363a. [DOI] [PubMed] [Google Scholar]
- Belova V.; Gorin D. A.; Shchukin D. G.; Möhwald H. Angew. Chem., Int. Ed. 2010, 49, 7129. 10.1002/anie.201002069. [DOI] [PubMed] [Google Scholar]
- Belova V.; Shchukin D. G.; Gorin D. A.; Kopyshev A.; Möhwald H. Phys. Chem. Chem. Phys. 2011, 13, 8015. 10.1039/c1cp20218a. [DOI] [PubMed] [Google Scholar]
- Bremond N.; Arora M.; Ohl C.-D.; Lohse D. J. Phys.: Condens. Matter 2005, 17, S3603. 10.1088/0953-8984/17/45/054. [DOI] [Google Scholar]
- Huang P.-H.; Xie Y.; Ahmed D.; Rufo J.; Nama N.; Chen Y.; Chan C. Y.; Huang T. J. Lab Chip 2013, 13, 3847. 10.1039/c3lc50568e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nama N.; Huang P.-H.; Huang T. J.; Costanzo F. Lab Chip 2014, 14, 2824. 10.1039/c4lc00191e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez Rivas D.; Prosperetti A.; Zijlstra A. G.; Lohse D.; Gardeniers H. J. Angew. Chem., Int. Ed. 2010, 49, 9699. 10.1002/anie.201005533. [DOI] [PubMed] [Google Scholar]
- Bortz P. D. S.; Wamhoff B. R. PLoS One 2011, 6, e26015. 10.1371/journal.pone.0026015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring M.; Offermann S.; Danker T.; Horst I.; Peterhansel C.; Stam M. Plant Methods 2007, 3, 11. 10.1186/1746-4811-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough R.; Rechsteiner M. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 90. 10.1073/pnas.81.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea A. K.; Nagarajan R. P.; Bilenky M.; Ballinger T. J.; D’Souza C.; Fouse S. D.; Johnson B. E.; Hong C.; Nielsen C.; Zhao Y. Nature 2010, 466, 253. 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G. D.; Morán-Mirabal J. M.; Branch D. W.; Craighead H. G. IEEE Sens. J. 2006, 6, 254. 10.1109/JSEN.2006.870172. [DOI] [Google Scholar]
- Hsu W.-T.; Feng G.-H.; Cho C.-L.; Chau L.-K. RSC Adv. 2013, 3, 16159. 10.1039/c3ra42368a. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.