

High expression of a gene that controls tomato shape is accompanied by dramatic shifts in gene expression and metabolite and hormone accumulation during the early stages of fruit development.
Abstract
SUN controls elongated tomato (Solanum lycopersicum) shape early in fruit development through changes in cell number along the different axes of growth. The gene encodes a member of the IQ domain family characterized by a calmodulin binding motif. To gain insights into the role of SUN in regulating organ shape, we characterized genome-wide transcriptional changes and metabolite and hormone accumulation after pollination and fertilization in wild-type and SUN fruit tissues. Pericarp, seed/placenta, and columella tissues were collected at 4, 7, and 10 d post anthesis. Pairwise comparisons between SUN and the wild type identified 3,154 significant differentially expressed genes that cluster in distinct gene regulatory networks. Gene regulatory networks that were enriched for cell division, calcium/transport, lipid/hormone, cell wall, secondary metabolism, and patterning processes contributed to profound shifts in gene expression in the different fruit tissues as a consequence of high expression of SUN. Promoter motif searches identified putative cis-elements recognized by known transcription factors and motifs related to mitotic-specific activator sequences. Hormone levels did not change dramatically, but some metabolite levels were significantly altered, namely participants in glycolysis and the tricarboxylic acid cycle. Also, hormone and primary metabolite networks shifted in SUN compared with wild-type fruit. Our findings imply that SUN indirectly leads to changes in gene expression, most strongly those involved in cell division, cell wall, and patterning-related processes. When evaluating global coregulation in SUN fruit, the main node represented genes involved in calcium-regulated processes, suggesting that SUN and its calmodulin binding domain impact fruit shape through calcium signaling.
Tomato (Solanum lycopersicum) is an important vegetable that is extensively studied for aspects related to plant and fruit development, fruit ripening, and quality (de Jong et al., 2011; Matas et al., 2011; Goulet et al., 2012; Ruan et al., 2012; Mazzucato et al., 2013; Osorio et al., 2013; Pan et al., 2013; Zhong et al., 2013; van der Knaap et al., 2014). As a member of the Solanaceae family, tomato is also an excellent genetic system because of its diploid and highly inbred nature as well as the availability of genetic and genomic resources, such as a reference genome sequence (Tomato Genome Consortium, 2012). Fruit development initiates at anthesis (flower opening), which in tomato, is marked by the release of pollen and pollination. After successful fertilization of the ovules, fruit growth commences. In tomato and many other plants, the initial growth stages are characterized by cell division followed by the cell expansion of the maternally derived fruit tissues (Gillaspy et al., 1993; Xiao et al., 2009). Cross talk among the various hormones plays key roles during the initial stages of fruit growth (Gillaspy et al., 1993; Ozga et al., 2003; Montoya et al., 2005; de Jong et al., 2009).
Growth of plant organs, including the fruit, occurs along three axes: the proximal-distal, mediolateral, and abaxial-adaxial axes. Fruit length is determined by the degree of growth along the proximal-distal axis, whereas fruit width is determined by the degree of growth along the mediolateral axis. The degree of the pericarp thickness and other internal tissues are determined along the abaxial-adaxial axis. These growth axes are defined much earlier, namely during the formation of the floral meristem and gynoecium primordia in the developing flower. Correct initiation of the gynoecium requires the specification of organ and tissue identity as well as the establishment of the boundaries between primordia to ensure that the appropriate identities and division patterns are initiated and maintained throughout organ growth (Dinneny et al., 2005; Balanzá et al., 2006; Girin et al., 2009; van der Knaap et al., 2014). Hormones, such as auxin, play a critical role in setting up the patterns of cell division by regulating the expression of transcription factors that control ovary development along the three axes of growth (Ståldal and Sundberg, 2009; Nole-Wilson et al., 2010; Wang et al., 2011).
The role of patterning genes in the initiation and growth of organ primordia is relatively well understood. Except in fruit ripening (Vrebalov et al., 2009), the role of patterning genes in fruit ontogeny is largely unknown. Soon after initiation, the different tissue types within the developing gynoecium arise by reactivation of the cell identity and cell division in the so-called quasi-meristems (zones of active cell proliferation within an organ; Girin et al., 2009). The quasi-meristems give rise to medial tissues, such as the ovules, septum, style, stigma, and a structure specific to fruit of members in the Brassicaceae family, the replum (Girin et al., 2009). Other than the embryo and the placenta surrounding the developing seeds, fruit development is not typically marked by newly arising tissues. However, rates of cell division and cell expansion are enhanced after fertilization (Gillaspy et al., 1993; Xiao et al., 2009), and it is, therefore, conceivable that the patterning genes also play important roles during the initial stages of fruit development.
The tomato fruit shape gene SUN regulates proximal-distal patterning by controlling the elongation of the fruit (Xiao et al., 2008). The mutation resulted from an interchromosomal gene duplication event, whereby the coding region was placed in a different genome context, resulting in high expression of the derived version of SUN in tissues and at developmental time points when the ancestral version of this gene is barely expressed (Xiao et al., 2009; Wu et al., 2011). SUN encodes a protein of the IQ Domain (IQD) family characterized by a central domain of 67 conserved amino acid residues (referred to as the IQ67 domain), which is defined by a unique and repetitive arrangement of the three consensus calmodulin (CaM) recruitment motifs (Levy et al., 2005; Xiao et al., 2008). CaM binding proteins have extremely diverse functions in plants, including metabolism regulation and hormone signaling (Kim et al., 2009). The effect on fruit shape by SUN is noticeable at anthesis but most dramatic 7 to 10 d post anthesis (DPA; van der Knaap and Tanksley, 2001; Xiao et al., 2009; Wu et al., 2011). SUN does not control fruit weight but instead, controls the redistribution of fruit mass, and the degree of elongation is positively correlated to the level of gene expression (Xiao et al., 2008; Wu et al., 2011). Throughout development from the ovary at anthesis until the breaker stage, SUN is expressed highly in the SUN mutant compared with wild-type fruit (Xiao et al., 2009). The cellular basis of this elongated shape is clearly visible at 7 DPA, when an increase in cell number is found in columella and septum tissues in the proximal-distal direction and a decrease is found in the mediolateral direction in plants carrying the SUN mutation compared with the wild type. Cell size is not significantly different in SUN compared with wild-type fruit. Based on these results, it was proposed that SUN results in changes of the plane of cell division toward more cells in the proximal-distal direction (Wu et al., 2011). Little is known about the function of the IQD family, despite their widespread occurrence in plant genomes (Abel et al., 2005; Huang et al., 2013). In Arabidopsis (Arabidopsis thaliana), increased expression of AtIQD1 leads to higher glucosinolate levels, which coincide with increased expression of certain enzymes in the shared auxin/glucosinolate pathway involving the intermediate metabolite indole-3-acetaldoxime (Levy et al., 2005). In tomato, however, glucosinolates are not produced, and the indole-3-acetaldoxime pathway is only found in the Brassicaceae family (Glawischnig et al., 2004; Sugawara et al., 2009; Zhao, 2010; Won et al., 2011), suggesting that the role of these proteins is not specific to glucosinolate production. In support of that notion, AtIQD1 was found to localize to microtubules in planta and physically interact with a kinesin light chain-related protein1 (Bürstenbinder et al., 2013). These results imply that members of the IQD family may provide a scaffold for protein transport along the microtubules through kinesin motor proteins (Abel et al., 2013; Bürstenbinder et al., 2013). In tomato, SUN overexpression leads to extreme phenotypes, such as seedless fruit with a pointed shape, twisted stems and leaf rachis, and altered vascular patterning as well as increased leaf serration (Wu et al., 2011). Combined, these traits are associated with altered auxin homeostasis in tomato and other plants (Bouchard et al., 2006; de Jong et al., 2009; Molesini et al., 2009; Wu et al., 2011; Sawchuk et al., 2013; Ge et al., 2014). SUN expression does not lead to extensive changes in expression of AUX/IAA or Auxin Response Factor; therefore, SUN has been proposed to function in auxin-regulated processes by either increasing its biosynthesis or altering polar transport (Xiao et al., 2009; Wu et al., 2011). However, direct links between SUN expression and the auxin pathway have not been established.
The goal of this study was to investigate the molecular mechanisms by which SUN controls fruit shape in tomato. To do so, we carried out a network analyses approach to gain insights into the role of SUN in fruit development. Three tissues at different stages of fruit development were collected from wild-type and SUN plants and evaluated for gene expression, primary metabolite, and hormone profiles. By clustering gene expression based on similar profiles in the different tissues over time and evaluating metabolite and hormone accumulation, putative regulatory networks were identified that accompanied the early stages of fruit development when the changes in fruit shape mediated by SUN became apparent. The results showed that the early stages of tomato fruit development were highly dynamically controlled. Moreover, clusters of genes that were codifferentially expressed in SUN compared with the wild type share common promoter elements, which might explain the codifferential expression patterns. The findings provided unique information as to how SUN may regulate elongated fruit shape, while also providing a framework for the metabolite, hormone, and gene networks that are operating during the early stages of tomato fruit development.
RESULTS
Clustering of Coregulated Genes in Developing Fruit Tissues
Fruit growth after pollination consists of a rapid increase in cell division and the initiation of seed development followed by a period of cell expansion and organ enlargement (Gillaspy et al., 1993; Xiao et al., 2009). The effect of SUN on fruit shape is most dramatic immediately after pollination, and in cultivated tomato, the shape is final at 10 to 12 DPA (Fig. 1; van der Knaap and Tanksley, 2001). The tissue that is most affected in the change in shape is the central part of the fruit, namely the columella and septum (Wu et al., 2011). To identify key processes that accompanied the initial stages of fruit development after pollination, we first evaluated global variation in gene expression in wild-type fruits in the following tissues at these time points: pericarp, columella, and the combined seed/placenta tissues at 4, 7, and 10 DPA (Supplemental Tables S1 and S2). Using fuzzy C means, we selected a subset of clusters of 100 based on the most dynamic expression changes in a spatial and temporal manner (Fig. 2; Supplemental Table S3). The selected clusters represented dynamic developmental changes in expression programs during early fruit development and included enrichment of different bin ontologies. Cell division (Fig. 2A; 604 genes) exhibited peak expression in 7-DPA pericarp tissue and shares a similar pattern with RNA (Fig. 2C; 224 genes), a cluster enriched for transcription factors. Showing peak expression in 4- and 7-DPA pericarp, the cluster enriched for secondary metabolism (Fig. 2D; 396 genes) was also enriched for lipid metabolism, cell division, and transport (Supplemental Table S4A). The cell wall-enriched (Fig. 2B; 188 genes) gene cluster showed peak expression at 7 DPA in all three fruit tissues. Interestingly, a cluster enriched for transport/signaling showed peak expression in 4-DPA columella and decreased as development of this tissue proceeded (Fig. 2F; 224 genes). This cluster’s expression program was in contrast to a cluster enriched for transport/hormone, which showed peak expression in 10-DPA columella (Fig. 2E; 353 genes), implying that signaling led to hormonal and transport responses during columella development. A cluster of genes enriched for photosynthesis (Fig. 2G; 201 genes) showed peak expression in 4- and 7-DPA columella. This cluster was also enriched for tricarboxylic acid cycle, carbohydrate metabolism, oxidative pentose phosphate pathway, and protein, showing an emphasis on metabolism in columella tissue during the earliest stages of fruit development. At 10 DPA, the latest developmental stage that we assayed, a cluster of protein-related enriched genes showed peak expression in all fruit tissues, presumably setting the stage for the next phase of fruit development.
Figure 1.

Tomato fruit growth after pollination. A, Wild-type fruit at anthesis and 4, 7, 10, and 16 DPA. B, SUN fruit at anthesis and 4, 7, 10, and 16 DPA. Bar = 1 cm. C, Expression of SUN in wild-type and SUN fruit tissues and at developmental time points. col, Columella; per, pericarp; se, seed and placenta; WT, wild type.
Figure 2.

Dynamically expressed and coregulated genes during early fruit development in wild-type tomato. A, Six hundred four genes represented by cell cycle- and cell division-related processes. B, One hundred eighty-eight genes represented by cell wall-related processes. C, Two hundred twenty-four genes represented by RNA- and transcription-related processes. D, Three hundred ninety-six genes represented by secondary metabolism-related processes. E, Three hundred fifty-three genes related to transport- and hormone-related processes. F, Two hundred twenty-four genes represented by transport- and signaling-related processes. G, Two hundred one genes represented by photosynthesis-related processes. H, Two hundred eighty-four genes represented by protein- and translation-related processes. Gene expression is on a relative mean-centered scale, showing the median expression level for all members in the respective cluster in SUN and wild-type fruit. Error bars indicate se for the genes in the respective cluster. Col.4, Columella 4 DPA; Col.7, columella 7 DPA; Col.10, columella 10 DPA; Per.4, pericarp 4 DPA; Per.7, pericarp 7 DPA; Per.10, pericarp 10 DPA; Se.7, seed 7 DPA; Se.10, seed 10 DPA.
Collectively, the temporal and spatial clusters showed a coordinated program of gene expression that was dynamically executed during early fruit development. To investigate whether SUN expression altered overall gene expression programs, we next evaluated those that were differentially regulated in the same tissues and time points.
Clustering of the Codifferentially Expressed Genes in SUN and Wild-Type Fruit
To provide insights into how SUN regulates shape and whether this is accompanied with shifts in transcript profiles, we identified differentially expressed genes in eight pairwise comparisons of SUN and wild-type fruit tissues and time points. The total number of significant differentially expressed genes in at least one comparison was 3,154 based on DESeq analysis and multiple Bonferroni corrections (Anders and Huber, 2010; Anders et al., 2013). Seven genes showed robust differential expression in all tissues at all developmental time points. Three of the genes were expected, namely SUN itself (Solyc10g079240; Fig. 1C) and the two defensin genes (DEFENSIN-LIKE1 [DEFL1] and DEFL2; Solyc07g007760 and Solyc07g007750, respectively), which had expression that was perturbed by the retrotransposition of SUN from chromosome 10 to chromosome 7 (Xiao et al., 2008; Jiang et al., 2009). Of the other four genes, three had lower expression in SUN fruit, and one had higher expression. The higher expressed gene, Solyc07g064380, encoded a Ser/Thr phosphatase. Of the three genes that showed lower expression in SUN, two encoded proteins that were involved in secondary metabolism. Solyc06g035940 was a likely paralog of PROTODERMAL FACTOR2, encoding a transcription factor regulating cutin biosynthesis (Nadakuduti et al., 2012). Solyc07g006670 encoded a hydroxycinnamoyl CoA quinate transferase participating in chlorogenic acid synthesis.
The differential gene expression appeared dynamic over the course of fruit development and in different tissues (Supplemental Table S5). To further investigate how elevated SUN expression impacts overall pathways in the developing tomato fruit, we evaluated the enrichment of gene ontology (GO) categories in the significantly differentially expressed gene data set (Supplemental Table S4B). Collectively, the enrichment for genes involved in cell wall, photosynthesis, hormone metabolism, transport, and biotic stress as well as secondary metabolism, including lipid metabolism, metal handling, and cell division, indicated that these processes were most dramatically affected by high expression of SUN. Linear factorial modeling of the data with genotype and genotype × time point interactions in the model also supported the importance of many of the same processes in differential fruit growth that is conditioned by SUN (Supplemental Tables S4C and S6).
We clustered the differentially expressed genes based on the log2 fold change of the wild type and SUN using fuzzy C means. This analysis resulted in the identification of several gene regulatory networks (GRNs), of which 14 exhibited dynamic differences in expression between SUN and wild-type fruit in the developmental time points and tissue types (Supplemental Fig. S1; Supplemental Table S7). GRN8 showed one of the highest significant enrichments for only one category, namely cell division (Supplemental Table S4D). This cluster included 9 cyclins and 17 cell organization genes, including 15 kinesin/microtubule motor genes (Supplemental Table S7). Of the several networks that were enriched for cell wall, GRN4 was most significantly enriched for genes in this category (16% of all genes in the network; Supplemental Tables S4D and S7). The network included genes involved in cell wall synthesis, degradation, modification, UDP GLUCOSYL and GLUCORONYL TRANSFERASES, INVERTASES, PECTATE LYASES, and PECTIN ESTERASES. Another interesting cluster was represented by GRN10 and GRN14, showing similar dynamics in differential expression (Supplemental Fig. S1). Combined, they represented genes, such as three PINFORMED1-like (PIN1-like), involved in auxin transport and enrichment for genes encoding transport-related proteins, cell wall, hormone metabolism, several Leu-rich repeat receptor-like kinases, and transcription factors putatively involved in patterning. Additional GRNs showed genes enriched in the secondary metabolism category and included those involved in the isoprenoid, polypropanoid, and lignin pathways (GRN2; Supplemental Table S4D). GRN3 had the most genes related to calcium, particularly those involved in its transport and signaling, and included a member of the IQD family, SlSUN25. GRN9 was enriched for genes involved in lipid, hormone, and secondary metabolism. In this cluster, four were predicted to act in the brassinosteroid pathway, and four other genes were predicted to act in the steroid metabolism pathway. Several genes involved in isoprenoid and phenylproponoid pathways were found in this network as well.
The log2 fold (SUN/the wild type) profiles of the total set of 3,154 differentially expressed genes were plotted in the principal components analysis (PCA) space (Fig. 3A). The highlighting of selected GRNs showed that they were tightly codifferentially regulated in SUN fruit in distinct programs (Fig. 3B). The most intriguing codifferentially regulated cluster was GRN8 enriched for cell division-related genes (Fig. 3E). This cluster showed decreased expression in SUN fruit in 7-DPA pericarp and increased expression in 7-DPA columella. At 10 DPA, the genes in this cluster were not differentially expressed between SUN and wild-type fruit, coinciding with when the shape controlled by SUN became finalized. GRN2, enriched for secondary metabolism, showed reduced expression in SUN fruit in the pericarp at 4 DPA and increased expression in SUN fruit in seed and columella at 7 DPA (Fig. 3C). The opposite regulation was found in GRN10 and GRN14 enriched for cell wall/RNA and hormone metabolism/development/transport, respectively, showing increased expression in SUN fruit in pericarp at 4 DPA, decreased expression in SUN fruit in seed at 7 DPA, and increased expression in SUN fruit in 4-DPA columella (Fig. 3G). GRN9, enriched for lipid and hormone metabolism, showed increased expression in SUN fruit at 7 DPA in seed and columella and at 10 DPA in seed and pericarp (Fig. 3F).
Figure 3.

GRN expression dynamics in developing fruit. A, PCA of all differentially expressed genes using the log2 SUN/wild-type expression over all tissue types and time points sampled. B, Selected GRNs in PCA space. C to G, Log2 SUN/the wild type for selected GRN per tissue type and time point. C, GRN2. D, GRN3. E, GRN8. F, GRN9. G, GRN10/GRN14. col.4, Columella 4 DPA; col.7, columella 7 DPA; col.10, columella 10 DPA; PC1, principal component 1; PC2, principal component 2; per.4, pericarp 4 DPA; per.7, pericarp 7 DPA; per.10, pericarp 10 DPA; se.7, seed 7 DPA; se.10, seed 10 DPA; WT, wild type.
To investigate in more detail the level of coexpression among selected genes, we focused on those that were known from other studies to participate in shared pathways or those that were involved in patterning. For brassinosteroid biosynthesis, eight tomato putative orthologs were higher expressed in SUN compared with the wild type in seed and columella at 7 DPA and seed, columella, and pericarp at 10 DPA (Supplemental Fig. S2A). The putative ortholog of the auxin biosynthesis gene YUCCA4, auxin influx gene AUX1, and auxin efflux gene PIN1 were higher expressed in 4-DPA columella and pericarp, and the putative auxin conjugation gene JASMONATE RESISTANT1 showed an opposite expression pattern, similar to findings from other systems (Supplemental Fig. S2B). Calcium-related genes were correlated with SlSUN25 and higher expressed in 4- and 7-DPA columella tissue in SUN fruit while being lower expressed in 7-DPA pericarp (Supplemental Fig. S2C). GROWTH-REGULATING FACTOR1 (GRF1), GRF-INTERACTING FACTOR1 (GIF1), and 12 cyclin genes were also higher expressed in 4-DPA pericarp and 7-DPA columella tissues in SUN fruit (Supplemental Fig. S2D).
Among the patterning genes, the Arabidopsis genes PIN1 and KANADI2 are involved in regulating organ polarity and coregulated with one another (Izhaki and Bowman, 2007). In SUN compared with wild-type tomato fruit, the putative orthologs of PIN1 and KANADI2 showed similar differential expression dynamics, with a strong increased expression in 4-DPA columella tissue (Supplemental Fig. S3A). JACKDAW and MAGPIE control SHORTROOT (SHR) activity in Arabidopsis roots (Welch et al., 2007). The putative tomato orthologs showed a similar expression dynamic as in Arabidopsis: when JACKDAW and MAGPIE were not differentially expressed, SHR was differentially expressed in fruit tissues (Supplemental Fig. S3B). The putative orthologs of the transcription factors REPLUMLESS (RPL), AGAMOUS (AG), SHOOT MERISTEMLESS (STM), and PHABULOSA (PHB) showed coregulated expression in SUN fruit (Supplemental Fig. S3C). RPL and APETALA2 (AP2) showed an opposite differential expression pattern in tomato fruit, where RPL was most differentially expressed at 4 and 7 DPA in the columella (Supplemental Fig. S3D). RPL and AG were expected to be coregulated, because they are both repressed by AP2 (Drews et al., 1991).
Promoter Analysis of Codifferentially Expressed Genes
To gain insights into the molecular mechanism that drives the regulation of the differentially expressed genes found in the same network, we evaluated the promoter sequences in a subset of the GRNs. The selected GRNs were enriched for genes related to the cell cycle (GRN8), RNA/cell wall (GRN10), and lipid/hormones (GRN9), and one network featured genes involved in calcium signaling and transport (GRN3). GRN10 contained several transcription factors representing those putatively involved in patterning, such as AINTEGUMENTA-LIKE5, STM, KANADI2, and PHB, as well as three PIN1-like genes and AUX1 involved in auxin transport. Commonly used motif-finding programs, such as MEME, Cosmo, and Wordseeker, resulted in few putative cis-elements, and most of them resembled microsatellite sequences (Blackwood et al., 2013). Therefore, we uniquely designed and implemented an exhaustive search and frequency-based analysis for 6-mers in 1 kb of promoter using Python scripts run on Hadoop (Hadoop, 2013). For each network and 6-mer, the occurrence of the motif was counted in the actual and shuffled promoters, yielding a promoter enrichment score (Supplemental Table S8). For the cell cycle network, 13 putative 6-mers were recognized based on the enrichment score of three or higher. These motifs were then clustered manually into four consensus motifs (Table I). For the RNA/cell wall network, two 6-mers were identified, which clustered into one consensus motif. For the lipid and hormone network, 18 6-mers were identified, resulting in six consensus motifs. In the calcium signaling and transport network, the five 6-mers identified were clustered into three consensus motifs (Table I). Some of the identified motifs were unknown, whereas others had been described previously to bind MYELOBLASTOSIS (MYB) or TEOSINTE BRANCHED1, CYCLOIDEA, and Proliferating Cell Nuclear Antigen gene-controlling element binding factor (TCP) family transcription factors, involve cell cycle regulation, or represent putative abscisic acid (ABA) response elements (Iwasaki et al., 1995; Abe et al., 1997; Ito et al., 1998; Ito, 2000; Schommer et al., 2008, 2014; Zhang et al., 2010; Supplemental Table S8).
Table I. Promoter enrichment scores for motifs found in four GRNs.
K corresponds to G or T, R corresponds to A or G, Y corresponds to C or T, S corresponds to C or G, and W corresponds to A or T.
| Data Set | GRN8 Motifs |
GRN3 Motifs |
GRN9 Motifs |
GRN10 Motif: TGGACCA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TRRCCGT | GCCCAA | CCAACGGYYA | CSGTYA | AGGTSATG | GGATGA | TTGSAGT | CAGAGGTG | CAYRTGG | ACACGGAG | CCACWC | WGKCTC | ACCTACC | ||
| GRN8 cell cycle cluster | 6.7 | 4.7 | 4.4 | 3.7 | 1.1 | 2.3 | 1.3 | 2 | 1.4 | 0.7 | 2.3 | 2.2 | 0.9 | 2.1 |
| GRN3 calcium signaling/transport cluster | 0 | 0 | 0.2 | 0.2 | 3.4 | 3.3 | 3.4 | 0 | 1 | 0 | 2 | 0 | 0.8 | 0 |
| GRN9 lipid/hormone cluster | 1.2 | 2 | 0.9 | 1.2 | 1.9 | 1.4 | 1.7 | 6.5 | 5.4 | 5 | 5 | 4.4 | 4.5 | 3 |
| GRN10 RNA/cell wall cluster | 0.5 | 2 | 0.8 | 0.6 | 1.1 | 1.8 | 1.4 | 2.3 | 2.6 | 0.2 | 2.7 | 1.3 | 1.1 | 3.8 |
To test the validity of the computationally identified 6-mers, the enrichment score for each was evaluated in the promoters of the genes in the other three networks. For the cell cycle cluster, all four consensus elements were occurring at much higher enrichment scores in the cell cycle than in any of the other networks (Table I). In fact, two of its consensus motifs, TRRCCGT and CCACGGYYA, were 22- to 5-fold higher enriched in the cell cycle network than in any of the others. However, the single-consensus motif found in the RNA/cell wall network, TGGACCA, was highest in its own network but only slightly lower in the lipid/hormone network. This result implied that the genes in the RNA/cell wall and lipid/hormone networks may be coregulated through the same putative 6-mer. Indeed, the RNA/cell wall GRN was represented by several genes involved in auxin transport. All six consensus elements from the lipid/hormone network showed higher enrichments scores compared with the other three networks (a 25- to 3-fold-higher representation). Finally, the three consensus motifs from the calcium network were 3- to 2-fold higher enriched in this compared with the other three networks.
The genes with promoters that carried the cell cycle network consensus motif (CCAACGGYYA expected to recognize MYB transcription factors or the mitotic-specific activator sequences, the RNA/cell wall consensus motif TGGACCA possibly recognized by TCP transcription factors, the lipid/hormone consensus motif CAYRTGG expected to be an ABA response element, and the unknown calcium/transport consensus motif AGGTSATG) were further investigated (Fig. 4). The expression profiles of the genes carrying these motifs in their promoters clustered according to genotype (SUN or the wild type), clearly showing that these genes were codifferentially expressed. To validate these results, we analyzed a random set of differentially expressed genes, which did not cluster based on genotype (Fig. 4, C and D). Thus, SUN expression is associated with extensive reprogramming of genes in the developing fruit that is coinciding with the changes in fruit growth patterns.
Figure 4.

PCA of the expression profile of genes with detected motifs in the promoter region. A, Fifty-eight genes in the cell cycle cluster with the CCAACGGYYA consensus motif (GRN8). B, Twenty-four genes in the RNA and cell wall cluster with the TGGACCA consensus motif (GRN10). C, A group of 45 random genes. D, Thirty-one genes in the lipid and hormone cluster with the CAYRTGG consensus motif (GRN9). E, Twenty-eight genes in the calcium signaling and transport cluster with the AGGTSATG consensus motif (GRN3). F, A second set of 45 random genes. PC1, principal component 1; PC2, principal component 2; WT, wild type.
SUN Modulates Metabolite Networks to Form Unique Associations with the Tricarboxylic Acid Cycle and Abolishes Associations with Amino Acids
To investigate whether metabolite accumulation also differed in SUN and wild-type fruit, we used the same tissues that were analyzed for gene expression analysis and evaluated the metabolite levels using gas chromatography-time-of-flight-mass spectrometry (GC-MS). Pairwise comparisons were made between SUN and wild-type fruit and placed into a global metabolic context by constructing a pathway map of significant changes using a relaxed statistical threshold (P < 0.05; Fig. 5). The pathway map of significant changes between SUN and the wild type showed that columella tissue at 7 and 10 DPA exhibited the most significant changes (Fig. 5). Globally, decreased accumulation of intermediates in the tricarboxylic acid cycle and increased accumulation in certain sugars and amino acids were observed in SUN, although these changes were less than 2-fold. Certain metabolites also showed significant changes between SUN and the wild type in seed tissue at 10 DPA in the same pattern as columella tissue at 10 DPA (Supplemental Fig. S4). However, these findings should be interpreted with caution, because pairwise comparisons may oscillate around the mean, which could lead to false-positive and -negative results, and the fold differences are relatively low.
Figure 5.

Metabolite changes in wild-type and SUN tomato fruit. Pairwise comparisons of metabolite accumulation between SUN and wild-type fruit in the pericarp tissue 7 and 10 DPA and septum tissue 4, 7, and 10 DPA. Highlighted boxes reflect a P < 0.05 using Student’s t test. The color legend indicates fold change of SUN/wild-type fruit. Positive values indicate values higher in SUN fruit. Negative values indicate values higher in wild-type fruit. GABA, γ-aminobutyric acid; PEP, phosphoenolpyruvate; 3PGA, 3-phosphoglyceric acid.
We next evaluated correlations of the metabolites with one another in SUN and wild-type fruit. The relative accumulation for each primary metabolite that was evaluated in SUN and wild-type tissues showed that many did not differ dramatically, except certain sugars and organic acids. (Supplemental Fig. S5). Suc levels were lower in SUN fruit in 4-, 7-, and 10-DPA columella. In contrast, Glc-6-P was higher in 10-DPA columella in SUN fruit. Another sugar, trehalose, was higher in 10-DPA pericarp in SUN fruit. The organic acids, fumaric acid and succinic acid, were lower in 7- and 10-DPA columella, whereas fumaric acid was lower in 10-DPA pericarp. Pairwise correlations of each metabolite were conducted separately. The analysis suggested positive and negative correlations between metabolites that were only found in SUN (Supplemental Fig. S6; Supplemental Table S9). Conversely, positive and negative correlations were also found only in wild-type fruit (Supplemental Fig. S7; Supplemental Table S10). We subsequently extracted the unique and abolished metabolite connections and created subnetworks of those metabolites exhibiting 10 or more correlations (Supplemental Fig. S8). These hubs mirror the results of the pairwise comparisons, suggesting that these metabolites seemed to be affected in SUN fruit, at least in the later stages of fruit development that were assayed.
Profiling of Hormone Levels in SUN and Wild-Type Fruit
Hormone levels were profiled in SUN and wild-type fruit in 4-DPA columella/seed and pericarp tissue and 10-DPA columella, seed, and pericarp tissue (Supplemental Table S11). Eleven hormones were profiled, including auxin (IAA); GA3 (GA1 and GA4); brassinolide (BL); the cytokinins (CKs) transzeatin (tZ), dhihydrozeatin (DHZ), and isopentenyl adenine (iP); jasmonic acid (JA) and its activated conjugate JA-Ile (JA-Ile); salicylic acid (SA); and ABA. We identified those hormonal networks that were found only in SUN or the wild type (Supplemental Fig. S9, A and B). Two unique subnetworks emerged in SUN fruit (Supplemental Fig. S9, A and C). One was a network with ABA as the hub and positive correlations with IAA and the CKs iP, DHZ, and tZ. iP and tZ also were highly correlated in SUN compared with the wild type (Pearson’s correlation coefficient [PCC] > 0.98). Additionally, GA4 and JA-Ile had formed a unique subnetwork in SUN fruit. One network with DHZ as the hub was only found in the wild type and therefore, abolished in SUN fruit (Supplemental Fig. S9, B and D). Negative correlations between IAA and GA4 and between IAA and DHZ were only found in the wild type as well as negative correlations between DHZ and GA1.
Hierarchical clustering of hormone correlations showed three distinct groups in wild-type fruits: GA; the defense hormones JA, JA-Ile, SA, and ABA; and IAA, BL, and the CK (Supplemental Fig. S9D). In SUN compared with the wild type, the grouping broke down as ABA became correlated with IAA, iP, and tZ, and JA-Ile became correlated with GA (Supplemental Fig. S9C). However, overall hormone levels did not change dramatically in these experiments, and therefore, the results should be interpreted with caution.
DISCUSSION
Tomato Fruit Initiation and Associated Transcription Profiles
By conducting transcriptome analyses of separate tissues at an early stage of fruit development, we identified coregulated gene networks that differed in SUN and wild-type fruit. Using different approaches, a cluster of genes involved in cell cycle regulation and cell division (cell division cluster in Fig. 2A; GRN8 in Supplemental Fig. S1) was found to differ most dramatically during fruit development while also being differentially expressed in SUN and wild-type fruit. Genes in GRN8 showed the highest coregulation, the most striking genotype-specific regulation of all of the coregulated gene clusters, and the highest enrichment score of cluster-specific promoter motifs identified in this study. For example, six of eight cyclin genes in the cluster contain the promoter consensus motif CCAACGGYYA, including CYCLIN B1;2 (CYCB1;2; slyc01g009040, solyc10g080950, and solyc10g078330), CYCB2;4 (solyc02g082820 and solyc04g082430), and FIZZY-RELATED3 (solyc06g043150). Thus, this consensus motif represents a potential target for regulation of cell division patterns. The data show that the cell cycle is one of the key processes that is most dynamically altered by SUN, which is consistent with its effect on increasing cell number on the proximal-distal axis while decreasing cell number on the mediolateral axis (Wu et al., 2011). Prior studies into gene expression regulated by SUN were less informative, because the RNA was isolated from whole organs as opposed to separate tissue types (Xiao et al., 2009). This shows that expression analyses of specific tissues are more insightful than expression analysis of whole organs.
A second cluster of genes, encoding those involved in cell wall-related processes, is dynamically and differentially expressed in developing fruit tissues and highly enriched based on GO terms (cell wall cluster in Fig. 2B; GRN4, GRN10, and others in Supplemental Fig. S1; Supplemental Table S4). The shift in expression of the cell wall-related genes as early as 4 d after fruit initiation implied that high expression of SUN leads to changes in cell wall properties that may precede the differential expression of genes related to cell division. The cell wall, its composition, and its structure are known to vary during tissue growth (Mirabet et al., 2011; Hernández-Hernández et al., 2014). Two models explain organ growth that results in divergent shapes: (1) stress-based axiality, whereby genes that alter patterning lead to cellular stresses influencing the cytoskeleton, which then drive the changes in the orientation of growth; and (2) polarity-based axiality, whereby genes that affect the distribution of signaling molecules define the polarity within the tissue (Uyttewaal et al., 2010; Kennaway et al., 2011). In both cases, mechanical constraints within the tissue caused by the properties of cell walls together with stress and/or polarity-based drivers are thought to result in the final morphology of plant organs. Thus, it may be expected that changes in expression of cell wall-related genes are associated with changes in organ shapes.
Another group of differentially expressed genes was putative orthologs of patterning genes. The role of these genes during growth of plant organs after their initiation is largely unknown. Reactivation of meristematic cells in otherwise differentiated zones has been proposed for the formation of ovules and other tissues in the developing ovary (Girin et al., 2009). Whether this also applies to the reactivation of cell division in fruit tissues after pollination and fertilization is unclear. GRN10 was moderately enriched for RNA regulation (Supplemental Table S4D). Because there is no organ patterning category using the Mapman bin ontology, we annotated the set of tomato gene models using the Trinotate pipeline (http://trinotate.sourceforge.net/) and associated GO terms with gene models (data not shown). Using these Trinotate GO terms, GRN10 was enriched for regulation of meristem structural organization, polarity specification of the adaxial-abaxial axis, radial pattern formation, and asymmetric cell division. Overall, our data showed that putative organ patterning genes were strongly differentially regulated in SUN fruit. This might be directly causative to the change in fruit shape by modulating the expression of cell cycle-related genes. Thus, in addition to cell division and cell wall, SUN expression also leads to dramatic shifts in the expression of putative patterning-related genes.
The transcriptome data were also analyzed using linear factorial modeling approaches (van Leeuwen et al., 2007). Several GO term categories that were found in the GRNs were also identified using linear modeling. Importantly, the linear factorial modeling identified approximately 80% of the genes found in GRN10 (cell wall and RNA regulation) and GRN14 (most significantly enriched for development), validating the genes identified in the clusters (Supplemental Table S6). These two GRNs were specifically higher expressed in SUN fruit in 4-DPA pericarp and 4-DPA columella, indicating significant genotype × time point interactions. However, the factorial approach did not identify GRN8, the most dramatically altered cell division cluster (Supplemental Table S4). Because our experimental setup was not representative of a full factorial (e.g. 4-DPA columella and seed/placenta were combined), we were not able to evaluate the interaction of genotype × time point × tissue type for all of the tissues collected. Therefore, genes that are affected by SUN at only one level, which is the case for GRN8, would not be identified using the linear model approach. Instead, to correct for testing errors in pairwise comparisons, we conducted a multiple testing correction on eight comparisons on top of the multiple testing correction within each comparison and only selected the genes that passed both. With the understanding that few genes in the GRN clusters still represent false positives, the clustering based on the pairwise comparisons gave us excellent insights into GO term pathways that were most clearly affected by SUN in a dynamic interaction of genotype, time point, and tissue type.
Comparison between Metabolite and Transcriptome Profiling in Developing Tomato Fruit
Investigations into metabolite accumulation in SUN fruit showed that some tricarboxylic acid cycle metabolites were lower in columella tissues. To link these results with the transcriptome data, we found that GRN5 was enriched for genes encoding tricarboxylic acid cycle enzymes and included malate dehydrogenase, citrate synthase, and citrate lyase. In tomato, inhibition of 2-oxoglutarate dehydrogenase results in decreased respiration and increased accumulation of oxoglutarate and succinic acid but no difference in fruit size or biomass (Araújo et al., 2012). In contrast, inhibition of succinate dehydrogenase leads to increased photosynthate assimilation and fruit biomass (Araújo et al., 2011). Our data suggest that these changes in TCA intermediates are a product of differential flux through the pathway, possibly as an effect of increased cell division in the columella, resulting in altered fruit shape (Wu et al., 2011). Other links between metabolites and their biosynthesis pathways were not evident, suggesting that the pairwise comparisons were not sufficiently robust or that the overlap in pathways identified from gene expression and metabolites is generally low.
Genes, Metabolites, and Hormones Interact to Regulate Elongated Fruit Shape
To evaluate the combined effect of SUN on gene expression, metabolite, and hormone accumulation, we constructed a gene-metabolite-hormone interactome map (Fig. 6). Genes and metabolites were grouped into pathways to show how SUN-affected pathways interact during early fruit development. The size of each node represented how many edges were attributed to the pathway (i.e. how many significant correlations each pathway exhibited with other pathways; PCC > 0.90 and < −0.90). The most represented pathways were calcium regulation/transport, lipid metabolism, cell cycle and division, amino acid metabolism, auxin biosynthesis/transport/signaling, brassinosteroid metabolism, and ethylene biosynthesis/signaling. A zoomed-in view at the network (Fig. 6B) revealed that processes involved in calcium regulation/transport form the main hub of connections with other pathways that are involved in regulating elongated fruit shape that resulted from high expression of SUN. The calcium pathway was positively correlated with patterning, cell cycle/division, GRF-GIF gene expression, auxin regulation/transport, and the hormones IAA and BL. Tricarboxylic acid cycle genes as well as tricarboxylic acid cycle metabolites were negatively correlated with the gene pathways controlling cell division/patterning as well as auxin.
Figure 6.

Gene, metabolite, and hormone interactome in SUN fruit. The size of the nodes represents the number of edges to that node. Blue edges represent significant positive correlations. Red edges represent significant negative correlations. Nodes represent genes, metabolites, or hormones that participate in the pathway indicated. Squares represent genes. Diamonds represent metabolites. Hormones are represented as circles. Cell pattern indicates transcription factors that regulate organ polarity and patterning. IQDs are members of the SUN family. Calcium indicates genes involved in calcium transport, CaM binding, and proteins requiring calcium for function. Lipid indicates genes involved in lipid biosynthesis and regulation. A, Full interactome network with bundled edges. B, Subnetwork of calcium hub. BR, Genes involved in brassinosteroid homeostasis; celldiv, genes involved in cell division; GRF GIF, genes in the GRF family and GIF; TCA, enzymes that participate in the tricarboxylic acid cycle; aa, amino acid; MAX4, more axillary branching; met, metabolite.
CONCLUSION
It is likely that SUN sets up the pattern for differential fruit growth before anthesis, because ovary shape is slightly different at the time that the flower opens (Wu et al., 2011; van der Knaap et al., 2014). Thus, the fruit tissues used in this study represent later growth stages, when shape changes mediated by SUN are being executed. Inferring from the data generated in this study, we propose that the manner by which SUN regulates fruit shape is by impacting a calcium signaling cascade through its CaM domain-interacting motif to set up the pattern of organ growth. Based on the interaction of an SUN family member AtIQD1 (Bürstenbinder et al., 2013) with a kinesin motor protein, SUN may be involved in transport of cargo throughout the cell (van der Knaap et al., 2014), possibly of cell wall components. This then leads to dramatic shifts in the spatiotemporal gene expression of many cell wall, cell division, and patterning genes at the early stages of fruit development. Although plant phenotypes associated with high SUN expression suggest a role for auxin in regulating fruit shape and plant morphology (Xiao et al., 2008; Wu et al., 2011), there may be no direct role for any of the known plant hormones in the regulation of fruit shape mediated by SUN.
MATERIALS AND METHODS
Plant Materials
Nearly isogenic lines were constructed by repeated backcrosses of recombinants harboring approximately 70 kb of introgression of the sun locus from Solanum pimpinellifolium LA1589 (the wild type for the sun locus). Sun1642 harboring the mutation at the sun locus (SUN) was used as the recurrent parent, and backcrosses were monitored using background markers. Thus, in principle, these lines only differ for the allele of the tomato (Solanum lycopersicum) SUN gene (Wu et al., 2011). The nearly isogenic lines (the wild type; SUN) were grown in the greenhouse in 2-gallon pots and 16 h of daylight. Flowers at anthesis were tagged and self-pollinated on successive days. Pollination of flowers was staggered, such that fruit of all developmental stages would be harvested on the same day. Fruits at 4, 7, and 10 DPA were harvested on ice and brought to the laboratory for additional dissection. Columella, seed, and pericarp tissues were separated using a razor blade, immediately frozen in liquid nitrogen, and stored at −80°C. Four days post anthesis columella represents a mixture of columella, seed, and placenta tissues. Four days post anthesis pericarp represents a mixture of pericarp and exocarp tissue. Seven-- and 10-DPA pericarp samples were peeled and lack exocarp tissues. The seed tissues include the developing gel tissue surrounding the seed (placenta). Four harvest dates were carried out on 5 d (February 25–27, March 6, and March 7 in 2009) for a total of 359 samples. A subset of 116 samples was sent for metabolite and hormone analyses, representing a minimum of eight biological replicates per tissue type per genotype for 10 DPA and four biological replicates per tissue type per genotype for 7 and 4 DPA. The remaining samples were used for RNA extraction (see below). We collected two additional replicates for RNA extraction in the summer of 2012. These tissues were collected as described above, except that they were pooled for each replicate for each genotype, tissue type, and time point, and aliquots were used for RNA extraction. The two replicates were collected over 2 successive d.
For the metabolite and hormone analyses, samples were freeze dried and shipped to the RIKEN Center for Sustainable Resource Science Institute. They were divided for the hormone and metabolite using 9 to 450 mg dry weight for the hormone (Growth Regulation Research Group) and 2.0 to 2.7 mg dry weight for the metabolite (Metabolomics Research Group) analyses. Metabolite extraction procedures (2.5 mg dry weight mL−1 extraction solution) and identification/annotation of metabolites using GC-MS were done as described previously (Schauer et al., 2005; Redestig et al., 2009). Hormone analyses were conducted as described previously (Yamamoto et al., 2007; Katsumata et al., 2011).
RNA Isolation, RNA-Seq Library Preparation, and Sequencing
Frozen tissues were ground in liquid nitrogen, and total RNA was extracted using the Trizol (Invitrogen Inc.) method as described by the manufacturer. RNA quantity and quality were assessed using a Qubit 2.0 Fluorometer RNA Assay Kit (Invitrogen Inc.) and an Agilent 2100 Bioanalyzer RNA 6000 Nano Kit (Agilent) housed at the Molecular and Cellular Imaging Center at Ohio State University, Wooster. Strand-specific RNA-seq libraries with insert size of approximately 250 bp were prepared using the protocol described previously (Zhong et al., 2011) using 10 μg of total RNA. Eight libraries with compatible barcodes were pooled and run on a single lane on a flow cell of the Illumina HiSeq2000 at the Genomics Resources Core Facility at Weill Cornell Medical College. Single-end reads of 51 bp were generated.
Alignment and Analysis of Illumina Reads
After sequencing, Illumina reads were quality checked, demultiplexed, and trimmed. The reads were aligned to a known ribosomal RNA database using Bowtie (Langmead and Salzberg, 2012) and allowing for two mismatches. The filtered reads with the ribosomal reads removed were aligned with TopHat2 (Kim et al., 2013) against the tomato genome annotation version 2.4, allowing for maximum intron lengths of 5,000 bp, segment lengths of 22 bp, and only one mismatch per segment. All other parameters were set to default. Aligned sequences were separated into sense and antisense and counted using an in-house Python script. Of four replicates collected from 16 samples, all but one showed a quality score of above 95%, with an average of 14.4 million reads per library that mapped to the genome (Supplemental Table S1). The correlations between replicates were, in general, very high, particularly the samples that were collected from wild-type fruit (Supplemental Table S2). Reads per kilobase of exon model per million mapped reads (RPKM) were calculated by the formula RPKMij = (109 × C)/(N × L), where C is the number of reads mapped to gene i in sample j, N is the total number of reads mapped (taken as the sum of reads mapped to annotated genes in sample j), and L is the number of 1,000 bp of exon in the gene model of gene i. The reads are archived at the Short Read Archive at the National Center for Biotechnology Information under accession number SRA065144.
Differential Gene Expression Analysis
Analysis of differentially expressed genes between SUN and wild-type fruit was conducted using the DESeq package (Anders et al., 2013). Raw counts data were normalized by library size and fit to a negative binomial model. We used the fit-only option of the SharingMode function using the method of estimating the dispersions. Briefly, we assumed that the variation in dispersions from the predicted fit line is caused by the sampling variance and not caused by the true endemic variant expression of the gene. P values were adjusted using the Benjamini-Hochberg correction for multiple testing. All four biological replicates were used in the analysis, and differentially expressed genes with an adjusted P < 0.05 were considered significant.
Differential Expression Analysis Using Linear Factorial Modeling
Using DESeq2 and the function nbinomLRT(), we tested three separate null hypotheses. Null hypothesis 1 tested whether each gene was significantly affected by genotype. Null hypothesis 2 tested whether each gene was affected by the interaction of genotype by time point. Because including time points and tissue types together in the model results in a model that is not full rank, for null hypothesis 2, we only tested genotype, time point, and the genotype × time-point interaction. Null hypothesis 3 tested whether each gene was affected by the interaction of genotype by tissue type.
Clustering of the Expression of Genes during Fruit Development Using Fuzzy C Means
The expression data set was filtered by excluding genes that were low expressed. This was done by calculating the RPKM values for all genes as described above and filtering using the criterion of at least four replicates being expressed above 2 RPKM. This filtering assures that, in at least one tissue type, time point, and genotype, we are confident that the gene is expressed. The filtering for low expression resulted in a reduced data set to 19,963 genes. Gene expression values expressed in RPKM were mean centered and converted to their Z score, which was calculated by dividing each mean-centered value by the sd of all of the values for each gene. This calculation can be illustrated by the following formula:
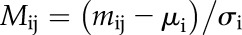 |
where Mij is the normalized value for gene i in sample j, mij is the nonnormalized value of gene i in sample j, µi is the mean of gene i in all samples, and σi is the sd of the gene i in all samples.
Expression profiles for wild-type fruit were clustered using fuzzy C means by using the Mfuzz package (Futschik and Carlisle, 2005) in R, with a C value of 100 to maximize dynamic differential clustering identification and core clustered at 0.70 membership probability. Dynamically regulated gene clusters were identified by inspecting plots of the normalized expression profiles of each cluster, which led to the identification of 10 clusters that showed a dynamic range of expression in one tissue over another as well as six ontogeny and spatially specific clusters. Based on genes found predominantly in each cluster and hypergeometric enrichment of Mapman ontology bins, the clusters were named accordingly: cell division, cell wall, RNA, secondary metabolism, transport/hormone, transport/signaling, photosynthesis, and protein.
Expression Networks of Differentially Expressed Genes Using Fold-Change SUN/Wild Type
For the network analysis, we filtered the data set further by only including those that showed significant differential expression in at least one pairwise comparison using DESeq. All genes that were significantly differentially expressed (Benjamini-Hochberg-adjusted P < 0.05) were chosen (total of 5,623). An additional multiple testing correction using Bonferroni was performed to account for eight pairwise comparisons. The final list consisted of 3,154 nonredundant genes, of which 1,455 were differentially expressed in more than one comparison. Any gene that was filtered out in a particular time point and tissue type because of low expression was set to 0 for that condition. Log-transformed fold changes of SUN relative to the wild type for each gene were estimated using DESeq in R (Anders et al., 2013). Briefly, the means for each gene at a particular time point, tissue type, and genotype were estimated based on all four biological replicates, and the estimated dispersions were based on the sample dispersions. These estimated means were used to calculate the fold-change SUN/the wild type. A log2 fold of 0 means no difference, a log2 fold of one means 2-fold or higher, a log2 fold of two means 4-fold or higher, and so on. Fold change for each sample was used as differential expression profiles to include the information of differences in expression in SUN fruit in the same analysis. These profiles were clustered using fuzzy C means, with a C of 60 using the Mfuzz package in R (Futschik and Carlisle, 2005). We determined that a C of 60 properly incorporated all patterns in the data without forming spurious or random clustering. The results showed patterns that changed slightly but were still unique (differences below 1 log2). The 14 clusters that showed the most dynamic response (i.e. clusters with log2 values above 1 and below −1) were chosen for additional analysis (Supplemental Fig. S1) and grouped together based on the median profile of their members.
Hypergeometric Enrichment Test of Gene Clusters
Enrichment of Mapman ontology bin codes represented in each gene cluster was done using a hypergeometric test using the webserver GeneProf (http://www.geneprof.org/GeneProf/tools/hypergeometric.jsp) and phyper() in R. P values were adjusted for multiple testing by Benjamini-Hochberg correction in R (false discovery rate [FDR] < 5%).
Promoter Motif Searches
We selected four sets of genes from the identified GRNs above: GRN3 (calcium signaling/transport), GRN8 (cell cycle), GRN9 (lipid/hormone), and GRN10 (RNA/cell wall). Clusters were named based on hypergeometric enrichment tests and observations of putative orthologs present in each cluster. For example, GRN10 is enriched for RNA regulation and contains a group of transcription factors that regulate patterns of cell proliferation. GRN clusters were further filtered for outliers by visually inspecting the log2 SUN/wild-type profile. Filtering resulted in 5% reduction for GRN3 (54 remaining), 27.6% reduction for GRN8 (118 remaining), 27.8% reduction for GRN9 (57 remaining), and 25% reduction for GRN10 (133 remaining).
To build the promoter region data sets, we wrote a Python script, which using functionality from the Biopython package (Cock et al., 2009), accessed the tomato genome and extracted 1,000 bases upstream of each gene. For each of four data sets, our script generated a text file with the corresponding promoter regions. These files were further used for identifying overrepresented motifs as described below.
For each of four promoter data sets, an exhaustive search of all potential 5- to 8-bp motifs was designed and implemented (Blackwood et al., 2013). This computationally demanding step was conducted in Hadoop (Hadoop, 2013), a platform for big data analysis that supports distributed processing of large data sets. We wrote Python scripts, including mapper and reducer scripts for Hadoop (Hadoop, 2013), which we ran on the Cloudera Hadoop (www.cloudera.com) on Oracle VM VirtualBox for Mac OS. For each of four data sets, these scripts computed the frequency of each possible motif of lengths 5 to 8. To investigate whether the motifs were occurring with similar frequencies in the randomly rearranged promoter sequences, the promoter regions were shuffled 100 times, and the averaged frequencies were computed from the shuffled promoters. The ratio of the frequency in the actual promoter region over the shuffled one was returned as the enrichment score. An enrichment score of one indicates that the corresponding motif is found at the same rate in the original and the shuffled promoters. Therefore, the larger the enrichment score, the more likely the corresponding motif is a true motif and not caused by chance. Furthermore, only putative motifs with an enrichment score of three or larger were recorded. This threshold ensured that only motifs occurring at least three times more frequently in the original promoter region were selected for the next step of the analysis. In the next step, unlikely motifs, such as microsatellite (e.g. ACACAC) or homopolymer runs (e.g. AAAAAA), were removed from the set of results. For the reasons explained below, the results obtained for motifs of lengths 5, 7, and 8 were not as good as the ones for 6-base-long motifs. Many of the 5-base-long motifs were found with similar frequencies in both the original and the shuffled promoter regions; in addition, many 5-base-long motifs with high enrichment scores were repeats. Few motifs of lengths 7 and 8 were identified, and these motifs were combinations of two or more motifs of length 6. Therefore, to include all potential motifs, for each of four data sets, we continued the analysis with the results of the 6-base-long motifs. In the next stage of our analysis, promoter motifs of length 6 that shared a common core element (e.g. 4 of 6 bases were conserved) were grouped together into a consensus motif.
To further evaluate the likelihood of a consensus motif being enriched in one cluster over another, the enrichment scores (ratio of actual to shuffled frequency) were computed for each consensus motif in all four data sets (Blackwood et al., 2013). More precisely, for each consensus motif s found in a particular data set Di (i = 1,…, 4), we first computed its relative frequency in that data set by using Equation 1, where s denotes a consensus motif, |s| denotes its number of (motif) components, c denotes a (motif) component, and 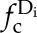 denotes the relative frequency of a component c in the data set
denotes the relative frequency of a component c in the data set 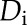 :
:
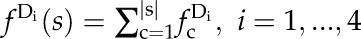 |
(1) |
Similarly, the frequency of a consensus motif s in the shuffled promoter region is computed by Equation 2:
 |
(2) |
Finally, the enrichment score for a consensus motif s is calculated as the ratio of the two frequencies computed above:
 |
(3) |
An enrichment score close to one indicates that the corresponding motif is equally represented in the original and the shuffled promoter regions and therefore, not likely to be a true motif for a given data set. An enrichment score of two or larger implies that the corresponding consensus motif is found more frequently in the original than in the shuffled data set, and therefore, such motifs are selected for additional analysis.
Investigation of Gene Expression of Those Containing CCAACGGYYA, CAYRTGG, TGGACCA, and AGGTSATG Consensus Motifs
Expression profiles from genes containing the motifs were extracted from SUN and wild-type fruit separately, and PCA was carried out using the prcomp() function in R. Loadings for the principal components were extracted for SUN and wild-type expression profiles and plotted using ggplot() in R (Supplemental Table S12). Gene sets included 58 for the CCAACGGYA motif-containing set, 31 for the CAYRTGG motif-containing set, 24 for the TGGACCA motif-containing set, and 28 for the AGGTSATG motif-containing set.
Metabolite Pairwise Comparisons
To compare levels of each metabolite in tissue types and time points, we performed pairwise Student's t tests between SUN and wild-type fruit. A relaxed significance threshold was used of P < 0.10 to highlight patterns of change in SUN fruit. Average fold change of SUN/the wild type was used to construct a primary metabolite pathway heat map that was constructed based on the work by Do et al. (2010) using pairwise comparisons with P < 0.10.
Metabolite and Hormone Data Analysis
To compare metabolite and hormone accumulation while taking into account large differences in magnitude and variance, the values for each were first mean centered by subtracting the mean value for a metabolite across all samples from the value for each sample. The data were further normalized for unequal variance by converting it into its Z score as described above.
Metabolite and Hormone Correlation Networks Comparison between Genotypes
To compare metabolite and hormone networks between wild-type and SUN fruit, the values were reclustered separately, and networks for only SUN were inferred. Basically, clustering was first constructed with fruit samples collected from SUN plants. These networks were then evaluated in wild-type fruit for significant correlations. Unique correlations in SUN fruit (adjusted P < 0.05) that were not significant in wild-type fruit were considered as unique associations in SUN. Correlations that were significant in wild-type fruit (adjusted P < 0.05) and not significant in SUN fruit were considered abolished associations in SUN fruit. All P values were adjusted for multiple testing by Benjamini-Hochberg correction (FDR < 5%).
Interactome Network Analysis of Genes, Metabolites, and Hormones
Hormone data were not available for 7 DPA, and therefore, the genes-metabolite-hormone interactome network was generated using 4- and 10-DPA data (five conditions). Hormone values were normalized to their Z score as described above. Normalized values from hormone and metabolite accumulation and gene expression were used as described previously only using SUN fruit. Because fewer conditions were used to calculate correlations, a higher correlation (<−0.90 and >0.90) was determined to be significant after multiple testing correction (adjusted P < 0.05). To assess how different pathways were interacting, genes were grouped into putative pathways based on closest Arabidopsis (Arabidopsis thaliana) ortholog and tomato genome annotation ITAG2.4 (www.solgenomics.net), metabolites were grouped into biochemical categories, and hormones were taken as hormone class. These identifiers were then used as the gene, metabolite, or hormone name when calculating correlations. When constructing the network, nodes represented a pathway, metabolite class, or hormone. The number of edges connecting the nodes was used to define the size of each node. The size of the nodes is indicative of the predominance of each pathway, metabolite category, or hormone in its interactions in SUN fruit and how each pathway is interacting with each other.
Visual Network Creation
Networks were constructed using Cytoscape software (Cline et al., 2007). Nodes are genes, metabolites, and hormones, and edges are the significant PCCs between the nodes. The layout of the nodes is based on the strength of the edge, and the proximity of two nodes is based on the strength of the correlation between the nodes.
Graphics and Statistical Analysis
PCCs were calculated using R and corrected for 5% FDR using the Benjamini-Hochberg correction for multiple testing. All PCA plots and jigger box plots were created in R using the ggplot2() package.
Sequence data from this article can be found in the Short Read Archive at the National Center for Biotechnology Information under accession number SRA065144.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. The log2 fold dynamics of 14 GRNs in eight fruit tissues and developmental time points.
Supplemental Figure S2. Codifferential expression of genes in plant growth pathways.
Supplemental Figure S3. Codifferential expression of the putative orthologs of patterning genes.
Supplemental Figure S4. Significant differences in metabolite accumulation in seed tissue at 10 DPA.
Supplemental Figure S5. Relative metabolite accumulation in wild-type and SUN fruit tissues.
Supplemental Figure S6. Unique metabolite network of associations in SUN fruit.
Supplemental Figure S7. Metabolite network of correlations in wild-type fruit.
Supplemental Figure S8. Subnetworks of metabolite correlations in wild-type fruit.
Supplemental Figure S9. Correlations of hormone levels in SUN and wild-type fruit.
Supplemental Table S1. Summary statistics for each of the RNA-seq libraries.
Supplemental Table S2. Correlations among samples in fractions.
Supplemental Table S3. Genes in eight coregulated clusters in wild-type fruit.
Supplemental Table S4. Hypergeometric GO term enrichments.
Supplemental Table S5. RPKM values and pairwise differential expression of all genes.
Supplemental Table S6. Genes found in the linear factorial modeling analyses.
Supplemental Table S7. Membership of genes in 14 GRNs.
Supplemental Table S8. Promoter motifs enriched in GRN3, GRN8, GRN9, and GRN10.
Supplemental Table S9. Unique metabolite correlations in SUN fruit.
Supplemental Table S10. Abolished metabolite correlations in SUN fruit.
Supplemental Table S11. Hormone levels in the different fruit tissues.
Supplemental Table S12. Loadings for all PCA analyses.
Supplementary Material
Acknowledgments
We thank Meghan Fisher and Jenny Moyseenko for greenhouse assistance and plant care and Yumiko Takebayashi and Makoto Kobayshi for technical support of hormone and GC-MS analysis, respectively.
Glossary
- CaM
calmodulin
- GC-MS
gas chromatography-time-of-flight-mass spectrometry
- GO
gene ontology
- PCA
principal components analysis
- PCC
Pearson’s correlation coefficient
- RPKM
reads per kilobase of exon model per million mapped reads
Footnotes
This work was supported by Ohio State University (Presidential Fellowship to J.P.C.) and the National Science Foundation (grant no. IOS 0922661 to S.V. and E.v.d.K.).
Articles can be viewed without a subscription.
References
- Abe H, Yamaguchi-Shinozaki K, Urao T, Iwasaki T, Hosokawa D, Shinozaki K (1997) Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 9: 1859–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel S, Bürstenbinder K, Müller J (2013) The emerging function of IQD proteins as scaffolds in cellular signaling and trafficking. Plant Signal Behav 8: e24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel S, Savchenko T, Levy M (2005) Genome-wide comparative analysis of the IQD gene families in Arabidopsis thaliana and Oryza sativa. BMC Evol Biol 5: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD (2013) Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786 [DOI] [PubMed] [Google Scholar]
- Araújo WL, Nunes-Nesi A, Osorio S, Usadel B, Fuentes D, Nagy R, Balbo I, Lehmann M, Studart-Witkowski C, Tohge T, et al. (2011) Antisense inhibition of the iron-sulphur subunit of succinate dehydrogenase enhances photosynthesis and growth in tomato via an organic acid-mediated effect on stomatal aperture. Plant Cell 23: 600–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araújo WL, Tohge T, Osorio S, Lohse M, Balbo I, Krahnert I, Sienkiewicz-Porzucek A, Usadel B, Nunes-Nesi A, Fernie AR (2012) Antisense inhibition of the 2-oxoglutarate dehydrogenase complex in tomato demonstrates its importance for plant respiration and during leaf senescence and fruit maturation. Plant Cell 24: 2328–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanzá V, Navarrete M, Trigueros M, Ferrándiz C (2006) Patterning the female side of Arabidopsis: the importance of hormones. J Exp Bot 57: 3457–3469 [DOI] [PubMed] [Google Scholar]
- Blackwood M, VanHorn H, Visa S, Van der Knaap E (2013) Using Hadoop to find transcription factor binding sites in the Solanum lycopersicum genome. In Conference for Undergraduate Research in Computer Science and Mathematics. Ohio Wesleyan, Delaware, OH: http://math.owu.edu/MCURCSM/papers/paper11.pdf (May 27, 2015) [Google Scholar]
- Bouchard R, Bailly A, Blakeslee JJ, Oehring SC, Vincenzetti V, Lee OR, Paponov I, Palme K, Mancuso S, Murphy AS, et al. (2006) Immunophilin-like TWISTED DWARF1 modulates auxin efflux activities of Arabidopsis P-glycoproteins. J Biol Chem 281: 30603–30612 [DOI] [PubMed] [Google Scholar]
- Bürstenbinder K, Savchenko T, Müller J, Adamson AW, Stamm G, Kwong R, Zipp BJ, Dinesh DC, Abel S (2013) Arabidopsis calmodulin-binding protein IQ67-domain 1 localizes to microtubules and interacts with kinesin light chain-related protein-1. J Biol Chem 288: 1871–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25: 1422–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong M, Wolters-Arts M, Feron R, Mariani C, Vriezen WH (2009) The Solanum lycopersicum auxin response factor 7 (SlARF7) regulates auxin signaling during tomato fruit set and development. Plant J 57: 160–170 [DOI] [PubMed] [Google Scholar]
- de Jong M, Wolters-Arts M, García-Martínez JL, Mariani C, Vriezen WH (2011) The Solanum lycopersicum AUXIN RESPONSE FACTOR 7 (SlARF7) mediates cross-talk between auxin and gibberellin signalling during tomato fruit set and development. J Exp Bot 62: 617–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinneny JR, Weigel D, Yanofsky MF (2005) A genetic framework for fruit patterning in Arabidopsis thaliana. Development 132: 4687–4696 [DOI] [PubMed] [Google Scholar]
- Do PT, Prudent M, Sulpice R, Causse M, Fernie AR (2010) The influence of fruit load on the tomato pericarp metabolome in a Solanum chmielewskii introgression line population. Plant Physiol 154: 1128–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews GN, Bowman JL, Meyerowitz EM (1991) Negative regulation of the Arabidopsis homeotic gene AGAMOUS by the APETALA2 product. Cell 65: 991–1002 [DOI] [PubMed] [Google Scholar]
- Futschik ME, Carlisle B (2005) Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol 3: 965–988 [DOI] [PubMed] [Google Scholar]
- Ge L, Peng J, Berbel A, Madueño F, Chen R (2014) Regulation of compound leaf development by PHANTASTICA in Medicago truncatula. Plant Physiol 164: 216–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillaspy G, Ben-David H, Gruissem W (1993) Fruits: a developmental perspective. Plant Cell 5: 1439–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girin T, Sorefan K, Ostergaard L (2009) Meristematic sculpting in fruit development. J Exp Bot 60: 1493–1502 [DOI] [PubMed] [Google Scholar]
- Glawischnig E, Hansen BG, Olsen CE, Halkier BA (2004) Camalexin is synthesized from indole-3-acetaldoxime, a key branching point between primary and secondary metabolism in Arabidopsis. Proc Natl Acad Sci USA 101: 8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet C, Mageroy MH, Lam NB, Floystad A, Tieman DM, Klee HJ (2012) Role of an esterase in flavor volatile variation within the tomato clade. Proc Natl Acad Sci USA 109: 19009–19014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadoop (2013) Hadoop. http://hadoop.apache.org/ (May 27, 2015)
- Hernández-Hernández V, Rueda D, Caballero L, Alvarez-Buylla ER, Benítez M (2014) Mechanical forces as information: an integrated approach to plant and animal development. Front Plant Sci 5: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Van Houten J, Gonzalez G, Xiao H, van der Knaap E (2013) Genome-wide identification, phylogeny and expression analysis of SUN, OFP and YABBY gene family in tomato. Mol Genet Genomics 288: 111–129 [DOI] [PubMed] [Google Scholar]
- Ito M. (2000) Factors controlling cyclin B expression. Plant Mol Biol 43: 677–690 [DOI] [PubMed] [Google Scholar]
- Ito M, Iwase M, Kodama H, Lavisse P, Komamine A, Nishihama R, Machida Y, Watanabe A (1998) A novel cis-acting element in promoters of plant B-type cyclin genes activates M phase-specific transcription. Plant Cell 10: 331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki T, Yamaguchi-Shinozaki K, Shinozaki K (1995) Identification of a cis-regulatory region of a gene in Arabidopsis thaliana whose induction by dehydration is mediated by abscisic acid and requires protein synthesis. Mol Gen Genet 247: 391–398 [DOI] [PubMed] [Google Scholar]
- Izhaki A, Bowman JL (2007) KANADI and class III HD-Zip gene families regulate embryo patterning and modulate auxin flow during embryogenesis in Arabidopsis. Plant Cell 19: 495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Gao D, Xiao H, van der Knaap E (2009) Genome organization of the tomato sun locus and characterization of the unusual retrotransposon Rider. Plant J 60: 181–193 [DOI] [PubMed] [Google Scholar]
- Katsumata T, Fukazawa J, Magome H, Jikumaru Y, Kamiya Y, Natsume M, Kawaide H, Yamaguchi S (2011) Involvement of the CYP78A subfamily of cytochrome P450 monooxygenases in protonema growth and gametophore formation in the moss Physcomitrella patens. Biosci Biotechnol Biochem 75: 331–336 [DOI] [PubMed] [Google Scholar]
- Kennaway R, Coen E, Green A, Bangham A (2011) Generation of diverse biological forms through combinatorial interactions between tissue polarity and growth. PLoS Comput Biol 7: e1002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MC, Chung WS, Yun DJ, Cho MJ (2009) Calcium and calmodulin-mediated regulation of gene expression in plants. Mol Plant 2: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Wang Q, Kaspi R, Parrella MP, Abel S (2005) Arabidopsis IQD1, a novel calmodulin-binding nuclear protein, stimulates glucosinolate accumulation and plant defense. Plant J 43: 79–96 [DOI] [PubMed] [Google Scholar]
- Matas AJ, Yeats TH, Buda GJ, Zheng Y, Chatterjee S, Tohge T, Ponnala L, Adato A, Aharoni A, Stark R, et al. (2011) Tissue- and cell-type specific transcriptome profiling of expanding tomato fruit provides insights into metabolic and regulatory specialization and cuticle formation. Plant Cell 23: 3893–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucato A, Willems D, Bernini R, Picarella ME, Santangelo E, Ruiu F, Tilesi F, Soressi GP (2013) Novel phenotypes related to the breeding of purple-fruited tomatoes and effect of peel extracts on human cancer cell proliferation. Plant Physiol Biochem 72: 125–133 [DOI] [PubMed] [Google Scholar]
- Mirabet V, Das P, Boudaoud A, Hamant O (2011) The role of mechanical forces in plant morphogenesis. Annu Rev Plant Biol 62: 365–385 [DOI] [PubMed] [Google Scholar]
- Molesini B, Pandolfini T, Rotino GL, Dani V, Spena A (2009) Aucsia gene silencing causes parthenocarpic fruit development in tomato. Plant Physiol 149: 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya T, Nomura T, Yokota T, Farrar K, Harrison K, Jones JD, Kaneta T, Kamiya Y, Szekeres M, Bishop GJ (2005) Patterns of dwarf expression and brassinosteroid accumulation in tomato reveal the importance of brassinosteroid synthesis during fruit development. Plant J 42: 262–269 [DOI] [PubMed] [Google Scholar]
- Nadakuduti SS, Pollard M, Kosma DK, Allen C Jr, Ohlrogge JB, Barry CS (2012) Pleiotropic phenotypes of the sticky peel mutant provide new insight into the role of CUTIN DEFICIENT2 in epidermal cell function in tomato. Plant Physiol 159: 945–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nole-Wilson S, Azhakanandam S, Franks RG (2010) Polar auxin transport together with aintegumenta and revoluta coordinate early Arabidopsis gynoecium development. Dev Biol 346: 181–195 [DOI] [PubMed] [Google Scholar]
- Osorio S, Vallarino JG, Szecowka M, Ufaz S, Tzin V, Angelovici R, Galili G, Fernie AR (2013) Alteration of the interconversion of pyruvate and malate in the plastid or cytosol of ripening tomato fruit invokes diverse consequences on sugar but similar effects on cellular organic acid, metabolism, and transitory starch accumulation. Plant Physiol 161: 628–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozga JA, Yu J, Reinecke DM (2003) Pollination-, development-, and auxin-specific regulation of gibberellin 3β-hydroxylase gene expression in pea fruit and seeds. Plant Physiol 131: 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bradley G, Pyke K, Ball G, Lu C, Fray R, Marshall A, Jayasuta S, Baxter C, van Wijk R, et al. (2013) Network inference analysis identifies an APRR2-Like gene linked to pigment accumulation in tomato and pepper fruits. Plant Physiol 161: 1476–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redestig H, Fukushima A, Stenlund H, Moritz T, Arita M, Saito K, Kusano M (2009) Compensation for systematic cross-contribution improves normalization of mass spectrometry based metabolomics data. Anal Chem 81: 7974–7980 [DOI] [PubMed] [Google Scholar]
- Ruan YL, Patrick JW, Bouzayen M, Osorio S, Fernie AR (2012) Molecular regulation of seed and fruit set. Trends Plant Sci 17: 656–665 [DOI] [PubMed] [Google Scholar]
- Sawchuk MG, Edgar A, Scarpella E (2013) Patterning of leaf vein networks by convergent auxin transport pathways. PLoS Genet 9: e1003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer N, Steinhauser D, Strelkov S, Schomburg D, Allison G, Moritz T, Lundgren K, Roessner-Tunali U, Forbes MG, Willmitzer L, et al. (2005) GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett 579: 1332–1337 [DOI] [PubMed] [Google Scholar]
- Schommer C, Debernardi JM, Bresso EG, Rodriguez RE, Palatnik JF (2014) Repression of cell proliferation by miR319-regulated TCP4. Mol Plant 7: 1533–1544 [DOI] [PubMed] [Google Scholar]
- Schommer C, Palatnik JF, Aggarwal P, Chételat A, Cubas P, Farmer EE, Nath U, Weigel D (2008) Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol 6: e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståldal V, Sundberg E (2009) The role of auxin in style development and apical-basal patterning of the Arabidopsis thaliana gynoecium. Plant Signal Behav 4: 83–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara S, Hishiyama S, Jikumaru Y, Hanada A, Nishimura T, Koshiba T, Zhao Y, Kamiya Y, Kasahara H (2009) Biochemical analyses of indole-3-acetaldoxime-dependent auxin biosynthesis in Arabidopsis. Proc Natl Acad Sci USA 106: 5430–5435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyttewaal M, Traas J, Hamant O (2010) Integrating physical stress, growth, and development. Curr Opin Plant Biol 13: 46–52 [DOI] [PubMed] [Google Scholar]
- van der Knaap E, Chakrabarti M, Chu YH, Clevenger JP, Illa-Berenguer E, Huang Z, Keyhaninejad N, Mu Q, Sun L, Wang Y, et al. (2014) What lies beyond the eye: the molecular mechanisms regulating tomato fruit weight and shape. Front Plant Sci 5: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap E, Tanksley SD (2001) Identification and characterization of a novel locus controlling early fruit development in tomato. Theor Appl Genet 103: 353–358 [Google Scholar]
- van Leeuwen H, Kliebenstein DJ, West MA, Kim K, van Poecke R, Katagiri F, Michelmore RW, Doerge RW, St Clair DA (2007) Natural variation among Arabidopsis thaliana accessions for transcriptome response to exogenous salicylic acid. Plant Cell 19: 2099–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrebalov J, Pan IL, Arroyo AJ, McQuinn R, Chung M, Poole M, Rose J, Seymour G, Grandillo S, Giovannoni J, et al. (2009) Fleshy fruit expansion and ripening are regulated by the Tomato SHATTERPROOF gene TAGL1. Plant Cell 21: 3041–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu B, Wang H, Li J, Huang H, Xu L (2011) YUCCA genes are expressed in response to leaf adaxial-abaxial juxtaposition and are required for leaf margin development. Plant Physiol 157: 1805–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch D, Hassan H, Blilou I, Immink R, Heidstra R, Scheres B (2007) Arabidopsis JACKDAW and MAGPIE zinc finger proteins delimit asymmetric cell division and stabilize tissue boundaries by restricting SHORT-ROOT action. Genes Dev 21: 2196–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won C, Shen X, Mashiguchi K, Zheng Z, Dai X, Cheng Y, Kasahara H, Kamiya Y, Chory J, Zhao Y (2011) Conversion of tryptophan to indole-3-acetic acid by TRYPTOPHAN AMINOTRANSFERASES OF ARABIDOPSIS and YUCCAs in Arabidopsis. Proc Natl Acad Sci USA 108: 18518–18523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Xiao H, Cabrera A, Meulia T, van der Knaap E (2011) SUN regulates vegetative and reproductive organ shape by changing cell division patterns. Plant Physiol 157: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Jiang N, Schaffner E, Stockinger EJ, van der Knaap E (2008) A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319: 1527–1530 [DOI] [PubMed] [Google Scholar]
- Xiao H, Radovich C, Welty N, Hsu J, Li D, Meulia T, van der Knaap E (2009) Integration of tomato reproductive developmental landmarks and expression profiles, and the effect of SUN on fruit shape. BMC Plant Biol 9: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Ichida H, Matsui M, Obokata J, Sakurai T, Satou M, Seki M, Shinozaki K, Abe T (2007) Identification of plant promoter constituents by analysis of local distribution of short sequences. BMC Genomics 8: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liang W, Yang X, Luo X, Jiang N, Ma H, Zhang D (2010) Carbon starved anther encodes a MYB domain protein that regulates sugar partitioning required for rice pollen development. Plant Cell 22: 672–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. (2010) Auxin biosynthesis and its role in plant development. Annu Rev Plant Biol 61: 49–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Vrebalov J, McQuinn R, Gapper N, Liu B, Xiang J, et al. (2013) Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol 31: 154–159 [DOI] [PubMed] [Google Scholar]
- Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, Xiang JZ, Fei Z, Giovannoni JJ (2011) High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc 2011: 940–949 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.