

Abstract
Exposure to environmental pollutants, such as polycyclic aromatic hydrocarbons (PAHs) found in coal tar mixtures and tobacco sources, is considered a significant risk factor for the development of heart disease in humans. The goal of this study was to determine the influence of PAHs present at a Superfund site on human coronary artery endothelial cell (HCAEC) phospholipase A2 (PLA2) activity and apoptosis. Extremely high levels of 12 out of 15 EPA high-priority PAHs were present in both the streambed and floodplain sediments at a site where an urban creek and its adjacent floodplain were extensively contaminated by PAHs and other coal tar compounds. Nine of the 12 compounds and a coal tar mixture (SRM 1597A) activated group IVC PLA2 in HCAECs, and activation of this enzyme was associated with histone fragmentation and poly (ADP) ribose polymerase (PARP) cleavage. Genetic silencing of group IVC PLA2 inhibited both 3H-fatty acid release and histone fragmentation by PAHs and SRM 1597A, indicating that individual PAHs and a coal tar mixture induce apoptosis of HCAECs via a mechanism that involves group IVC PLA2. Western blot analysis of aortas isolated from feral mice (Peromyscus leucopus) inhabiting the Superfund site showed increased PARP and caspase-3 cleavage when compared to reference mice. These data suggest that PAHs induce apoptosis of HCAECs via activation of group IVC PLA2.
Keywords: Atherosclerosis; Lipid signaling; Phospholipase A2; Apoptosis; Anthracene; Benzo(a)pyrene; Acenapthalene; Benzo(a)anthracene; Benzo(b)fluoranthene; Benzo(k)fluoranthene; Benzo(g, h, i)perylene
Introduction
Coal tar, a byproduct of anoxic combustion of coal to produce coal gas and coke, is composed of monocyclic, polycyclic, and heterocyclic aromatic hydrocarbons (Dabestani et al. 1999; Mackay et al. 1992; Mueller et al. 1989). Extensive coal tar contamination of soil, sediments, and water has occurred at many former manufactured gas plant sites across the United States where coal tar was produced, including one site in Chattanooga, TN (Mackay et al. 1992). A 4 km stretch of urban Chattanooga Creek and its surrounding floodplain were contaminated by wastes from a nearby manufactured gas plant that operated from 1918 to 1987 (US 1999; Davis 1992). This stretch of the creek has been designated as a Superfund site by the US Environmental Protection Agency (US EPA) because of concerns about the health risks for people living in or near the floodplain (Davis 1992). Between 1997 and 1998, the US EPA dredged the contaminated streambed sediments from 1.6 km of the creek, down to the bedrock (IT Corporation 1999). Dredging of the remaining 2.4 km of the contaminated streambed is currently underway. The creek floodplain was not a part of any remedial action. Within the 100-year floodplain of the contaminated portion of the creek are numerous single-family residences, a high school, and several elementary schools. No studies to date have addressed the health hazards related to exposure to polycyclic aromatic hydrocarbons (PAHs) in the creek or floodplain, but it is clear that there is potential for residents to come in contact with high levels of PAHs at this site and at other sites where coal tar was disposed of in creeks or floodplains.
Linked to industrial pollution, atherosclerosis and subsequent acute myocardial infarction are the leading cause of death in industrialized nations (Donaldson et al. 2001; Howard et al. 1998). PAHs are important environmental pollutants that have been implicated in atherosclerosis associated with urban pollution (Donaldson et al. 2001). Treatment of mice with benzo(a)pyrene (B[a]P) or dimethylbenzanthracene induces bioactivation of drug-metabolizing enzymes and irreversible binding of hydrocarbon metabolites to endothelial cells (EC). These data suggest that PAHs may augment atherosclerosis by a process that involves EC damage (Granberg et al. 2003), possibly via apoptosis. The importance of apoptotic cell death to the atherosclerotic process is well documented (Dimmeler et al. 1998; Geng and Libby 1995; Isner et al. 1995). Apoptosis has been demonstrated to predominate in atherosclerotic plaques of humans, cholesterol-fed rabbits, and hyperlipidemic knockout mice and has been shown to be essential for the development of plaque instability and subsequent atherothrombosis, a process that is critical in acute myocardial infarction. Apoptosis is a gene-directed form of cell death characterized by events that include cellular shrinkage, phosphatidylserine externalization, nuclear condensation, DNA fragmentation, and protein cleavage (Dimmeler et al. 1998; Soldani and Scovassi 2002). Several of these biochemical events differentiate apoptosis from necrosis. For example, cleavage of poly (ADP)ribose polymerase (PARP) to 89- and 24-kD fragments and histone fragmentation are both useful markers of apoptosis that result from the activities of proteases and endonucleases, respectively (Soldani and Scovassi 2002).
We have demonstrated recently that 3 PAHs induce apoptosis of human coronary artery endothelial cells (HCAEC): 1-methylanthracene, B(a)P, and phenanthrene (PHEN) induced apoptosis of HCAECs by activation of the phospholipase A2 (PLA2)/arachidonic acid (AA) cascade (Tithof et al. 2002), a pro-inflammatory pathway linked to apoptosis as well as atherosclerotic vascular disease (Atsumi et al. 1998, 2000; Capper and Marshall 2001). PLA2s comprise a large and diverse family of enzymes exhibiting different substrate specificities, cofactor requirements, subcellular localizations, and cellular functions. PLA2s hydrolyze membrane phospholipids at the sn-2 position to release AA or other fatty acids. AA serves as a substrate for the production of over 100 biologically active lipid mediators. In addition, several fatty acids also act as second messengers important in apoptosis (Capper and Marshall 2001).
In the present study, sediments in the creek and soils in the floodplain of Chattanooga Creek were sampled for the presence of 15 EPA priority PAHs. These low molecular weight PAHs have typically been ignored for their toxic potential despite the fact that the EPA considers them to be high priority. The explanation for this is that genotoxicity has been considered to be the predominant cause of toxicity of PAHs. These small molecular weight compounds are not genotoxic but rather have epigenetic effects. Upham et al. (1998) published the first paper demonstrating that small 3 ring PAHs induced inhibition of gap junctional communication, a process linked to tumor-promoting activities that does not involve genotoxicity. Subsequent to this, three additional papers were published that extended these observations to 2–6 ring PAHs (Blaha et al. 2002; Upham et al. 1998; Weis et al. 1998).
The current study focuses on the PAHs found in the highest concentrations in the Chattanooga Superfund site. They were evaluated for their effects on PLA2 activation and apoptosis of HCAECs in vitro as individual PAHs. To explore the effect of mixtures of organic compounds in coal tar on HCAEC PLA2 activation and apoptosis, a coal tar sample from the National Institute for Standards and Technology (NIST, Gaithersburg, MD, USA; sample SRM 1597A) was also evaluated. To investigate whether PAHs at the Chattanooga Creek Superfund site were biologically available to mammals and whether on-site PAH exposure could result in vascular apoptosis, liver and aortic tissue was collected from feral mice (Peromyscus leucopus) trapped along the floodplain of the Superfund site and compared to mice from a control upstream site where no PAHs were found.
Materials and methods
Materials
PAHs were obtained from Aldrich (Milwaukee, WI, USA). 1-palmitoyl-2-[arachidonoyl-1-14C] phosphatidylcholine, [5,6,8,9,11,12,14,15-3H], 3H-oleic acid, and 3H-arachidonic acid were purchased from American Radiolabeled Chemicals (St. Louis, MO, USA). Methylarachidonoyl fluorophosphonate (MAFP) came from Biomol (Plymouth Meeting, PA, USA). Anti-PARP antibodies were obtained from Pharmingen (San Jose, CA, USA) and Cell Signaling (Beverly, MA, USA), and Cell Death Detection ELISAplus apoptosis kits were obtained from Roche (Indianapolis, IN, USA). Anti-caspase 3 antibody was obtained from Cell Signaling (Beverly, MA, USA). Anti-cPLA2 antibody was obtained from Santa Cruz (Santa Cruz, CA, USA). Qiagen (Valencia, CA, USA) was the source of control and targeting siRNA oligonucleotides. G3PDH primers were purchased from Clontech (Palo Alto, CA, USA). All chemical standards, including surrogates and spikes, were purchased from either Ultra Scientific (North Kingstown, RI, USA) or AccuStandard (New Haven, CT, USA) and were used without further purification. All soil extraction solvents were Optimagrade and were purchased from Fisher Scientific. The following reagents were used to measure PAH exposure in the feral mouse liver; all were purchased from Sigma–Aldrich (St. Louis, MO, USA): bovine serum albumin (BSA), resorufin, 7-ethyoxyresorufin, sodium phosphate, potassium phosphate, Sepharose gel, HPLC-grade methanol, fluorescamine, and reduced nicotinamide adenine dinucleotide phosphate (NADPH).
Methods
Creek sediment and floodplain soil sample extractions for PAHs
Streambed sediment grab samples were collected at depths of up to 15 cm at six sites along the Superfund-designated portion of the creek (i.e., the highly contaminated portion) and at two sites upstream of the Chattanooga Creek Superfund site. Using coring devices, floodplain samples were collected from the upper 10 cm of the soils at two locations on either side of the creek in the Superfund site. All samples were transferred to a freezer pending hot solvent extractions of organic compounds as described below. Extraction of PAHs was performed on homogenized subsamples using a commercially available Accelerated Solvent Extractor (ASE 300, Dionex, Sunnyvale, CA, USA). The extraction protocols and quality-control procedures closely followed US EPA Method 3545A (US 2004b). The final solvent extract was concentrated to 2 ml and stored at −20°C pending gas chromatograph/mass spectrometer (GC/MS) analyses. Method blanks and laboratory controls were carried out using the same extraction protocol but with the clean analog floodplain soil from Ooltewah, TN, USA.
Sample extracts were analyzed for 15 US EPA-designated priority PAHs using an Agilent Gas Chromatographer (Model 6890) equipped with an MS (Model 5973N). All PAH compounds were analyzed according to US EPA Method 8270D [IT Corporation]. The peak area for each PAH was calculated using Agilent Chemstation software. Subsequently, the concentration of individual compounds was estimated from their areas under the chromatographic peaks using the internal standard peaks as instrument reference as described in EPA Method 8270D (IT Corporation).
Cell culture
HCAECs were purchased from Clonetics (San Diego, CA, USA) and maintained in microvascular endothelial cell growth medium (Clonetics, San Diego, CA, USA) containing 5% fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2/95%O2.
PLA2 activity/AA release
HCAECs were seeded into 6-well plates (105 cells/well) in growth medium, and at 75% confluence, the cells were pre-labeled for 24 h with 0.25 μCi/ml of 3H-arachidonic acid (3H-AA) or 3H-oleic acid (3H-OA). On the day of the experiments, the cells were washed twice with Hanks’ balanced salt solution (HBSS) and equilibrated for 45 min in HBSS + 0.1% BSA. Radiolabeled cells were exposed to various concentrations of individual PAHs found in the contaminated section of the Chattanooga creek. The cells were exposed to PAHs or vehicle for 60 min. Cumulative release of 3H-AA or 3H-OA into the medium was measured by scintillation counting and the data expressed as percent of total cellular radioactivity. Total cellular uptake of 3H-fatty acids was greater than 75% as determined in preliminary studies. To examine the effects of a mixture of organic compounds, cells were treated with SRM 1597A, a sample of coal tar obtained from NIST, or vehicle for 60 min. Before treatment with SRM 1597A, toluene was removed via evaporation under nitrogen gas, and the residue was reconstituted in dimethyl sulfoxide (DMSO).
Cell-free PLA2 activity
PLA2 activity obtained with intact cells was verified with cell-free PLA2 activity measured in whole cell sonicates as described previously. The conditions of this assay are optimum for activity of group IV PLA2 enzymes. The cells were washed with Ca2+-free PBS containing 5 mM EDTA and 1 mM PMSF, resuspended in cold homogenizing buffer (50 mM Tris HCl, pH 7.4, 2 mM EGTA, 0.5 mM Dithiothreitol, 20% glycerol, 1 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM phenylmethylsulfonylchloride), placed on ice, and sonicated 2 times for 10 s. Light microscopy was used to determine whether the cells were broken. The substrate 1-palmitoyl-2-[arachidonoyl-1-14C] phosphatidylcholine (14C-AA-PC) was used as a substrate. The substrate was dried under nitrogen and resuspended by sonication in assay buffer (80 mM glycine pH 9.0, 2 mM dithiothreitol, 1 mg/ml BSA) to a final optimum concentration as determined in preliminary experiments. To determine the role of calcium in activation of PLA2, assays were performed in the presence of either 5 mM CaCl2 or 5 mM EGTA. Experiments were also performed in the presence and absence of MAFP, an inhibitor of group IV and VI cytosolic PLA2 enzymes, or BEL, an inhibitor of group VI enzymes (Lio et al. 1996; Stewart et al. 2002; Zupan et al. 1993). The experiments were terminated by the addition of chloroform: methanol, 2:1 (v/v), extraction of the chloroform layer, and separation of lipids by thin-layer chromatography. Chromatography was performed in a neutral lipid solvent (hexane: diethyl ether: glacial acetic acid, 7:3:0.2) and the lipids visualized with I2 vapor. The zones corresponding to fatty acid and phospholipid were cut out and radioactivity determined by scintillation counting.
RT-PCR for group IV PLA2 gene transcripts
Total cellular RNA was isolated from HCAECs (10–20 × 106 cells) using NucleoSpin nucleic acid purification kits (Clontech, Palo Alto, CA, USA) according to the instructions of the manufacturer. Reverse transcription of total RNA (2 μg) was performed using the Superscript first-strand synthesis system. The reverse transcription reaction was carried out at 42°C for 50 min and then terminated by heating at 70°C for 15 min. PCR was performed in a total volume of 50 μl containing 4 μl of the reverse transcription reaction mixture, 0.2 mM of dNTPs, 0.2 μM of each primer, and 1.25 units of Taq polymerase in PCR buffer (20 mM Tris–HCl, pH 8.4, 50 mM KCl, 1.5 mM MgCl2). The conditions for PCR were an initial denaturation step at 94°C for 5 min, followed by 34 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 30 s, and a final extension step at 72°C for 10 min. As a positive control, glyceraldehyde 3-phosphate dehydrogenase (G3PDH) was also amplified. Additional negative control reactions were performed to ensure that amplification of a gene was not generated from contaminated genomic DNA: one was performed without RNA and the other under conditions to inhibit reverse transcriptase activity. Primers used for PCR were human PLA2 group IVA (sense, 5′-CCAAAATGTCATTTATAGATCC-3′ and antisense, 5′-CATGAACTATGCTTTGGGTTTAC-3′: GeneBank accession number-NM_024420); human PLA2 group IVB (sense, 5′-ACTGAGTGCCCTGCCCTCTGGTCAAG-3′ and antisense, 5′-TGCCCCATAAAGAACTCGGAGCCAAAGA-3′: Gene-Bank accession number-XM_009980); and human PLA2 group IVC (sense, 5′-AGAAAGAAGAAAAGGCGGCCGTGGAGAGAC-3′ and antisense, 5′-CGGCACTGAAGTCGAAGGAGAGGATGAGGT-3′: GeneBank accession number-XM_009119). The PCR products were analyzed on a 1.5% agarose gel.
Group IVC PLA2 Gene Silencing
iRNA sense [r(GAUAAUGAGCAGCCGGAAG)d(TT)] and antisense [r(CUUCCGGCUGCUCAUUAUC)d(TT)] strands specific for group IVC PLA2 were designed using batch analysis against a target sequence (AAGATAA TGAGCAGCCGGAAG) and synthesized by Qiagen, (Valencia, CA). Oligonucleotides were suspended in serum-free medium with SuperFect reagent and incubated at room temperature for 5–10 min. SuperFect reagent alone and untargeted siRNA served as controls. Cells were washed in PBS and incubated with 3.5 ml/plate of oligonucleotide mixer in medium/10% FBS for 3 h. At the end of the incubation period, the cells were washed with PBS and placed in medium with 10% FBS. To ensure that the protein of interest was silenced, Western blots were performed using an antibody specific for group IVC PLA2 as described below. To ensure that other cytosolic PLA2 enzymes were not silenced, two commercially available antibodies that recognize group IVA PLA2 and group VIA PLA2 were also used. 3H-OA release and apoptosis assays were performed in cells treated with PAHs, SRM 1597A, or vehicle; transfected with siRNA; and results compared to responses obtained with vehicle-, PAH-, or SRM 1597A-treated cells transfected with SuperFect reagent alone.
Animal studies
Feral mice (Peromyscus leucopus) were trapped along the floodplain of the Chattanooga Superfund site; and in a control site, traps were set each evening and checked each morning. The trapped animals were transported to the laboratory and anesthetized with CO2. The mice were exsanguinated by cardiac puncture after thoracotomy, and the distal aorta, heart, and lungs were excised and perfused with phosphate-buffered saline (PBS) to remove clotted blood. The mice were weighed and the liver, heart, lungs and aorta removed and frozen in liquid nitrogen for Western blot analysis.
PAH exposure analysis
Microsomes were prepared from liver tissue following methods of Trudeau and Maisonneuve (2001). Briefly, approximately 215 mg of liver was homogenized in phosphate buffer (20% 0.1 M sodium phosphate, 80% 0.1 M potassium phosphate, adjusted to pH 7.4 at 25°C) equal to four times the mass of the sample. This homogenate was then centrifuged at 9,000× g for 15 min at 4°C to remove cell debris, nuclei, and mitochondria. The supernatant was then filtered through a Sepharose gel column to isolate liver microsomes.
Cytochrome P450 activity was assayed in microsomes following the method of Trudeau and Maisonneuve (2001). BSA and resorufin were used as standards to determine the total protein content and total cytochrome P450 activity, respectively, in each liver sample. The 7-ethoxyresorufin dealkylation activity was kinetically measured by quantifying the formation of resorufin via fluorescence (excitation and emission were 530 and 590, respectively). The resultant values were protein normalized, and ethoxyresorufin-O-deethylase (EROD) activity was expressed as pmol/min/mg protein.
Apoptosis
For in vitro studies, Western analysis and ELISA were performed for detection of the cleavage products of PARP and histone fragmentation, respectively. For aortic tissue from mice trapped in the floodplain of the Chattanooga Creek, Western blot analysis of tissue homogenates was evaluated using antibodies to PARP and caspase-3.
PARP cleavage was evaluated after treatment with individual PAHs, SRM 1597A, or vehicle (DMSO) for 2 h by preparing crude extracts of protein and suspending them in ice-cold sample buffer (62.5% Tris–HCl, pH 6.8, 6 M urea, 10% glycerol, 2% SDS, 0.003% bromophenol blue, 5% β-mercaptoethanol) containing protease inhibitors. The cells were lysed by sonication on ice for 20 s, 40% duty, and the lysates subjected (40 μg of protein) to 8% SDS-polyacrylamide gel electrophoresis. The proteins were transferred to nitrocellulose membranes via a semi-dry blotting apparatus, and the membrane was blocked in 5% nonfat dry milk in PBS/0.1% Tween 20 and incubated overnight at 4°C with two different anti-PARP antibodies, one that recognizes both the 89-kD fragment and parent protein (Cell Signaling, Beverly, MA, USA) and another that recognizes only the 89-kD cleavage product (Pharmingen, San Jose, CA, USA). Visualization was enhanced by chemiluminescence (ECL).
To quantify the apoptotic effect and to determine the kinetics of PAH- and SRM-1597-A-induced apoptosis, a Cell Death Detection ELISAplus kit (Roche, Indianapolis, IN, USA) for detecting histone fragmentation was used. This assay is based upon a quantitative sandwich-enzyme immunoassay using mouse monoclonal antibodies against DNA and histones that allow for specific quantitative determination of cytoplasmic histone-associated DNA fragments in cell lysates. We have demonstrated previously that histone fragmentation correlates well with morphological measurements of apoptosis (Tithof et al. 2002). The cells were treated at 80–90% confluence with 30 μM PAHs, 0.8 mg/mL SRM 1597, or vehicle for 2 h and histone fragmentation determined. The cells were washed with warm PBS, lysed and the lysates incubated with antihistone-biotin and anti-DNA-POD at room temperature for 2 h. After washing with incubation buffer, 100 μl of substrate solution was added to develop the color, and measurements were taken at 405 nm using a plate reader.
Apoptosis of aortic tissues collected from mice captured in the floodplain of Chattanooga Creek was evaluated by caspase-3 and PARP cleavage. The tissues were lysed in homogenizing buffer (100 mM NaCI, 50 mM Tris [pH 7.4], 0.5 mM Triton X-100, 1 mM Dithiotreitol, 50 mM NaF, 0.5 mM NaVO3, protease inhibitors) and the homogenates spun at 14,000 rpm for 10 min. Western blotting for caspase-3 and PARP cleavage was performed as described above.
Statistical analysis
Data were expressed as mean ± SEM. Analysis of variance was used to analyze the data, and group means compared using the Student–Newman–Keuls test. Appropriate transformations were performed on all data that did not follow a normal distribution (e.g. percent data). If transformation failed to normalize the data, nonparametric statistics (Mann–Whitney rank sum test) were used for data analysis. For comparing the EROD activity between sites, a Student t test was employed. The criterion for statistical significance was P < 0.05 for all studies.
Results
PAH levels in streambed and floodplain sediments from Chattanooga Creek
Extremely high levels of all PAHs were observed in the streambed sediments. PAHs were also present in soil samples from the adjacent floodplain, but at lower concentrations than in the streambed sediments (Table 1). Typically, the streambed sediments were black in color and consisted of immiscible-phase coal tar mixed with silty sediments. There was no evidence of coal tar in the silty floodplain soils, and it appeared that the PAHs were sorbed to the soils. Concentrations of PHEN, fluoranthene (FLUO), and pyrene (PYRE) were very high in all streambed samples with average values of 2,770, 2,190, and 1,720 mg/g of sediment, respectively. Average concentrations of 4–6 ring compounds, Benzo(a)anthracene (B[a]A), B(a)P, Benzo (b)fluoranthene (B[b]F), Indeno(1,2,3-c,d) pyrene, (I[1,2]P), and benzo(g,h,i) perylene (B[g]P) were also high and ranged between 268 and 878 mg/g of sediment. In most cases, these concentrations exceed the EPA’s preliminary remediation goals for residential and industrial soils (US 2004a).
Table 1.
Levels of PAHs measured in the upstream and contaminated creekbed sediments and the contaminated floodplain (upper 10 cm)
| PAH | Creekbed sediments (mg/g sediment)
|
Contaminated floodplain (mg/g sediment) | |
|---|---|---|---|
| Upstream | Contaminated | ||
| Acenaphthylene | NDa | 164 | 1.32 |
| Acenaphthene | ND | 40.4 | 0.18 |
| Fluorene | ND | 656 | 0.19 |
| Phenanthrene | ND | 2,770 | 1.24 |
| Anthracene | ND | 678 | 1.04 |
| Fluoranthene | 2.24 | 2,190 | 5.27 |
| Pyrene | 2.1 | 1,720 | 2.64 |
| Benzo[a]anthracene | 5.01 | 749 | 4.48 |
| Chrysene | 0.27 | 524 | 4.55 |
| Benzo[b]fluoranthene | 1.88 | 878 | 5.72 |
| Benzo[k]fluoranthene | ND | 268 | 2.68 |
| Benzo[a]pyrene | ND | 678 | 5.50 |
| Indeno[1,2,3-c,3]pyrene | ND | 489 | 2.76 |
| Dibenz[a,h]anthracene | ND | 29.3 | 0.72 |
| Benzo[g,h,i]perylene | 6.01 | 345 | 3.87 |
ND Not detected
The PAH concentrations were approximately 2–3 orders lower in the floodplain soils relative to PAHs in the streambed sediments. The higher PAH concentrations in the streambed sediments were due to the presence of immiscible phase coal tar within the creek. Similarly, high concentrations in these sediments were reported in other studies (IT Corporation 1999; Vulava et al. 2004). The PAHs in the floodplain were most likely derived from sediments that came in contact with the streambed coal tar and were subsequently eroded and deposited in the floodplain during seasonal flood events (Vulava et al. 2004). The effects of 12 PAH compounds found in highest concentrations in the soil on endothelial cell PLA2 activity and apoptosis were determined.
PAH- and coal tar-induced PLA2 activation and 3H-fatty acid release from HCAECs
Figures 1 and 2 show the effects of 3-ring (Figs. 1a, 2a), 4-ring (Figs. 1b, 2b), 5-ring (Figs. 1c, 2c), and 6-ring PAHs (Figs. 1d, 2d) and SRM 1597A (Fig. 1e) on 3H-AA and 3H-OA release, respectively, from HCAECs. Little 3H-AA or 3H-OA release was observed from unstimulated HCAECs or from cells exposed to anthracene (ANTH [Figs. 1a, 2a]), chrysene (CHRY), or B(a)A (Figs. 1b, 2b); however, all other individual compounds (acenapthalene [ACEN], PHEN, FLUO, PYRE, B[a]P, B[b]F, B[k]F, I[1,2]P, B[g]P) and the coal tar mixture SRM1597A caused concentration-dependent release of 3H-AA (Fig. 1a–e) and 3H-OA (Fig. 2a–d). 3H-fatty acid release caused by SRM 1597A was of greater magnitude than that caused by individual compounds.
Fig. 1.
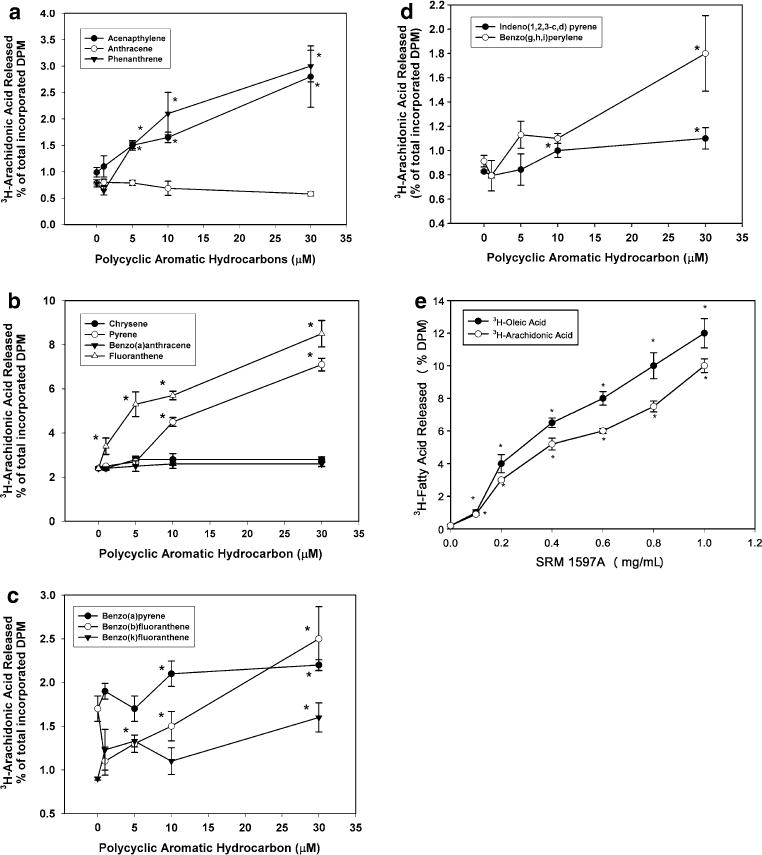
3H-arachidonic acid (3H-AA) and/or 3H-oleic acid (3H-OA; Fig. 1e) release from human coronary artery endothelial cells induced by 3-, 4-, 5-, or 6-ring PAHs. Cells were pre-labeled with 3H-fatty acid and stimulated for 1 h with vehicle or various concentrations of PAHs as described in “Materials and methods”. Cumulative release of 3H-AA into the medium was measured by scintillation counting. a 3-ring PAHs (acenapthalene, anthracene, phenanthrene). b 4-ring PAHs (chrysene, pyrene, benzo[a]anthracene, fluoranthene). c 5-ring compounds (benzo[a]pyrene, benzo[b]fluoranthene, benzo[k]fluoranthene). d 6-ring compounds (indeno[1,2,3-c,d] pyrene, benzo[g,h,i]perylene). e 3H-oleic acid and 3H-AA release from ECs treated with SRM 1597A. *Significantly different from vehicle-treated control, P < 0.05; n = 4
Fig. 2.

3H-oleic acid (3H-OA) release from human coronary artery endothelial cells induced by 3-, 4-, 5-, or 6-ring PAHs. Cells were pre-labeled with 3H-OA and stimulated for 1 h with vehicle or various concentrations of PAHs as described in “Materials and methods”. Cumulative release of 3H-OA into the medium was measured by scintillation counting. a 3-ring PAHs. b 4-ring PAHs. c 5-ring PAHs. d 6-ring PAHs. *Significantly different from vehicle-treated control, P < 0.05; n = 4
PAH- and coal tar-induced apoptosis
All nine compounds that caused 3H-fatty acid release (ACEN, PHEN, PYRE, FLUO, PYRE, B(a)P, B[b]F, B[k]F, I[1,2]P and B[g]P) also induced apoptosis as determined by histone fragmentation (Fig. 3a) or PARP cleavage (Fig. 3b–c). The compounds that failed to induce 3H-fatty acid release also failed to induce apoptosis (ANTH, CHRY and B[a]A; Fig. 3a). To seek further evidence that PAH-induced 3H-fatty acid release was linked to apoptosis, cells were treated with free AA or OA (10 μM) for 2 h and apoptosis determined using histone fragmentation (Fig. 3a) and PARP cleavage (Fig. 3b, c; lanes 11, 12). Both AA and OA induced significant apoptosis of HCAECs. Treatment with SRM 1597A (0.8 mg/mL) induced PARP cleavage (Fig. 3b, c, lane 13) and significant histone fragmentation that was similar in magnitude to the response obtained with AA and OA (Fig. 3a).
Fig. 3.
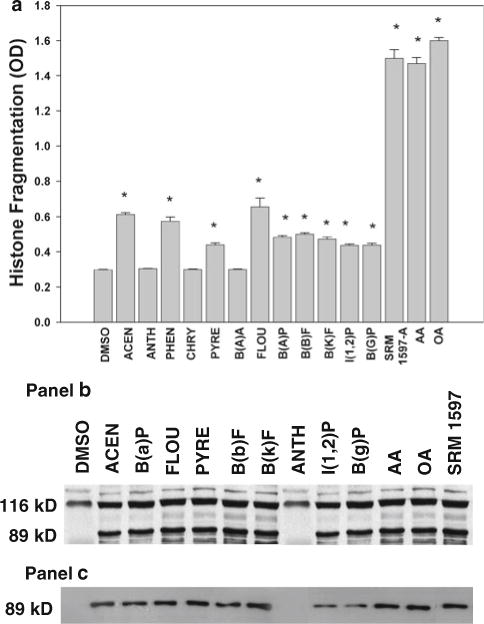
Apoptosis of human coronary artery endothelial cells induced by PAHs. Cells were treated with vehicle, 30 μM PAHs, 10 μM arachidonic acid (AA) or oleic acid (OA) or 0.8 mg/mL SRM-1597A for 2 h and apoptosis determined by histone fragmentation (a) or by detection of PARP cleavage as described in “Materials and methods”. *Significantly different from vehicle-treated control P < 0.05; n = 4. Two antibodies were used for detection of PARP cleavage: one that recognizes both the 116-kD and the 89-kD fragments of PARP (b) and one that recognizes only the 89-kD cleavage product (c)
RT-PCR and Cell-free PLA2 assays
To characterize further the PLA2 isoforms functioning in HCAECs, RT-PCR and cell-free PLA2 assays were performed. As seen in Fig. 4a, HCAECs expressed mRNA for at least three group IV PLA2 isoforms: A, B, and C. PLA2 activity in HCAEC whole-cell sonicates in the presence and absence of calcium was 14.7 ± 1.3 and 11.8 ± 1.3 mmol/μg protein/min, respectively, and these values were not significantly different from each other. The group IV and VI inhibitor MAFP attenuated PLA2 activity in the presence and absence of calcium; however, the group VI inhibitor BEL did not (Fig. 4b).
Fig. 4.

Characterization of groups IVA, B, and C PLA2 isoform expression and activity in human coronary artery endothelial cells. a Group IVA, B, and C are expressed by endothelial cells. RT-PCR analysis of total RNA from human coronary artery endothelial cells using PLA2 primers was performed as described in “Materials and methods”. A 10-μL aliquot of the PCR was analyzed on a 1.5% agarose gel containing ethidium bromide. Amplification of glyceraldehyde 3-phosphate dehydrogenase (G3PDH) was performed as a control using the same reaction conditions. DNA size markers are in bp. b Endothelial cells exhibit significant group IVC PLA2 activity. Whole cell homogenates were incubated with 14C-AA-PC in the presence of calcium or EGTA. Cells were pretreated with the group IV and VI inhibitor methyl arachidonoyl fluorophosphonate (MAFP; 10 μM), the group VI inhibitor bromoenol lactone suicide substrate (BEL; 1 μM), or vehicle and release of 14C-labeled arachidonic acid measured as described in “Materials and methods”. *Significantly different from results obtained in the absence of inhibitor. P < 0.05; n = 3
Silencing of group IVC PLA2 inhibits 3H-OA release and apoptosis of HCAECs
The results of the cell-free assays suggested that the predominant PLA2 activity when 14C-AA-PC was used as a substrate was calcium independent and inhibited by MAFP. Furthermore, the lack of acyl chain selectivity is consistent with activation of group IVC PLA2 by PAHs. To test this hypothesis, cells were transfected with siRNA against group IVC PLA2 and 3H-OA release and histone fragmentation determined. As shown in Fig. 5a (top blot), transfection with siRNA against group IVC inhibited translation of group IVC PLA2, but not the group IVA PLA2 (middle blot) or group VIA PLA2 (bottom blot). Silencing also attenuated significantly both 3H-OA release (Fig. 5b) and histone fragmentation (Fig. 5c).
Fig. 5.
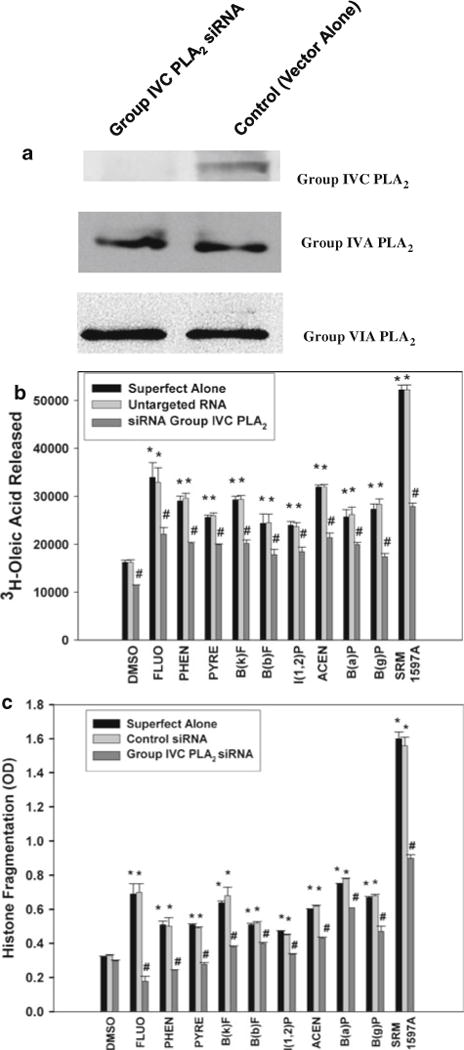
Silencing of group IVC PLA2 inhibits 3H-OA release and histone fragmentation induced by PAHs or SRM1597A. a Western blot analysis of lysates from human coronary artery endothelial cells (HCAEC) transfected with siRNA against group IVC PLA2 (lane 1) or control siRNA (lane 2). Silencing RNA against group IVC PLA2 did not silence the other cytosolic PLA2 enzymes. b 3H-OA release from HCAECs transfected with Superfect alone, untargeted RNA or siRNA group IVC PLA2 and treated with PAHs listed in the legend to Fig. 3 or SRM 1597A. c Histone fragmentation in HCAECs transfected with Superfect alone, untargeted RNA or siRNA group IVC PLA2 and treated with PAHs or SRM 1597A. *Significantly different from vehicle, #Significantly different from response obtained in the absence of siRNA, P < 0.05 n = 3
Feral mouse studies
We quantified biological markers indicative of PAH exposure in indigenous mice collected from a contaminated (Superfund) and relatively clean site (control). All of the mice (Peromyscus leucopus) used in this study were adults as defined by size and weight. The weights of the mice trapped along the floodplain of the Superfund site were not different from the weights of mice from the reference site (27.2 ± 1.1 gm vs. 27.0 ± 1.1 gm, respectively). The mice from the contaminated sites had significantly higher cytochrome P450 concentrations than those from the reference site: 59 ± 5 pmol/min/mg compared to 38 ± 8, respectively (P < 0.05 as indicated by increased EROD activity).
Aortic tissue from mice from the contaminated floodplain exhibited an increase in the cleavage product of PARP (Fig. 6b) and caspase-3 (Fig. 6c; lanes 1–3) when compared to mice from the uncontaminated reference area (Fig. 6a and c, lanes 4–6, respectively). Increased expression of a protein of approximately 61 kD was also observed in aortic tissue from mice from the Superfund site when compared to control mice (data not shown).
Fig. 6.

Western blot analysis of PARP and caspase-3 cleavage in aorta from feral mice trapped from the Superfund site or control site. PARP cleavage was assessed from mice taken from the control site (a) and from the Superfund site (b). c demonstrates the cleavage of caspase-3 from mice inhabiting the superfund site vs. the control site. Lanes 1–3 are representative of aortic tissue from mice from the Superfund site, and lanes 4–6 are from control mice. These blots are representative of 10 mice from each site
Discussion
This study confirms that significant levels of PAHs are present in the Chattanooga Creek Superfund site and the adjacent floodplain as a result of industrial contamination. Nine of the 12 compounds analyzed induced 3H-fatty acid release in a concentration-dependent manner and apoptosis. One of the PLA2 isoforms activated by PAHs was the group IVC PLA2 enzyme. This conclusion is based upon the ability of siRNA against this enzyme, but not untargeted sequences, to attenuate both 3H-fatty acid release and apoptosis. This is the first report of a role for this enzyme in PAH-induced apoptosis. These data provide compelling evidence that PAHs cause endothelial cell death by a mechanism that involves release of fatty acids from the sn-2 position of membrane phospholipids.
The coal tar mixture SRM 1597A also activated group IVC PLA2 and induced apoptosis of HCAECs. The effects of this mixture were substantially greater than the individual PAHs, indicating synergistic or additive responses to a complex mixture of PAHs in endothelial cells. Coal tar mixtures have been evaluated previously for genotoxicity and carcinogenicity (Mahadevan et al. 2005), but to our knowledge, this is the first report of a non-genotoxic effect of a coal tar mixture on endothelial cells.
All compounds that activated group IVC PLA2 and induced apoptosis possessed a bay or bay-like region on the molecule. A bay region is a nonlinear/angular condensation of benzo rings, whereas a bay-like region is formed from methyl substitution of linear-condensed molecules (Mackay et al. 1992). Previous studies suggest that bay or bay-like regions on PAHs are essential for their tumor-promoting activity via inhibition of gap junction communication (Rummel et al. 1999). Among the 15 compounds studied, only three (ANTH, PYRE, and B[a]A) failed to cause 3H-fatty acid release or apoptosis. Both ANTH and PYRE are linear compounds lacking the ability to activate PLA2 or to induce apoptosis. Interestingly, in previous studies, 1-methylanthracene was shown to activate PLA2 and to induce apoptosis (Tithof et al. 2002). This compound has a methyl group at the 1-position, which gives it a bay-like region. In contrast, 2-methylanthracene and anthracene, both linear compounds, were inactive. These data suggest that specific structural determinants, namely bay or bay-like regions, are necessary for activation of group IVC PLA2 and induction of apoptosis in endothelial cells. However, structural determinants alone cannot account for the apoptotic toxicity profile, since B(a)A possesses a bay region but did not activate PLA2 or induce apoptosis. One explanation for this discrepancy may be the bulkiness of this molecule when compared to the other compounds with bay or bay-like regions.
Group IVC PLA2 is constitutively expressed in HCAECs (Tithof et al. 2002). Gene silencing of group IVC PLA2 significantly attenuated both 3H-OA release and apoptosis, providing compelling evidence that this enzyme is involved in PAH-induced fatty acid release and apoptotic cell death. An increase in group IVC PLA2 protein expression was reported in macrophages exposed to Mycobacterium tuberculosis, and this increase in protein expression was associated with apoptotic cell death (Duan et al. 2001). In addition, group IVC PLA2 has been shown to associate with mitochondrial membranes, further supporting a role for this enzyme in the mitochondrial induction of apoptosis (Tucker et al. 2005).
A role for other PLA2 isoforms should not be ignored, as groups IVA PLA2 and VI iPLA2 have been linked to apoptosis (Atsumi et al. 1998, 2000; Capper and Marshall 2001). In a previous study, we showed that benzo(a)pyrene, phenanthrene, and 1-methylanthracene differentially activated three distinct PLA2 isoforms linked to apoptosis (Tithof et al. 2002). Moreover, phospholipase C has been shown to cause release of arachidonic acid that was linked to inhibition of gap junctional communication in cultured epithelial cells after treatment with 1-MA (Upham et al. 2008). The activation of other phospholipase enzymes by PAHs may, in fact, account for different levels of inhibition of 3H-OA release and apoptosis by different compounds.
The molecular mechanism by which PAHs activate group IVC PLA2 is not known. Many PAHs are known to bind the aromatic hydrocarbon receptor, an action that promotes oxidative stress and apoptosis (Chaloupka et al. 1995). Further, reactive oxygen species are produced during cellular metabolism of PAHs (Kerzee and Ramos 2000), and oxygen radicals induce apoptosis of endothelial cells (Hermann et al. 1997) and activate the group IVC enzyme via a tyrosine phosphorylation pathway (Asai et al. 2003). The ability of PAHs to interact with other receptors or proteins involved in the activation of PLA2 should be considered. For instance, PAHs bind steroid hormone receptors and glycine N-methyltransferase (Bhat and Bresnick 1997; Chang and Liao 1987). PLA2 activation and apoptosis may also be related to the lipophilicity of PAHs. Because of their fat solubility, PAHs may enhance the interaction of PLA2 enzymes with their respective substrates. Other lipophilic compounds, such as arachidonic acid, have been shown to facilitate assembly of protein complexes (Dana et al. 1994). Arachidonic acid induces a conformational change in NADPH oxidase, enabling proper assembly of the enzyme complex responsible for the production of superoxide anion (Dana et al. 1994; Tithof et al. 1998).
Cigarette smoke contains many of the PAHs commonly found in urban pollution. The Honolulu Heart Program, which began in 1968, provided a 20-year prospective study of 8,006 Japanese-American men aged 45–65. Of relevance was the finding that morbidity and mortality due to cardiovascular disease in this group of men was reduced by 50% in smokers who consumed diets rich in omega-3 fatty acids, compounds that inhibit the AA cascade at multiple levels (Rodriguez et al. 1996). The Honolulu Heart Project is one of several studies (Goodnight 1994; McCarty 1996; Ridker et al. 1997) linking toxic chemicals in cigarette smoke such as PAHs, PLA2/AA activation, and cardiovascular disease. This relationship is worthy of continued investigation.
Acknowledgments
Special thanks to Misty Bailey and Anik Vasington for excellent technical assistance. This work was supported by the Waste Management and Education Institute at the University of Tennessee, American Heart Association Grant #0160264B, and NIH grants HL34303 and HL61378.
Contributor Information
Patricia K. Tithof, Email: ptithof@utk.edu, Department of Pathobiology, College of Veterinary Medicine, University of Tennessee, Knoxville, TN 37996-4545, USA; Center for Environmental Biotechnology, University of Tennessee, Knoxville, TN 37996, USA.
Sean M. Richards, Department of Biological and Environmental Sciences, University of Tennessee, Chattanooga, TN 37403, USA
Mona A. Elgayyar, Department of Pathobiology, College of Veterinary Medicine, University of Tennessee, Knoxville, TN 37996-4545, USA
Fu-Minn Menn, Center for Environmental Biotechnology, University of Tennessee, Knoxville, TN 37996, USA.
Vijay M. Vulava, Center for Environmental Biotechnology, University of Tennessee, Knoxville, TN 37996, USA
Larry McKay, Center for Environmental Biotechnology, University of Tennessee, Knoxville, TN 37996, USA.
John Sanseverino, Center for Environmental Biotechnology, University of Tennessee, Knoxville, TN 37996, USA.
Gary Sayler, Center for Environmental Biotechnology, University of Tennessee, Knoxville, TN 37996, USA.
Dawn E. Tucker, Department of Pediatrics, National Jewish Medical and Research Center, Denver, CO 80206, USA
Christina C. Leslie, Department of Pediatrics, National Jewish Medical and Research Center, Denver, CO 80206, USA
Kim P. Lu, Department of Biology, Texas A&M University, College Station, TX 77843, USA
Kenneth S. Ramos, Department of Biochemistry and Molecular Biology, University of Louisville, Louisville, KY 40202, USA
References
- Asai K, Hirabayashi T, Houjou T, Uozumi N, Taguchi R, Shimizu T. Human group IVC phospholipase A2 (cPLA2gamma). Roles in the membrane remodeling and activation induced by oxidative stress. J Biol Chem. 2003;278:8809–8814. doi: 10.1074/jbc.M212117200. [DOI] [PubMed] [Google Scholar]
- Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2, but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273:13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. J Biol Chem. 2000;275:18248–18258. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- Bhat R, Bresnick E. Glycine N-methyltransferase is an example of functional diversity: role as a polycyclic aromatic hydrocarbon-binding receptor. J Biol Chem. 1997;272:21221–21226. doi: 10.1074/jbc.272.34.21221. [DOI] [PubMed] [Google Scholar]
- Blaha L, Machala M, Vondracek J, Breinekova K. Multiple oxidative stress parameters are modulated in vitro by oxygenated polycyclic aromatic hydrocarbons identified in river sediments. Adv Exp Med Biol. 2001;500:225–228. doi: 10.1007/978-1-4615-0667-6_32. [DOI] [PubMed] [Google Scholar]
- Blaha L, Kapplova P, Vondracek J, Upham B, Machala M. Inhibition of gap-junctional intercellular communication by environmentally occurring polycyclic aromatic hydrocarbons. Toxicol Sci. 2002;65:43–51. doi: 10.1093/toxsci/65.1.43. [DOI] [PubMed] [Google Scholar]
- Capper EA, Marshall LA. Mammalian phospholipase A(2): mediators of inflammation, proliferation and apoptosis. Prog Lipid Res. 2001;40:167–197. doi: 10.1016/s0163-7827(01)00002-9. [DOI] [PubMed] [Google Scholar]
- Chaloupka K, Steinberg M, Santostefano M, Rodriguez LV, Goldstein L, Safe S. Induction of cyp1a–1 and cyp1a–2 gene expression by a reconstituted mixture of polynuclear aromatic hydrocarbons in B6C3F1 mice. Chem Biol Ineract. 1995;96:207–221. doi: 10.1016/0009-2797(94)03586-w. [DOI] [PubMed] [Google Scholar]
- Chang CS, Liao SS. Topographic recognition of cyclic hydrocarbon and related compounds by receptors for androgens, estrogens and glucocorticoids. J Steroid Biochem. 1987;27(1–3):123–131. doi: 10.1016/0022-4731(87)90303-7. [DOI] [PubMed] [Google Scholar]
- Dana R, Malech HL, Levy R. The requirement for phospholipase A2 for activation of the assembled NADPH oxidase in human neutrophils. Biochem J. 1994;297(pt 1):217–223. doi: 10.1042/bj2970217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis WR. Chattanooga Creek sediment profile study. Chattanooga, Tennessee: United States environmental protection agency; 1992. region IV, April/August 1992. [Google Scholar]
- Dimmeler S, Hermann C, Zeiher AM. Apoptosis of endothelial cells. Contribution to the patho-physiology of atherosclerosis? Eur Cytokine Netw. 1998;9:697–698. [PubMed] [Google Scholar]
- Donaldson K, Stone V, Seaton A, MacNee W. Ambient particle inhalation and the cardiovascular system: potential mechanisms. Environ Health Perspect. 2001;109(suppl 4):523–527. doi: 10.1289/ehp.01109s4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Gan H, Arm J, Remold HG. Cytosolic phospholipase A2 participates with TNF-a in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J Immunol. 2001;166:7469–7476. doi: 10.4049/jimmunol.166.12.7469. [DOI] [PubMed] [Google Scholar]
- Geng YJ, Libby P. Evidence for apoptosis in advanced human atheroma: colocalization with interleukin-1β converting enzyme. Am J Pathol. 1995;147:251–266. [PMC free article] [PubMed] [Google Scholar]
- Goodnight SH. The vascular effects of ω-3 fatty acids. J Invest Dermatol. 1994;93:102S–106S. doi: 10.1111/1523-1747.ep12581218. [DOI] [PubMed] [Google Scholar]
- Granberg L, Brunstrom B, Brandt I. Formation of benzo[a]-pyrene and 7, 12-dimethylbenz[a]anthracene adducts in vascular endothelia of cytochrome P4501A-induced chicken embryos. Environ Toxicol Chem. 2003;22(10):2393–2399. doi: 10.1897/02-420. [DOI] [PubMed] [Google Scholar]
- Hermann C, Zeiher AM, Dimmeler S. Shear stress inhibits H2O2-induced apoptosis of human endothelial cells by modulation of the glutathione redox cycle and nitric oxide synthase. Arterioscler Thromb Vasc Biol. 1997;17:3588–3592. doi: 10.1161/01.atv.17.12.3588. [DOI] [PubMed] [Google Scholar]
- Howard G, Wagenknecht LE, Burke GL, Diez-Roux A, Evans GW, McGovern P, Nieto FJ, Tell GS. Cigarette smoking and progression of atherosclerosis: the atherosclerosis risk in communities (ARIC) study. J Am Med Assoc. 1998;279:119–124. doi: 10.1001/jama.279.2.119. [DOI] [PubMed] [Google Scholar]
- Isner JM, Kearney M, Bortman S, Passeri J. Apoptosis in human atherosclerosis and restenosis. Circulation. 1995;91:2703–2711. doi: 10.1161/01.cir.91.11.2703. [DOI] [PubMed] [Google Scholar]
- IT Corporation. Final report: removal action for the tennessee products superfund site. Chattanooga, Tennessee: US army corps of engineers; 1999. [Google Scholar]
- Kerzee JK, Ramos KS. Activation of c-Ha-ras by benzo(a)pyrene in vascular smooth muscle cells involves redox stress and aryl hydrocarbon receptor. Mol Pharmacol. 2000;58:152–158. doi: 10.1124/mol.58.1.152. [DOI] [PubMed] [Google Scholar]
- Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca(2+)-independent phospholipase A2 by methyl arachidonoyl fluorophosphonate. Biochim Biophys Acta. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- Mackay D, Shiu WY, Ma KC. Illustrated handbook of physical-chemical properties and environmental fate for organic chemicals. I–IV. Lewis Chelsea; MI: 1992. [Google Scholar]
- Mahadevan B, Marston C, Dashwood W, Yonghai L, Pererira C, Baird W. Effect of a standardized complex mixture derived from coal tar on the metabolic activation of carcinogenic polycyclic hydrocarbons in human cells in culture. Chem Res Toxicol. 2005;18:224–231. doi: 10.1021/tx0497604. [DOI] [PubMed] [Google Scholar]
- McCarty MF. Fish oil may be an antidote for the cardiovascular risk of smoking. Med Hypotheses. 1996;46:337–347. doi: 10.1016/s0306-9877(96)90183-8. [DOI] [PubMed] [Google Scholar]
- Mueller JG, Chapman PJ, Pritchard PH. Creosote-contaminated sites. Environ Sci Technol. 1989;23:1197–1201. [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- Rodriguez BL, Sharp DS, Abbott RD, Burchfiel CM, Masaki K, Chyou PH, Huang B, Yano K, Curb JD. Fish intake may limit the increase in risk of coronary heart disease morbidity and mortality among heavy smokers. The honolulu heart program. Circulation. 1996;94:952–956. doi: 10.1161/01.cir.94.5.952. [DOI] [PubMed] [Google Scholar]
- Rummel AM, Trosko JE, Wilson MR, Upham BL. Polycyclic aromatic hydrocarbons with bay-like regions inhibited gap junctional intercellular communication and stimulated MAPK activity. Toxicol Sci. 1999;49:232–240. doi: 10.1093/toxsci/49.2.232. [DOI] [PubMed] [Google Scholar]
- Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
- Stewart A, Ghosh M, Spencer DM, Leslie CC. Enzymatic properties of human cytosolic phospholipase A2γ. J Biol Chem. 2002;277:29526–29536. doi: 10.1074/jbc.M204856200. [DOI] [PubMed] [Google Scholar]
- Tithof PK, Peters-Golden M, Ganey PE. Distinct phospholipases A2 regulate release of arachidonic acid for eicosanoid production and superoxide anion generation in neutrophils. J Immunol. 1998;160:953–960. [PubMed] [Google Scholar]
- Tithof PK, Elgayyar M, Cho Y, Guan W, Fisher AB, Peters-Golden M. Polycyclic aromatic hydrocarbons present in cigarette smoke cause endothelial cell apoptosis by a phospholipase A2-dependent mechanism. FASEB J. 2002;16:1463–1464. doi: 10.1096/fj.02-0092fje. [DOI] [PubMed] [Google Scholar]
- Trudeau SF, Maisonneuve FJ. Headquarters Technical Report 339E. Canadian Wildlife Services; Hull, Quebec, Canada: 2001. A method to determine cytochrome P4501A activity in wildlife microsomes. [Google Scholar]
- Tucker DE, Stewart A, Nallan L, Bendale P, Ghomashchi F, Gelb MH, Leslie CC. Group IVC cytosolic phospholipase A2g is farnesylated and palmitoylated in mammalian cells. J Lipid Res. 2005;46:2122–2133. doi: 10.1194/jlr.M500230-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham BL, Weis LM, Trosko JE. Modulated gap junctional intercellular communication as a biomarker pf AH epigenetic toxicity: structure-function relationship. Eviron Health Perspect. 1998;106:975–981. doi: 10.1289/ehp.98106s4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham BL, Blaha L, Babica P, Park JS, Sovadinova I, idrotj C, Rummel AM, Weis M, Sai L, Tithof PK, Guzvic M, Vondracek J, Machala M, Trosko JE. Tumor promoting properties of cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidyl choline-specific phospholipase C. Cancer Sci. 2008;99:696–705. doi: 10.1111/j.1349-7006.2008.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US EPA. Remedial planning activities at selected uncontrolled hazardous substance disposal sites for EPA Region IV. United States environmental protection agency; 1999. Remedial investigation report for the Tennessee products site, Chattanooga, Tennessee. Region IV, EPA 68-W9-0056, February 1999. [Google Scholar]
- US EPA. Preliminary remediation goals. United States environmental protection agency; 2004a. region 9. http://www.epa.gov/region9/waste/sfund/prg/index.html. [Google Scholar]
- US EPA. SW-846 Manual: test methods for evaluating solid wastes, physical/chemical methods. United States environmental protection agency, office of solid waste; 2004b. http://www.epa.gov/epaoswer/hazwaste/test/sw846.htm. [Google Scholar]
- Vulava VM, McKay LD, McCarthy JF, Dickerson DS, Cooper L, Menn F. Transport and deposition of coal tar contaminated sediments in an urban freshwater stream. Presented at Fourth World Congress of the Society of environmental toxicology and chemistry; Portland, Oregon. Nov 14–19.2004. [Google Scholar]
- Weis LM, Rummel AM, Masten SJ, Trosko JE, Upham BL. Bay or baylike regions of polycylic aromatic hydrocarbons were potent inhibitors of gap junctional intercellular communication. Environ Health Perspect. 1998;106:17–22. doi: 10.1289/ehp.9810617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JJ, Tillitt DE. Ethoxyresorufin-O-deethylase (EROD) activity in fish as a biomarker of chemical exposure for use in the biomonitoring of environmental status and trends (BEST) program. Crit Rev Tox. 2000;30:347–570. doi: 10.1080/10408440091159239. [DOI] [PubMed] [Google Scholar]
- Zupan LA, Weiss RH, Hazen SL, Parnas BL, Aston DW, Lennon PJ, Getman DP, Gross RW. Structural determinants of haloenol lactone-mediated suicide inhibition of canine myocardial calcium-independent phospholipase A2. J Med Chem. 1993;36:95–100. doi: 10.1021/jm00053a012. [DOI] [PubMed] [Google Scholar]