

Abstract
Granulocyte Colony Stimulating Factor (G-CSF) and Granulocyte/Macrophage Colony Stimulating Factor (GM-CSF) are used widely to promote the production of granulocytes or antigen presenting cells (APC). The Food and Drug Administration approved G-CSF (filgrastim) for the treatment of congenital and acquired neutropenias and for mobilization of peripheral hematopoietic progenitor cells for stem cell transplantation. A polyethylene glycol modified (PEGylated) form of G-CSF is approved for the treatment of neutropenias. Clinically significant neutropenia, rendering an individual immunocompromised, occurs when their number is less than 1500/µl. Current guidelines recommend their use when the risk of febrile neutropenia is greater than 20%. GM-CSF (sargramostim) is approved for neutropenia associated with stem cell transplantation. Because of its promotion of APC function, GM-CSF is being evaluated as an immunostimulatory adjuvant in a number of clinical trials. More than 20 million persons have benefited worldwide, and more than $5 billion sales occur annually in the United States.
Introduction
Few physician-scientists have made as great an impact on our understanding of hematology or improved the lives of patients, estimated more than 20 million(1), with blood and cancer disorders as Don Metcalf, who died in December 2014. Laboring at the Walter and Eliza Hall Institute in Melbourne throughout his fifty-year career, Metcalf used semisolid medium and cell culture supernatants to discover hematopoietic progenitor cells (e.g., granulocyte/macrophage colonies) and their growth factors (e.g., granulocyte colony-stimulating factor, G-CSF, and granulocyte/macrophage colony stimulating factor, GM-CSF). Increased purification of these and related growth factors, sometimes from hundreds of mice injected with endotoxin, led to the molecular characterization and cloning of G-CSF, GM-CSF, macrophage colony stimulating factor (M-CSF), stem cell factor, and interleukin-3 (IL-3) in the 1980s(2).
Hematopoiesis is a highly proliferative (~1010 cells/day), dynamic process driven by multiple hematopoietic growth factors/cytokines (Figure 1A). The hematopoietic growth factors are multi-functional: critical for proliferation, survival, and differentiation of hematopoietic stem, progenitor, and precursor cells to a terminally differentiated, functional cell type. Colony forming assays identified the ability of first crude supernatants, then highly purified cytokines to drive muti-lineage and single lineage differentiation. After co-culturing for 7–14 days, colonies from mononuclear cells obtained from the mouse spleen or bone marrow were measured in semisolid medium. Based on characteristics of cells within a single colony, the lineage(s) governed by the cytokine was determined. Granulocytes comprise the majority of white blood cells in human circulation and play an integral role in innate and adaptive immunity. In granulopoiesis, their production is mediated by a number of different growth factors, especially G-CSF and GM-CSF (3, 4). Due to asymmetric division some daughter cells of the hematopoietic stem cell (HSC) remain as HSC, preventing the depletion of the stem cell pool(5). Multiparameter immunophenotyping has transformed our ability to identify different cell types in hematopoiesis. Murine HSC are characterized as lin−sca1+c−kit+, and human HSC display CD34+ in the absence of lineage markers. The differentiation pathway from HSC to granulocytes is dependent on G-CSF and, less so, GM-CSF. The HSC gives rise to a common myeloid progenitor (CMP) and common lymphoid progenitor (CLP) cell(6). The CMP cells differentiate into myeloblasts, erythrocytes, and megakaryocytes via at least two intermediates, the Granulocyte/Monocyte Progenitor cell and the Erythrocyte/Megakaryocyte Progenitor cell. In the granulocytic series, myeloblasts (15–20 µm) are the first recognizable cells by their scant cytoplasm, absence of granules, and fine nucleus with nucleoli in the bone marrow clearly committed to differentiation to granulocytes. Myeloblasts differentiate into promyelocytes, which are larger (20 µm) and begin to possess granules (Figure 1B). Promyelocytes give rise to neutrophilic, basophilic, and eosinophilic precursor cells. Cell division continues through the promyelocyte stage. Fine specific granules containing inflammatory-related proteins appear during myelocyte maturation. For neutrophils, their size an nuclei become increasingly more condensed as the cells mature through myelocyte, metamyelocyte, band, and the terminally differentiated neutrophil (polymorphonuclear and ~15 µm). During episodes of stress such as infection, band cells can be found in the peripheral blood and are used as a measure of inflammation. The above process is a complex and dynamic process orchestrated by multiple cytokines and their receptors, most notably G-CSF and GM-CSF.
Figure 1.

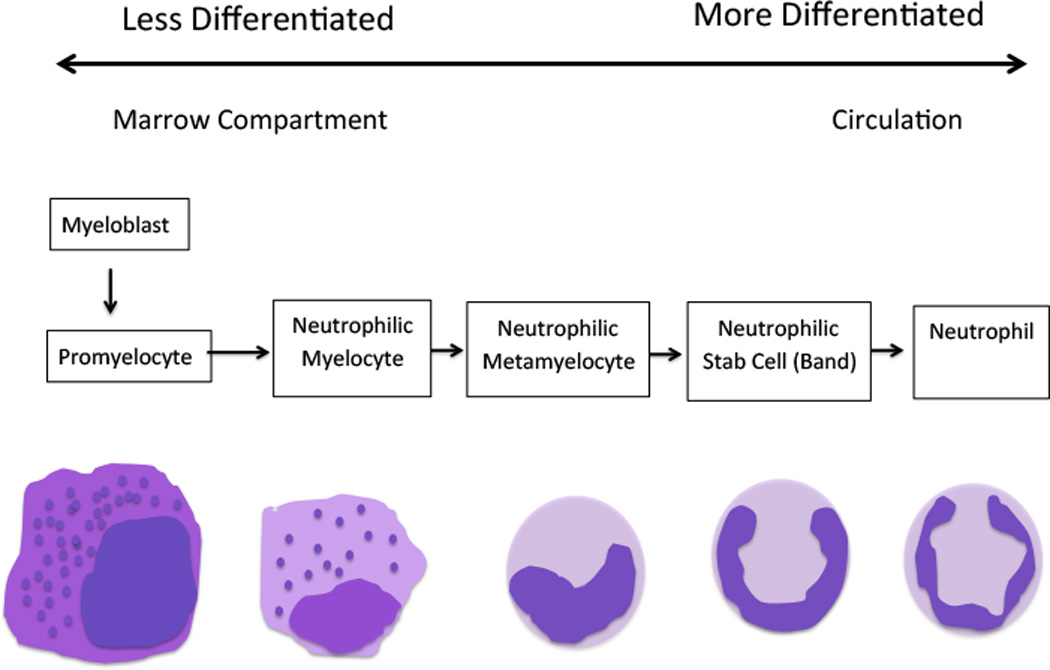
A. The scheme of hematopoiesis from the multipotential hematopoietic stem cell to fully differentiated cell types. Principal cytokines that determine differentiation patterns in red. Epo, Erythropoietin; FLT-3 ligand, FMS-like tyrosine kinase 3 ligand; G-CSF, Granulocyte-colony stimulating factor; GM-CSF, Granulocyte Macrophage-colony stimulating factor; IL, Interleukin; M-CSF, Macrophage-colony stimulating factor; SCF, Stem Cell Factor; SDF-1, Stromal cell-derived factor-1; TGFβ, Transforming growth factor beta; TNFα, Tumour necrosis factor-alpha; Tpo, Thrombopoietin; B. The stages of granulopoiesis from myeloblast to the mature granulocyte. During neutrophil maturation, which is driven primarily by G-CSF, granulocytic cells change shape, acquire primary and specific granules, and undergo nuclear condensation.
Following antigen stimulation or activation by cytokines such as IL-1, IL-6, and TNFα, macrophages, T cells, endothelial cells, and fibroblasts produce and secrete G-CSF and GM-CSF. Of unknown significance, a variety of tumor cells also produce these paracrine growth factors. Glycoproteins with a molecular weight of ~ 23 kDa, G-CSF and GM-CSF are now produced through recombinant technology in either E. coli or yeast. G-CSF induces the appearance of colonies containing only granulocytes, while GM-CSF gave colonies containing both granulocytes and macrophages. Generation of G-CSF (genomic nomenclature: Csf3) and G-CSF Receptor (Csf3r) knockout mice confirmed that G-CSF critically drives granulopoiesis(7). The cognate receptor for G-CSF is a single transmembrane receptor that homodimerizes upon G-CSF binding. Unlike G-CSF, GM-CSF functions via a two receptor system involving a specific α-chain and a common β-chain shared by IL-3 and IL-5(8). GM-CSF knockout mice however did not display a perturbation in hematopoiesis(9, 10). Both G-CSF and GM-CSF signal through pathways involving JAK/STAT, SRC family kinases, PI3K/AKT, and Ras/ERK1/2. The receptor complexes are characterized by high-affinity (apparent Kd ~ 100–500 pM) and low density (50–1000 copies/cell). Interestingly, human G-CSF is functionally active on murine myeloid cells, but human GM-CSF is not. The signaling specificity likely involves nuances in the proximal post-receptor phosphoprotein networks and the distal gene regulatory networks. The molecular pathways and their cross-interactions in determining lineage specificity are critical to development of more specific therapies.
Cloning of human GM-CSF and its expression in bacterial and eukaryotic cells was achieved in 1985 at Genetics Institute (11), and, a year later, G-CSF and its expression in E. coli at Amgen(12). Commercialized by these biotechnology start-ups, G-CSF and GM-CSF revolutionized the treatment of patients with congenital or acquired neutropenias and those undergoing stem cell transplantation. Sidelined from the treatment of neutropenias by its toxicity profile, GM-CSF is now undergoing a renaissance as an immunomodulatory agent.
G-CSF is approved by the United States Food and Drug Administration (FDA) for use to decrease the incidence of infection in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a significant incidence of severe neutropenia with fever; reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of patients with (AML) leukemia; reduce the duration of neutropenia and febrile neutropenia in patients with non-myeloid malignancies undergoing myeloablative chemotherapy followed by stem cell transplantation; mobilize hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis; and reduce the incidence and duration of complications of severe neutropenia in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia. Forms of G-CSF available worldwide include filgrastim, pegfilgrastim, and lenograstim.
GM-CSF is currently approved by the FDA to accelerate myeloid recovery in patients with non-Hodgkin’s lymphoma, acute lymphoblastic leukemia, and Hodgkin’s disease undergoing autologous stem cell transplantation; following induction chemotherapy in older adult patients with AML to shorten time to neutrophil recovery and reduce the incidence of life-threatening infections; to accelerate myeloid recovery in patients undergoing allogeneic stem cell transplantation from HLA- matched related donors; for patients who have undergone allogeneic or autologous stem cell transplantation in whom engraftment is delayed or failed; and to mobilize hematopoietic progenitor cells into peripheral blood for collection by leukapheresis. Forms of GM-CSF available worldwide are sargramostim and molgramostim.
The recommended dosage of G-CSF is 5 mcg/kg/day, and for GM-CSF, 250 mcg/m2/day. Both drugs may be given subcutaneously or intravenously, although randomized clinical trials demonstrate greater efficacy (i.e., decreased duration of neutropenia) without a difference in toxicity for the subcutaneous route(13). For chemotherapy-induced neutropenia, G-CSF is administered until there is >1000 neutrophils/µl. For congenital neutropenias, the goal is to maintain neutrophil counts ~ 750/µl. G-CSF is well tolerated. Transient fever and bone pain are more commonly observed in those receiving GM-CSF. Pleural and/or pericardial effusions can also occur in those receiving GM-CSF. Long-term side effects, such as osteopenia, of G-CSF administration are being monitored in patients with severe congenital neutropenia (SCN). One concern is that G-CSF may accelerate the transformation of SCN to myelodysplastic syndromes (MDS) or AML, associated with acquired mutations in the G-CSF Receptor.
G-CSF and GM-CSF signaling pathways and functional consequences
The receptors for both GM-CSF and G-CSF belong to the hematopoietin/cytokine receptor superfamily. The G-CSF Receptor (G-CSFR) acts as a homodimer, whereas the GM-CSF Receptor is a heterodimer with a shared β chain with the IL-3 Receptor and IL-5 Receptor complexes. The G-CSFR is expressed primarily on neutrophils and bone marrow precursor cells, which undergo proliferation and eventually differentiation into mature granulocytes. G-CSF binds to G-CSFR, resulting in its dimerization, with a stoichiometry of 2:2 and with a high affinity (KD = 500 pM)(14, 15). Among the activated downstream signal transduction pathways are Janus kinase (JAK)/signal transducer and activator of transcription (STAT), Src kinases such as Lyn, Ras/Extracellular Regulated Kinase (ERK), and phosphatidylinositol 3-kinase (PI3K)(16). The cytoplasmic domain of G-CSFR possesses four tyrosine residues (Y704, Y729, Y744, Y764), serving as phospho-acceptor sites (17, 18). Src homology 2 (SH2) containing proteins STAT5 and STAT 3 bind to Y704 and Gab2 to Y764..Grb2 couples to both Gab2 and to SOS, permitting signaling diversification, such as Ras/ERK, PI 3-kinase/Akt, and Shp2 (19, 20). An alternatively spliced isoform of G-CSFR elicits activation of a JAK-SHP2 pathway(15). The precise physiological roles of protein kinases and their downstream events in G-CSF-induced signaling remain unclear, although some clues are beginning to emerge (21, 22).
GM-CSF binds to the α chain of the GM-CSFR with a low affinity (KD = 0.2 – 100 nM), but a higher affinity (KD = 100 pM) occurs in the presence of both subunits. GM-CSF signaling involves formation of dodecameric supercomplex that is required for JAK activation (23). In addition to JAK/STAT pathway, GM-CSF also activates the ERK1/2, PI3K/Akt and IκB/NFκB pathways. Although the α-chain is considered primarily as ligand recognition units, it interacts with Lyn to activate JAK independent Akt activation of the survival pathway (24). Thus, differences in receptor expression patterns and known and unknown nuances in signaling pathway circuits account for the functional differences between G-CSF and GM-CSF.
G-CSF and GM-CSF are pleiotropic growth factors, with overlapping functions. GM-CSF also shares properties with and macrophage colony stimulating factor (M-CSF) on monocyte function. (25) Both GM-CSF and G-CSF increase chemotaxis and migration of neutrophils, but response kinetics may differ. GM-CSF may be considered to be more pro-inflammatory than G-CSF. As GM-CSF increases cytotoxic killing of C. albicans, surface expression of Fc- and complement-mediated cell-binding (FcγR1, CR-1 and CR-3), and adhesion receptor (ICAM-1) (14). Yet, both cytokines will promote neutrophil phagocytosis (26). More extensive reviews on G-CSF and GM-CSF function in neutrophils may be found (27, 28).
Acquired and Congenital Neutropenia
An absolute neutrophil count (ANC) less than 1,500/µl is defined as neutropenia, which is graded on the severity of decreased ANC (Table I). Causes for neutropenia may be congenital or, more commonly, acquired. Neutropenia may be asymptomatic until an infection occurs. Benign neutropenia exists, and the individuals are not at risk for serious infection. However, onset of fever with neutropenia, termed febrile neutropenia, commonly occurs as a potentially life-threatening complication of chemotherapy and involves considerable cost due to treatment with intravenous antibiotics and prolonged hospitalization. In addition, febrile neutropenia prevents continuation of chemotherapy until there is recovery from neutropenia. According to the Norton-Simon hypothesis(29), the efficacy of chemotherapy would be reduced if stopped midway. A pause in treatment allows recovery of the cancer cells and facilitates the emergence of chemoresistant clones(29–31). Neutropenia also occurs secondary to bone marrow infiltration with leukemic or myelodysplastic cells.
TABLE I.
Correlation of neutropenia with absolute neutrophil count
| Neutropenia Grade | Absolute neutrophil count |
|---|---|
| Grade 1 | ≥1.5 × 109/ml – < 2 ×109/ml |
| Grade 2 | ≥1 × 109/ml – < 1.5 ×109/ml |
| Grade 3 | ≥0.5 × 109/ml – < 1 ×109/ml |
| Grade 4 | < 0.5 ×109/ml |
Neutropenia results from a growing list of germline mutations in genes, such as ELANE, HAX1, GFI1, G6PC3, WAS, and CSF3R(32). Soon after birth, children with SCN develop a grade 4 neutropenia. SCN is a lifetime condition resulting from increased apoptosis of granulocytic progenitors in the marrow. Due to the severity and chronic nature of SCN, individuals are prone to recurrent infections, especially from the endogenous flora in the gut, mouth and skin. Most cases of SCN are due to de novo mutations. Transmission may be autosomal dominant, recessive, or X-linked. The most common mutation involves ELANE and is autosomal dominant (33, 34). Mutations in ELANE encode the neutrophil elastase (NE), a serine protease. ELANE is expressed during ganulopoiesis, maximally at the promyelocyte stage. It is hypothesized that mutations in ELANE cause neutropenia via improper folding of the protein that triggers the unfolded protein response (UPR). UPR-generated stress drives apoptosis due to an overload of unfolded protein, and an arrest in differentiation at the promyelocyte stage is observed. Fascinatingly, ELANE mutations are also associated with cyclic neutropenia. Cyclic neutropenia is characterized by granulocyte nadirs of less than 200/µl occurring every 21 days.
Patients with SCN are always at risk for life-threatening infections. Early phase 1 clinical trials held in 1989(35, 36) evaluated G-CSF therapy for SCN and cyclic neutropenia. Both trials demonstrated at least a 10-fold increase in neutrophil counts, reducing the severity of the neutropenia from grade 4 to grade 1 to normal counts. Reduction in days of cyclic neutropenia from 21 to 14 days was observed and in SCN a consistent increase in ANC was observed. In 1990 two studies explored the benefit of G-CSF versus GM-CSF in treating congenital neutropenia. Grey collie dogs with cyclic neutropenia due to mutations in the endocytosis gene AP3B1(37) were studied with three cytokines, G-CSF, GM-CSF and IL-3. GM-CSF and G-CSF showed an expansion of neutrophil counts, but only G-CSF prevented the cycling of hematopoiesis(10). Similar to the dog study, G-CSF therapy increased ANC, whereas GM-CSF therapy increased eosinophil counts, but not neutrophil counts (38). Following the beneficial effects of G-CSF in the above phase 1/2 studies, a phase 3 clinical trial was performed in 1993(39). Patients with SCN, cyclic neutropenia, and idiopathic neutropenia (n=123) were included in the study. Patients were randomly treated immediately or after a four-month observation period. Almost all of the patients (108 of 120) receiving G-CSF therapy displayed a restoration of ANC from grade 4 to normal levels. The increase in ANC resulted from increased production of neutrophils in bone marrow. Infection related incidents were reduced by ~50% (P < 0.05) and a reduction by 70% in antibiotic use.
One particular form of inherited neutropenia is the WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome(40). Myelokathexis refers to a build-up of mature neutrophils in the bone marrow. Mutations in CXCR4 result in the syndrome(41). CXCR4 and its ligand SDF-1 mediate the retention of neutrophils. G-CSF administration leads to upregulation of SDF-1 and subsequent release of neutrophils into the peripheral circulation(42). A recently published phase I study demonstrated the safety and efficacy of low-dose plerixafor, a CXCR4 antagonist(43). One widely-used indication for G-CSF to mobilize and harvest hematopoietic progenitor cells into the periphery for stem cell transplantation(44), and concomitant use of plerixafor enhances the mobilization(45).
Neutropenia associated with aplastic anemia
Severe aplastic anemia (SAA) is a disease where stem cells residing in the bone marrow are damaged leading to a deficiency in all hematopoietic cell lines. SAA has a high mortality rate, but the five-year mortality rate is reduced to less than 10% with matched sibling stem cell transplantation or 30% with immunosuppressive therapy (IST)(46). IST includes antithymocyte globulin, cyclosporine, and glucocorticoids. The addition of G-CSF to IST has been studied in a number of randomized studies and showed that G-CSF reduces the number of infectious complications and hospital days when compared to standard therapy alone. However, its addition did not affect a difference in overall survival rates(47, 48). While treatment with G-CSF or GM-CSF results in a neutrophil response, a sustained tri-lineage response was uncommon when used alone or in combination with other hematopoietic growth factors (49, 50). The response to G-CSF may have prognostic value. Patients treated with IST plus G-CSF who did not achieve a white blood cell count of at least 5,000/µl had a low probability of response and high mortality(51–53). Similarly GM-CSF has been studied as a potential adjunct to IST with similar results (48). These finding suggest G-CSF and GM-CSF may be useful adjuncts to standard IST for SAA.
Neutropenia associated with chemotherapy for solid tumors
The FDA approved in 1991 the use of recombinant human G-CSF (filgrastim) to treat cancer patients undergoing myelotoxic chemotherapy. Multiple factors affect the severity of neutropenia, most important being the type and severity of chemotherapy dosage and the underlying disease(54, 55). In 1994 the American Society of Clinical Oncology (ASCO) recommended primary prophylaxis with G-CSF or GM-CSF for expected incidence of neutropenia of ≥40% (56). The purpose of the guidelines was to reduce the incidence and length of neutropenia and thus time of hospitalization, which would reduce costs significantly. Three prospective, randomized, placebo controlled trials formed the basis of the recommendations. The first phase 3 trial tested the applicability of G-CSF as an adjunct to chemotherapy in patients treated for small cell lung cancer with cyclophosphamide, doxorubicin, and etoposide (CDE)(57). A major outcome of the study identified a significant reduction of at least one episode of febrile neutropenia occurring at 77% in placebo versus 40% in G-CSF group (P < 0.001). A reduction in median duration of grade 4 neutropenia was observed in all cycles of chemotherapy (1–day G-CSF group versus 6-days placebo group). From a cost-benefit perspective, the data translated into reduction of 50% incidence of infection, antibiotic treatment, and days of hospitalization with G-CSF treatment versus placebo. A similar study performed in Europe for small cell lung cancer also found that prophylactic G-CSF treatment reduced the incidence of febrile neutropenia (53% placebo group versus 26% G-CSF group)(58). A significant reduction in hospitalization and antibiotic treatment was observed. The study also identified a benefit of G-CSF treatment in adherence to the chemotherapy regimen. A reduction in chemotherapy dose by 15% was indicated in 61% of the placebo group versus 29% of the G-CSF group. A gap of two or more days in the chemotherapy treatment group was observed for 47% patients of the placebo group and 29% of G-CSF group. The third trial investigated G-CSF therapy in non-Hodgkin lymphoma (NHL) treated with vincristine, doxorubicin, prednisolone, etoposide, cyclophosphamide, and bleomycin (VAPEC-B)(59). Incidence of neutropenia was reduced for the G-CSF group (23%) versus placebo group (44%), with fewer delays and shorter duration of treatment in G-CSF-treated group. In comparison, GM-CSF trials provided less convincing data. In a trial for cyclophosphamide, vincristine, procarbazine, bleomycin, prednisolone, doxorubicin, and mesna (COP-BLAM) administered as therapy for NHL, use of molgramostim (GM-CSF) resulted in faster recovery from neutropenia and reduced hospitalization, but the benefit was limited to only 72% of the patients that could tolerate GM-CSF(60). Another trial with small cell lung cancer did not show any significant effect with molgramostim treatment (61).
Development of better chemotherapeutic regimens that were less myelotoxic, provided more cost effective options compared to colony stimulating factor therapy. The incidence of neutropenia in many cases was reduced to ≤ 10%. However, the advantage of colony stimulating factor therapy in both increasing the intensity and maintenance of dose were actively debated versus the cost of the growth factors. The 2000 ASCO guidelines noted the lack of colony stimulating factor therapy in improving survival benefits with newer chemotherapeutic regimens. In 2003, a large randomized study showed benefit of G-CSF therapy for a dose-dense chemotherapy (cyclophosphamide, paclitaxel, and doxorubicin) in patients with node-positive breast cancer (62). Significantly improved disease-free survival (RR 0.74, p=0.01) and overall survival (RR 0.69, p=0.013) was observed in patients receiving G-CSF. Fewer patients reported grade 4 neutropenia in G-CSF group (6%) versus non-G-CSF group (33%). In 2004, two additional studies with old (60–75) and young (<60 years) NHL patients observed a reduction of chemotherapy regimens from 3 to 2 weeks combined with an improved the rate of progressive disease and overall survival (63, 64). In 2005 two trials emerged that brought about significant support for G-CSF support and reduced the threshold for recommended CSF therapy from 40% to 20%(65, 66). The first study compared the effect of antibiotics (A) versus antibiotics + G-CSF (A + G) in small cell lung cancer patients undergoing CDE treatment. A significant reduction in incidence of febrile neutropenia was observed for A + G group (10%) versus antibiotics only group (24%) (66). The second study investigated effect of pegfilgrastim in breast cancer patients treated with docetaxel (65). Approved in 2001, pegfilgrastim was developed to improve the renal clearance rate(67) and a single dose provided similar or greater improvement in the ANC after chemotherapy compared to daily filgrastim doses (68). The randomized, placebo-controlled trial conducted with 928 patients demonstrated a lower incidence of febrile neutropenia in patients receiving pegfligrastim (1%) versus placebo (17%). Hospitalization was also reduced in pegfilgrastim group (1%) versus placebo group (14%). In 2005 and 2006, the National Comprehensive Cancer Network (http://www.nccn.org) and ASCO adopted guidelines that reduced the thresholdfrom 40% to 20% for the risk of neutropenia to be treated with growth factors as an adjuvant to chemotherapy (69). The issues of use of myeloid growth factors, their cost-effectiveness, and the duration of their use during chemotherapy remain of great interest to clinical oncologists. A randomized phase 3 study with a non-inferiority design demonstrated the efficacy of G-CSF prophylaxis against febrile neutropenia in women with breast cancer for the entirety of their myelosuppressive treatment (70). Current guidelines from American Society of Clinical Oncology, National Comprehensive Cancer Network, and the European Organisation for Research and Treatment of Cancer recommend the use of myeloid growth factors when the risk of febrile neutropenia is 20% or greater (71, 72).
Neutropenia associated with leukemia
Neutropenia in patients with leukemia results from both the underlying disease and aggressive chemotherapy. The ASCO guidelines developed in 1994, like for solid tumors, considered data obtained from three large randomized trials. Unlike the solid tumor trials, two of the three trials used GM-CSF versus G-CSF. The two GM-CSF trials reported conflicting findings, with some statistical significance in recovery of ANC, but no significant reduction in hospitalization or incidence of serious infections(73, 74). The G-CSF trial showed a recovery in ANC, reduction in days of neutropenia, and a trend towards better recovery rates. However, like the GM-CSF trials no improvement in days of hospitalization or usage of antibiotics was observed(75). Thus a beneficial response by the growth factors was not observed in case of leukemia at this time. However at the time of ASCO’s 2000 guidelines, newer placebo-controlled trials demonstrated a reduction in neutrophil recovery time from 6 days to 2 days and reduced hospitalization times in the setting of induction chemotherapy(76). The 2000 ASCO guidelines also identified a potential benefit for growth factor therapy in consolidation chemotherapy. The 2006 update did not introduce any significant changes and recommended the application of CSF therapy post-induction and consolidation therapy(69).
Unlike chemotherapy-induced neutropenia, congenital neutropenia patients experience neutropenia for life and require long-term treatment with G-CSF. Long-term effects of G-CSF therapy have become important in management of congenital neutropenia. Patients receiving G-CSF therapy for as long as eight years were evaluated for safety and efficacy(77). Neutrophil counts were maintained without exhaustion of myelopoiesis. A significant improvement in the quality of life was achieved by reduction in antibiotic treatment and hospitalization time allowing for normal growth, development, and participation in normal daily activities. The SCN international registry (SCNIR) was formed in 1994 to further assess the progress of SCN patients being treated with G-CSF. A ten year report that followed patients with SCN (n=526) being treated with G-CSF was released in 2006 (78). Consistent with previous reports, an increase with maintenance of ANC was observed in majority of the patients with an overall improvement in quality of life.
Leukemia transformation is significantly higher in SCN patients, and the SCNIR reported 21% of patients with SCN developed leukemia while being treated with G-CSF. Although leukemic transformation have been reported in SCN patients before the development of G-CSF therapy(79), the precise role of G-CSF therapy in leukemic transformation remains unknown. Almost all SCN patients undergo G-CSF therapy, and thus it is difficult to assess leukemic transformations in the absence of G-CSF treatment. However, patients who require higher doses of G-CSF are at a higher risk of developing MDS/AML(80).
Germline mutations in CSF3R, which encodes the G-CSFR, are infrequent causes for SCN, and result in refractoriness to filgrastim(81). Acquired nonsense mutations in CSF3R have been observed in ~80% of SCN patients who progressed to secondary MDS/AML (82–86). The nonsense mutations result in deletion of the C-terminus of the G-CSFR, resulting in the loss of one to all four tyrosine residues and the inability to undergo normal ligand-induced internalization and endosomal routing (87, 88). The truncated receptor mutants produce a phenotype of enhanced proliferation and impaired differentiation in response to G-CSF. Furthermore, knock-in mice harboring a similar mutation showed hyperproliferative responses to G-CSF administration and strongly prolonged activation of STAT5, implicated in increased hematopoietic progenitor stem cell expansion in vivo (89). This prediction was validated in a patient with SCN who developed secondary AML concomitant with a nonsense mutation of G-CSFR. Upon discontinuation of G-CSF and without chemotherapy, the blast count in the blood and bone marrow disappeared, although the mutation remained detectable (90). The tight correlation between the acquisition of G-CSFR mutations and progression of SCN to secondary MDS/AML and the abnormal signaling features in vitro and in vivo strongly suggested that mutated CSF3R could be a driver of myelodysplasia. Recent studies reveal that CSF3R T595I mutation is the most prevalent mutation found in chronic neutrophilic leukemia and that treatment with the Jak2 inhibitor ruxolitinib resulted in marked clinical improvement support the hypothesis that mutations in G-CSFR are indeed drivers of myeloproliferative disease (91, 92). A low frequency of CSF3R mutations also occurs in AML and chronic myelomonocytic leukemia(93).
Future Developments
More than 20 years after its registration by the FDA, biosimilars (94) of G-CSF are being developed(95). (Amgen lost its patent protection for filgrastim in 2008, after developing worldwide sales of ~$4.5 billion.) The European Medicine Agency approved six biosimiliars to G-CSF, and in February 2015, the FDA approved the first biosimilar, filgrastim-sndz, in the United States since passage of the Biologics Price Competition and Innovation Act. Teva's tbo-filgrastim had been approved in the United States in 2012 before that legislation. These drugs must demonstrate “high similarity” in the molecular characterization, purity, stability, pharmacokinetics, pharmacodynamics, clinical efficacy, tolerability, and safety as the original agent(69). Pricing analysis suggests that use of biosimilars to filgrastim, bevacizumab, trastuzumab, and rituximab may save up to $44.2 billion over the next 10 years(96).
G-CSF and/or GM-CSF may improve chemotherapy and immunotherapy of hematologic malignancies and non-blood cancers. For instance, these myeloid growth factors can recruit quiescent leukemic cells into the cell cycle for enhanced killing from cell cycle-specific chemotherapy(74, 97). As a pro-inflammatory cytokine, GM-CSF is being used to promote dendritic cell activity in a variety of anti-cancer trials. Indeed, GM-CSF is approved as part of the sipuleucel-T regimen for the treatment of hormone-resistant prostate cancer. There, dendritic cells are incubated with a fusion protein consisting of prostatic acid phosphatase and GM-CSF. While sipuleucel-T has been underused, in part due to its expense, GM-CSF is being studied in the context of other immunotherapeutic interventions (clinicaltrials.gov).
G-CSF has immunomodulatory effects on immune cells. G-CSF enhances antibody-dependent cellular cytotoxicity and cytokine production in neutrophils(98). However, it also inhibits Toll Like Receptor-induced pro-inflammatory cytokines produced by monocytes and macrophages(99). CD34+ monocytes that inhibit graft-versus-host disease are mobilized in response to G-CSF(100). In addition, G-CSF inhibits LPS-induced IL-12 production from bone-marrow derived dendritic cells cultured in vitro(101). Interestingly, administration of GM-CSF has the opposite effect, inducing cytokine production in the circulation in response to LPS(102).
GM-CSF pathways may be high-value targets in autoimmune diseases. For example, inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gastrointestinal tract caused by a combination of environmental and genetic factors. Crohn’s disease and ulcerative colitis can be difficult to treat and relapse of disease can occur at any time. Biochemical markers identifying patients at risk for relapse are currently lacking. GM-CSF signaling has recently been implicated in the pathogenesis of Crohn’s disease. GM-CSF is required for myeloid cell antimicrobial functions and homeostatic responses to tissue injury in the intestine(103). Preliminary studies have found that GM-CSF reduces chemically-induced gut injury in mice(104). In human studies, higher concentrations of circulating antibodies against GM-CSF are found in patients with active IBD as compared with those with inactive disease(103). There are currently several studies and clinical trials looking at the use of GM-CSF in the treatment of IBD and anti-GM-CSF antibody for the monitoring of disease activity and assessing risk of recurrence(105, 106).
Pulmonary alveolar proteinosis (PAP) is a rare disorder characterized by accumulation of periodic acid-schiff-positive lipoproteinaceous material in the alveoli of the lung leading to impaired gas exchange, respiratory insufficiency, and in severe cases, respiratory failure(107). Autoimmune PAP (aPAP) accounts for 90% of cases and is due to the presence of autoantibodies against GM-CSF. Hereditary PAP (hPAP) is caused by mutations in the genes CSF2RA and CSF2RB that code for the α and β subunit of the GM-CSF receptor respectively. (108, 109). In aPAP, the presence of anti-GM-CSF antibodies leads to aberrant in vivo GM-CSF signaling that is required for macrophage-mediated clearance, but not uptake, of pulmonary surfactant. This results in the progressive accumulation of foamy surfactant laden macrophages and intra-alveolar surfactant in the alveoli of the lung(110). The gold standard of therapy has been whole lung lavage. Although an effective therapy, it often needs to be repeated due to re-accumulation of lipoproteinaceous sediment and is not without complications. Newer therapies have been studied including pulmonary macrophage transplantation, plasmapheresis to remove the GM-CSF autoantibody, and inhaled GM-CSF(111–113). Inhaled GM-CSF is of particular interest as it has been shown in animal studies and phase I and II clinical trials to be safe and effective (114, 115).
Acknowledgments
Disclosures
S.J.C receives research support from the National Institutes of Health, American Society of Hematology, Leukemia and Lymphoma Society, and Cures Within Reach Foundation.
References
- 1.Hilton DJ, Nicola NA, Alexander WS, Roberts AW, Dunn AR. Donald Metcalf (1929–2014) Cell. 2015;160:361–362. doi: 10.1016/j.cell.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 2.Metcalf D. The Molecular Control of Blood Cells. Cambridge, MA: Harvard University Press; 1988. [Google Scholar]
- 3.Kawamoto H, Ikawa T, Masuda K, Wada H, Katsura Y. A map for lineage restriction of progenitors during hematopoiesis: the essence of the myeloid-based model. Immunol Rev. 2010;238:23–36. doi: 10.1111/j.1600-065X.2010.00959.x. [DOI] [PubMed] [Google Scholar]
- 4.Theilgaard-Monch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, Nielsen FC, Cowland JB, Borregaard N. The transcriptional program of terminal granulocytic differentiation. Blood. 2005;105:1785–1796. doi: 10.1182/blood-2004-08-3346. [DOI] [PubMed] [Google Scholar]
- 5.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 6.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 8.Geijsen N, Koenderman L, Coffer PJ. Specificity in cytokine signal transduction: lessons learned from the IL-3/IL-5/GM-CSF receptor family. Cytokine Growth Factor Rev. 2001;12:19–25. doi: 10.1016/s1359-6101(00)00019-8. [DOI] [PubMed] [Google Scholar]
- 9.Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammond WP, Boone TC, Donahue RE, Souza LM, Dale DC. A comparison of treatment of canine cyclic hematopoiesis with recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF), G-CSF interleukin-3, and canine G-CSF. Blood. 1990;76:523–532. [PubMed] [Google Scholar]
- 11.Wong GG, Witek JS, Temple PA, Wilkens KM, Leary AC, Luxenberg DP, Jones SS, Brown EL, Kay RM, Orr EC, et al. Human GM-CSF: molecular cloning of the complementary DNA and purification of the natural and recombinant proteins. Science. 1985;228:810–815. doi: 10.1126/science.3923623. [DOI] [PubMed] [Google Scholar]
- 12.Souza LM, Boone TC, Gabrilove J, Lai PH, Zsebo KM, Murdock DC, Chazin VR, Bruszewski J, Lu H, Chen KK, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232:61–65. doi: 10.1126/science.2420009. [DOI] [PubMed] [Google Scholar]
- 13.Paul M, Ram R, Kugler E, Farbman L, Peck A, Leibovici L, Lahav M, Yeshurun M, Shpilberg O, Herscovici C, Wolach O, Itchaki G, Bar-Natan M, Vidal L, Gafter-Gvili A, Raanani P. Subcutaneous versus intravenous granulocyte colony stimulating factor for the treatment of neutropenia in hospitalized hemato-oncological patients: randomized controlled trial. Am J Hematol. 2014;89:243–248. doi: 10.1002/ajh.23622. [DOI] [PubMed] [Google Scholar]
- 14.Horan T, Wen J, Narhi L, Parker V, Garcia A, Arakawa T, Philo J. Dimerization of the extracellular domain of granuloycte-colony stimulating factor receptor by ligand binding: a monovalent ligand induces 2:2 complexes. Biochemistry. 1996;35:4886–4896. doi: 10.1021/bi9525841. [DOI] [PubMed] [Google Scholar]
- 15.Mehta HM, Futami M, Glaubach T, Lee DW, Andolina JR, Yang Q, Whichard Z, Quinn M, Lu HF, Kao WM, Przychodzen B, Sarkar CA, Minella A, Maciejewski JP, Corey SJ. Alternatively spliced, truncated GCSF receptor promotes leukemogenic properties and sensitivity to JAK inhibition. Leukemia. 2014;28:1041–1051. doi: 10.1038/leu.2013.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Touw IP, van de Geijn GJ. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci. 2007;12:800–815. doi: 10.2741/2103. [DOI] [PubMed] [Google Scholar]
- 17.Hermans MH, van de Geijn GJ, Antonissen C, Gits J, van Leeuwen D, Ward AC, Touw IP. Signaling mechanisms coupled to tyrosines in the granulocyte colony-stimulating factor receptor orchestrate G-CSF-induced expansion of myeloid progenitor cells. Blood. 2003;101:2584–2590. doi: 10.1182/blood-2002-07-2062. [DOI] [PubMed] [Google Scholar]
- 18.Akbarzadeh S, Ward AC, McPhee DO, Alexander WS, Lieschke GJ, Layton JE. Tyrosine residues of the granulocyte colony-stimulating factor receptor transmit proliferation and differentiation signals in murine bone marrow cells. Blood. 2002;99:879–887. doi: 10.1182/blood.v99.3.879. [DOI] [PubMed] [Google Scholar]
- 19.Zhu QS, Robinson LJ, Roginskaya V, Corey SJ. G-CSF-induced tyrosine phosphorylation of Gab2 is Lyn kinase dependent and associated with enhanced Akt and differentiative, not proliferative, responses. Blood. 2004;103:3305–3312. doi: 10.1182/blood-2003-06-1861. [DOI] [PubMed] [Google Scholar]
- 20.Futami M, Zhu QS, Whichard ZL, Xia L, Ke Y, Neel BG, Feng GS, Corey SJ. G-CSF receptor activation of the Src kinase Lyn is mediated by Gab2 recruitment of the Shp2 phosphatase. Blood. 2011;118:1077–1086. doi: 10.1182/blood-2009-12-261636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jack GD, Zhang L, Friedman AD. M-CSF elevates c-Fos and phospho-C/EBPalpha(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood. 2009;114:2172–2180. doi: 10.1182/blood-2008-11-191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 23.Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, Ramshaw H, Woodcock JM, Xu Y, Guthridge M, McKinstry WJ, Lopez AF, Parker MW. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134:496–507. doi: 10.1016/j.cell.2008.05.053. [DOI] [PubMed] [Google Scholar]
- 24.Perugini M, Brown AL, Salerno DG, Booker GW, Stojkoski C, Hercus TR, Lopez AF, Hibbs ML, Gonda TJ, D'Andrea RJ. Alternative modes of GM-CSF receptor activation revealed using activated mutants of the common beta-subunit. Blood. 2010;115:3346–3353. doi: 10.1182/blood-2009-08-235846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34:81–89. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Pitrak DL. Effects of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor on the bactericidal functions of neutrophils. Curr Opin Hematol. 1997;4:183–190. doi: 10.1097/00062752-199704030-00005. [DOI] [PubMed] [Google Scholar]
- 27.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 28.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 29.Simon R, Norton L. The Norton-Simon hypothesis: designing more effective and less toxic chemotherapeutic regimens. Nat Clin Pract Oncol. 2006;3:406–407. doi: 10.1038/ncponc0560. [DOI] [PubMed] [Google Scholar]
- 30.Norton L, Simon R, Brereton HD, Bogden AE. Predicting the course of Gompertzian growth. Nature. 1976;264:542–545. doi: 10.1038/264542a0. [DOI] [PubMed] [Google Scholar]
- 31.Norton L, Simon R. The Norton-Simon hypothesis revisited. Cancer Treat Rep. 1986;70:163–169. [PubMed] [Google Scholar]
- 32.Glaubach T, Minella AC, Corey SJ. Cellular stress pathways in pediatric bone marrow failure syndromes: many roads lead to neutropenia. Pediatr Res. 2014;75:189–195. doi: 10.1038/pr.2013.197. [DOI] [PubMed] [Google Scholar]
- 33.Gilman PA, Jackson DP, Guild HG. Congenital agranulocytosis: prolonged survival and terminal acute leukemia. Blood. 1970;36:576–585. [PubMed] [Google Scholar]
- 34.Welte K, Dale D. Pathophysiology and treatment of severe chronic neutropenia. Ann Hematol. 1996;72:158–165. doi: 10.1007/s002770050156. [DOI] [PubMed] [Google Scholar]
- 35.Hammond WPt, Price TH, Souza LM, Dale DC. Treatment of cyclic neutropenia with granulocyte colony-stimulating factor. N Engl J Med. 1989;320:1306–1311. doi: 10.1056/NEJM198905183202003. [DOI] [PubMed] [Google Scholar]
- 36.Bonilla MA, Gillio AP, Ruggeiro M, Kernan NA, Brochstein JA, Abboud M, Fumagalli L, Vincent M, Gabrilove JL, Welte K, et al. Effects of recombinant human granulocyte colony-stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med. 1989;320:1574–1580. doi: 10.1056/NEJM198906153202402. [DOI] [PubMed] [Google Scholar]
- 37.Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, Meade-White K, Williams K, Acland GM, Niemeyer G, Lothrop CD, Horwitz M. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet. 2003;35:90–96. doi: 10.1038/ng1224. [DOI] [PubMed] [Google Scholar]
- 38.Welte K, Zeidler C, Reiter A, Muller W, Odenwald E, Souza L, Riehm H. Differential effects of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in children with severe congenital neutropenia. Blood. 1990;75:1056–1063. [PubMed] [Google Scholar]
- 39.Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, Wang W, Jakubowski A, Winton E, Lalezari P, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496–2502. [PMC free article] [PubMed] [Google Scholar]
- 40.Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR., Jr WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91:368–376. [PubMed] [Google Scholar]
- 41.Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 42.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17:413–423. doi: 10.1016/s1074-7613(02)00424-7. [DOI] [PubMed] [Google Scholar]
- 43.McDermott DH, Liu Q, Velez D, Lopez L, Anaya-O'Brien S, Ulrick J, Kwatemaa N, Starling J, Fleisher TA, Priel DA, Merideth MA, Giuntoli RL, Evbuomwan MO, Littel P, Marquesen MM, Hilligoss D, DeCastro R, Grimes GJ, Hwang ST, Pittaluga S, Calvo KR, Stratton P, Cowen EW, Kuhns DB, Malech HL, Murphy PM. A phase 1 clinical trial of long-term, low-dose treatment of WHIM syndrome with the CXCR4 antagonist plerixafor. Blood. 2014;123:2308–2316. doi: 10.1182/blood-2013-09-527226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenbaum AM, Link DC. Mechanisms of G-CSF-mediated hematopoietic stem and progenitor mobilization. Leukemia. 2011;25:211–217. doi: 10.1038/leu.2010.248. [DOI] [PubMed] [Google Scholar]
- 45.To LB, Levesque JP, Herbert KE. How I treat patients who mobilize hematopoietic stem cells poorly. Blood. 2011;118:4530–4540. doi: 10.1182/blood-2011-06-318220. [DOI] [PubMed] [Google Scholar]
- 46.Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185–1196. doi: 10.1182/blood-2011-12-274019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tichelli A, Schrezenmeier H, Socie G, Marsh J, Bacigalupo A, Duhrsen U, Franzke A, Hallek M, Thiel E, Wilhelm M, Hochsmann B, Barrois A, Champion K, Passweg JR. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434–4441. doi: 10.1182/blood-2010-08-304071. [DOI] [PubMed] [Google Scholar]
- 48.Jeng MR, Naidu PE, Rieman MD, Rodriguez-Galindo C, Nottage KA, Thornton DT, Li CS, Wiang WC. Granulocyte-macrophage colony stimulating factor and immunosuppression in the treatment of pediatric acquired severe aplastic anemia. Pediatr Blood Cancer. 2005;45:170–175. doi: 10.1002/pbc.20278. [DOI] [PubMed] [Google Scholar]
- 49.Kojima S. Use of hematopoietic growth factors for treatment of aplastic anemia. Bone Marrow Transplant. 1996;18(Suppl 3):S36–S38. [PubMed] [Google Scholar]
- 50.Marsh JC. Hematopoietic growth factors in the pathogenesis and for the treatment of aplastic anemia. Semin Hematol. 2000;37:81–90. doi: 10.1016/s0037-1963(00)90032-5. [DOI] [PubMed] [Google Scholar]
- 51.Bacigalupo A, Broccia G, Corda G, Arcese W, Carotenuto M, Gallamini A, Locatelli F, Mori PG, Saracco P, Todeschini G, et al. Antilymphocyte globulin, cyclosporin, and granulocyte colony-stimulating factor in patients with acquired severe aplastic anemia (SAA): a pilot study of the EBMT SAA Working Party. Blood. 1995;85:1348–1353. [PubMed] [Google Scholar]
- 52.Bacigalupo A, Bruno B, Saracco P, Di Bona E, Locasciulli A, Locatelli F, Gabbas A, Dufour C, Arcese W, Testi G, Broccia G, Carotenuto M, Coser P, Barbui T, Leoni P, Ferster A. Antilymphocyte globulin, cyclosporine, prednisolone, and granulocyte colony-stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT study on 100 patients. European Group for Blood and Marrow Transplantation (EBMT) Working Party on Severe Aplastic Anemia and the Gruppo Italiano Trapianti di Midolio Osseo (GITMO) Blood. 2000;95:1931–1934. [PubMed] [Google Scholar]
- 53.Kojima S, Matsuyama T, Kodera Y, Nishihira H, Ueda K, Shimbo T, Nakahata T. Measurement of endogenous plasma granulocyte colony-stimulating factor in patients with acquired aplastic anemia by a sensitive chemiluminescent immunoassay. Blood. 1996;87:1303–1308. [PubMed] [Google Scholar]
- 54.Bodey GP, Buckley M, Sathe YS, Freireich EJ. Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med. 1966;64:328–340. doi: 10.7326/0003-4819-64-2-328. [DOI] [PubMed] [Google Scholar]
- 55.Talcott JA, Siegel RD, Finberg R, Goldman L. Risk assessment in cancer patients with fever and neutropenia: a prospective, two-center validation of a prediction rule. J Clin Oncol. 1992;10:316–322. doi: 10.1200/JCO.1992.10.2.316. [DOI] [PubMed] [Google Scholar]
- 56.Oncology, A. S. o. C. American Society of Clinical Oncology. Recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. J Clin Oncol. 1994;12:2471–2508. doi: 10.1200/JCO.1994.12.11.2471. [DOI] [PubMed] [Google Scholar]
- 57.Crawford J, Ozer H, Stoller R, Johnson D, Lyman G, Tabbara I, Kris M, Grous J, Picozzi V, Rausch G, et al. Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med. 1991;325:164–170. doi: 10.1056/NEJM199107183250305. [DOI] [PubMed] [Google Scholar]
- 58.Trillet-Lenoir V, Green J, Manegold C, Von Pawel J, Gatzemeier U, Lebeau B, Depierre A, Johnson P, Decoster G, Tomita D, et al. Recombinant granulocyte colony stimulating factor reduces the infectious complications of cytotoxic chemotherapy. Eur J Cancer. 1993;29A:319–324. doi: 10.1016/0959-8049(93)90376-q. [DOI] [PubMed] [Google Scholar]
- 59.Pettengell R, Gurney H, Radford JA, Deakin DP, James R, Wilkinson PM, Kane K, Bentley J, Crowther D. Granulocyte colony-stimulating factor to prevent dose-limiting neutropenia in non-Hodgkin's lymphoma: a randomized controlled trial. Blood. 1992;80:1430–1436. [PubMed] [Google Scholar]
- 60.Gerhartz HH, Engelhard M, Meusers P, Brittinger G, Wilmanns W, Schlimok G, Mueller P, Huhn D, Musch R, Siegert W, et al. Randomized, double-blind, placebo-controlled, phase III study of recombinant human granulocyte-macrophage colony-stimulating factor as adjunct to induction treatment of high-grade malignant non-Hodgkin's lymphomas. Blood. 1993;82:2329–2339. [PubMed] [Google Scholar]
- 61.Bajorin DF, Nichols CR, Schmoll HJ, Kantoff PW, Bokemeyer C, Demetri GD, Einhorn LH, Bosl GJ. Recombinant human granulocyte-macrophage colony-stimulating factor as an adjunct to conventional-dose ifosfamide-based chemotherapy for patients with advanced or relapsed germ cell tumors: a randomized trial. J Clin Oncol. 1995;13:79–86. doi: 10.1200/JCO.1995.13.1.79. [DOI] [PubMed] [Google Scholar]
- 62.Citron ML, Berry DA, Cirrincione C, Hudis C, Winer EP, Gradishar WJ, Davidson NE, Martino S, Livingston R, Ingle JN, Perez EA, Carpenter J, Hurd D, Holland JF, Smith BL, Sartor CI, Leung EH, Abrams J, Schilsky RL, Muss HB, Norton L. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21:1431–1439. doi: 10.1200/JCO.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 63.Pfreundschuh M, Trumper L, Kloess M, Schmits R, Feller AC, Rube C, Rudolph C, Reiser M, Hossfeld DK, Eimermacher H, Hasenclever D, Schmitz N, Loeffler M G. German High-Grade Non-Hodgkin's Lymphoma Study. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood. 2004;104:634–641. doi: 10.1182/blood-2003-06-2095. [DOI] [PubMed] [Google Scholar]
- 64.Pfreundschuh M, Trumper L, Kloess M, Schmits R, Feller AC, Rudolph C, Reiser M, Hossfeld DK, Metzner B, Hasenclever D, Schmitz N, Glass B, Rube C, Loeffler M G. German High-Grade Non-Hodgkin's Lymphoma Study. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of young patients with good-prognosis (normal LDH) aggressive lymphomas: results of the NHL-B1 trial of the DSHNHL. Blood. 2004;104:626–633. doi: 10.1182/blood-2003-06-2094. [DOI] [PubMed] [Google Scholar]
- 65.Vogel CL, Wojtukiewicz MZ, Carroll RR, Tjulandin SA, Barajas-Figueroa LJ, Wiens BL, Neumann TA, Schwartzberg LS. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double-blind, placebo-controlled phase III study. J Clin Oncol. 2005;23:1178–1184. doi: 10.1200/JCO.2005.09.102. [DOI] [PubMed] [Google Scholar]
- 66.Timmer-Bonte JN, de Boo TM, Smit HJ, Biesma B, Wilschut FA, Cheragwandi SA, Termeer A, Hensing CA, Akkermans J, Adang EM, Bootsma GP, Tjan-Heijnen VC. Prevention of chemotherapy-induced febrile neutropenia by prophylactic antibiotics plus or minus granulocyte colony-stimulating factor in small-cell lung cancer: a Dutch Randomized Phase III Study. J Clin Oncol. 2005;23:7974–7984. doi: 10.1200/JCO.2004.00.7955. [DOI] [PubMed] [Google Scholar]
- 67.Yang BB, Lum PK, Hayashi MM, Roskos LK. Polyethylene glycol modification of filgrastim results in decreased renal clearance of the protein in rats. J Pharm Sci. 2004;93:1367–1373. doi: 10.1002/jps.20024. [DOI] [PubMed] [Google Scholar]
- 68.Johnston E, Crawford J, Blackwell S, Bjurstrom T, Lockbaum P, Roskos L, Yang BB, Gardner S, Miller-Messana MA, Shoemaker D, Garst J, Schwab G. Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol. 2000;18:2522–2528. doi: 10.1200/JCO.2000.18.13.2522. [DOI] [PubMed] [Google Scholar]
- 69.Smith TJ, Khatcheressian J, Lyman GH, Ozer H, Armitage JO, Balducci L, Bennett CL, Cantor SB, Crawford J, Cross SJ, Demetri G, Desch CE, Pizzo PA, Schiffer CA, Schwartzberg L, Somerfield MR, Somlo G, Wade JC, Wade JL, Winn RJ, Wozniak AJ, Wolff AC. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol. 2006;24:3187–3205. doi: 10.1200/JCO.2006.06.4451. [DOI] [PubMed] [Google Scholar]
- 70.Aarts MJ, Grutters JP, Peters FP, Mandigers CM, Dercksen MW, Stouthard JM, Nortier HJ, van Laarhoven HW, van Warmerdam LJ, van de Wouw AJ, Jacobs EM, Mattijssen V, van der Rijt CC, Smilde TJ, van der Velden AW, Temizkan M, Batman E, Muller EW, van Gastel SM, Joore MA, Borm GF, Tjan-Heijnen VC. Cost effectiveness of primary pegfilgrastim prophylaxis in patients with breast cancer at risk of febrile neutropenia. J Clin Oncol. 2013;31:4283–4289. doi: 10.1200/JCO.2012.48.3644. [DOI] [PubMed] [Google Scholar]
- 71.Crawford J, Armitage J, Balducci L, Becker PS, Blayney DW, Cataland SR, Heaney ML, Hudock S, Kloth DD, Kuter DJ, Lyman GH, McMahon B, Rugo HS, Saad AA, Schwartzberg LS, Shayani S, Steensma DP, Talbott M, Vadhan-Raj S, Westervelt P, Westmoreland M, Dwyer M, Ho M n. National comprehensive cancer. Myeloid growth factors. J Natl Compr Canc Netw. 2013;11:1266–1290. doi: 10.6004/jnccn.2013.0148. [DOI] [PubMed] [Google Scholar]
- 72.Dinan MA, Hirsch BR, Lyman GH. Management of chemotherapy-induced neutropenia: measuring quality, cost, and value. J Natl Compr Canc Netw. 2015;13:e1–e7. doi: 10.6004/jnccn.2015.0014. [DOI] [PubMed] [Google Scholar]
- 73.Rowe JM, Andersen JW, Mazza JJ, Bennett JM, Paietta E, Hayes FA, Oette D, Cassileth PA, Stadtmauer EA, Wiernik PH. A randomized placebo-controlled phase III study of granulocyte-macrophage colony-stimulating factor in adult patients (> 55 to 70 years of age) with acute myelogenous leukemia: a study of the Eastern Cooperative Oncology Group (E1490) Blood. 1995;86:457–462. [PubMed] [Google Scholar]
- 74.Stone RM, Berg DT, George SL, Dodge RK, Paciucci PA, Schulman P, Lee EJ, Moore JO, Powell BL, Schiffer CA. Granulocyte-macrophage colony-stimulating factor after initial chemotherapy for elderly patients with primary acute myelogenous leukemia. Cancer and Leukemia Group B. N Engl J Med. 1995;332:1671–1677. doi: 10.1056/NEJM199506223322503. [DOI] [PubMed] [Google Scholar]
- 75.Ohno R, Tomonaga M, Kobayashi T, Kanamaru A, Shirakawa S, Masaoka T, Omine M, Oh H, Nomura T, Sakai Y, et al. Effect of granulocyte colony-stimulating factor after intensive induction therapy in relapsed or refractory acute leukemia. N Engl J Med. 1990;323:871–877. doi: 10.1056/NEJM199009273231304. [DOI] [PubMed] [Google Scholar]
- 76.Ozer H, Armitage JO, Bennett CL, Crawford J, Demetri GD, Pizzo PA, Schiffer CA, Smith TJ, Somlo G, Wade JC, Wade JL, Winn RJ, Wozniak AJ, Somerfield MR f. t. A. S. o. C. O. G. F. E. Panel. 2000 Update of Recommendations for the Use of Hematopoietic Colony-Stimulating Factors: Evidence-Based, Clinical Practice Guidelines. J Clin Oncol. 2000;18:3558–3585. doi: 10.1200/JCO.2000.18.20.3558. [DOI] [PubMed] [Google Scholar]
- 77.Bonilla MA, Dale D, Zeidler C, Last L, Reiter A, Ruggeiro M, Davis M, Koci B, Hammond W, Gillio A, et al. Long-term safety of treatment with recombinant human granulocyte colony-stimulating factor (r-metHuG-CSF) in patients with severe congenital neutropenias. Br J Haematol. 1994;88:723–730. doi: 10.1111/j.1365-2141.1994.tb05110.x. [DOI] [PubMed] [Google Scholar]
- 78.Dale DC, Bolyard AA, Schwinzer BG, Pracht G, Bonilla MA, Boxer L, Freedman MH, Donadieu J, Kannourakis G, Alter BP, Cham BP, Winkelstein J, Kinsey SE, Zeidler C, Welte K. The Severe Chronic Neutropenia International Registry: 10-Year Follow-up Report. Support Cancer Ther. 2006;3:220–231. doi: 10.3816/SCT.2006.n.020. [DOI] [PubMed] [Google Scholar]
- 79.Rosen RB, Kang SJ. Congenital agranulocytosis terminating in acute myelomonocytic leukemia. J Pediatr. 1979;94:406–408. doi: 10.1016/s0022-3476(79)80581-8. [DOI] [PubMed] [Google Scholar]
- 80.Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Fier C, Freedman M, Kannourakis G, Kinsey S, Schwinzer B, Zeidler C, Welte K, Dale DC R. Severe Chronic Neutropenia International. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107:4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinha S, Zhu QS, Romero G, Corey SJ. Deletional mutation of the external domain of the human granulocyte colony-stimulating factor receptor in a patient with severe chronic neutropenia refractory to granulocyte colony-stimulating factor. J Pediatr Hematol Oncol. 2003;25:791–796. doi: 10.1097/00043426-200310000-00010. [DOI] [PubMed] [Google Scholar]
- 82.Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood. 2007;109:93–99. doi: 10.1182/blood-2006-02-004275. [DOI] [PubMed] [Google Scholar]
- 83.Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333:487–493. doi: 10.1056/NEJM199508243330804. [DOI] [PubMed] [Google Scholar]
- 84.Dong F, Dale DC, Bonilla MA, Freedman M, Fasth A, Neijens HJ, Palmblad J, Briars GL, Carlsson G, Veerman AJ, Welte K, Lowenberg B, Touw IP. Mutations in the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Leukemia. 1997;11:120–125. doi: 10.1038/sj.leu.2400537. [DOI] [PubMed] [Google Scholar]
- 85.Dong F, Hoefsloot LH, Schelen AM, Broeders CA, Meijer Y, Veerman AJ, Touw IP, Lowenberg B. Identification of a nonsense mutation in the granulocyte-colony-stimulating factor receptor in severe congenital neutropenia. Proc Natl Acad Sci U S A. 1994;91:4480–4484. doi: 10.1073/pnas.91.10.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dong F, van Paassen M, van Buitenen C, Hoefsloot LH, Lowenberg B, Touw IP. A point mutation in the granulocyte colony-stimulating factor receptor (G-CSF-R) gene in a case of acute myeloid leukemia results in the overexpression of a novel G-CSF-R isoform. Blood. 1995;85:902–911. [PubMed] [Google Scholar]
- 87.Hunter MG, Avalos BR. Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood. 1999;93:440–446. [PubMed] [Google Scholar]
- 88.Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood. 1999;93:447–458. [PubMed] [Google Scholar]
- 89.Hermans MH, Antonissen C, Ward AC, Mayen AE, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189:683–692. doi: 10.1084/jem.189.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jeha S, Chan KW, Aprikyan AG, Hoots WK, Culbert S, Zietz H, Dale DC, Albitar M. Spontaneous remission of granulocyte colony-stimulating factor-associated leukemia in a child with severe congenital neutropenia. Blood. 2000;96:3647–3649. [PubMed] [Google Scholar]
- 91.Maxson JE, Gottlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, Bottomly D, Wilmot BM, McWeeney SK, Tognon CE, Pond JB, Collins RH, Goueli B, Oh ST, Loriaux MM, Druker BJ, Tyner JW. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013 doi: 10.1056/NEJMoa1214514. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, Ketterling RP, Maxson JE, Tyner JW, Tefferi A. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilc leukemia. Leukemia. 2013 doi: 10.1038/leu.2013.122. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mehta HM, Glaubach T, Long A, Lu H, Przychodzen B, Makishima H, McDevitt MA, Cross NC, Maciejewski J, Corey SJ. Granulocyte colony-stimulating factor receptor T595I (T618I) mutation confers ligand independence and enhanced signaling. Leukemia. 2013;27:2407–2410. doi: 10.1038/leu.2013.164. [DOI] [PubMed] [Google Scholar]
- 94.Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, Gravanis I, Heim HK, Heinonen E, Ho K, Moreau A, Narayanan G, Kruse NA, Reichmann G, Thorpe R, van Aerts L, Vleminckx C, Wadhwa M, Schneider CK. Biosimilars: what clinicians should know. Blood. 2012;120:5111–5117. doi: 10.1182/blood-2012-04-425744. [DOI] [PubMed] [Google Scholar]
- 95.Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19:411–419. doi: 10.1093/annonc/mdm345. [DOI] [PubMed] [Google Scholar]
- 96.Sun D, Andayani TM, Altyar A, MacDonald K, Abraham I. Potential Cost Savings From Chemotherapy-Induced Febrile Neutropenia With Biosimilar Filgrastim and Expanded Access to Targeted Antineoplastic Treatment Across the European Union G5 Countries: A Simulation Study. Clin Ther. 2015 doi: 10.1016/j.clinthera.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 97.Lowenberg B, van Putten W, Theobald M, Gmur J, Verdonck L, Sonneveld P, Fey M, Schouten H, de Greef G, Ferrant A, Kovacsovics T, Gratwohl A, Daenen S, Huijgens P, Boogaerts M G. Dutch-Belgian Hemato-Oncology Cooperative, and R. Swiss Group for Clinical Cancer. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med. 2003;349:743–752. doi: 10.1056/NEJMoa025406. [DOI] [PubMed] [Google Scholar]
- 98.Carulli G. Effects of recombinant human granulocyte colony-stimulating factor administration on neutrophil phenotype and functions. Haematologica. 1997;82:606–616. [PubMed] [Google Scholar]
- 99.Kim SO, Sheikh HI, Ha SD, Martins A, Reid G. G-CSF-mediated inhibition of JNK is a key mechanism for Lactobacillus rhamnosus-induced suppression of TNF production in macrophages. Cell Microbiol. 2006;8:1958–1971. doi: 10.1111/j.1462-5822.2006.00763.x. [DOI] [PubMed] [Google Scholar]
- 100.D'Aveni M, Rossignol J, Coman T, Sivakumaran S, Henderson S, Manzo T, Santos ESP, Bruneau J, Fouquet G, Zavala F, Alegria-Prevot O, Garfa-Traore M, Suarez F, Trebeden-Negre H, Mohty M, Bennett CL, Chakraverty R, Hermine O, Rubio MT. G-CSF mobilizes CD34+ regulatory monocytes that inhibit graft-versus-host disease. Sci Transl Med. 2015;7:281ra242. doi: 10.1126/scitranslmed.3010435. [DOI] [PubMed] [Google Scholar]
- 101.Martins A, Han J, Kim SO. The multifaceted effects of granulocyte colony-stimulating factor in immunomodulation and potential roles in intestinal immune homeostasis. IUBMB life. 2010;62:611–617. doi: 10.1002/iub.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brissette WH, Baker DA, Stam EJ, Umland JP, Griffiths RJ. GM-CSF rapidly primes mice for enhanced cytokine production in response to LPS and TNF. Cytokine. 1995;7:291–295. doi: 10.1006/cyto.1995.0035. [DOI] [PubMed] [Google Scholar]
- 103.Dabritz J, Bonkowski E, Chalk C, Trapnell BC, Langhorst J, Denson LA, Foell D. Granulocyte macrophage colony-stimulating factor auto-antibodies and disease relapse in inflammatory bowel disease. Am J Gastroenterol. 2013;108:1901–1910. doi: 10.1038/ajg.2013.360. [DOI] [PubMed] [Google Scholar]
- 104.Egea L, McAllister CS, Lakhdari O, Minev I, Shenouda S, Kagnoff MF. GM-CSF produced by nonhematopoietic cells is required for early epithelial cell proliferation and repair of injured colonic mucosa. J Immunol. 2013;190:1702–1713. doi: 10.4049/jimmunol.1202368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roth L, MacDonald JK, McDonald JW, Chande N. Sargramostim (GM-CSF) for induction of remission in Crohn's disease: a cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis. 2012;18:1333–1339. doi: 10.1002/ibd.22973. [DOI] [PubMed] [Google Scholar]
- 106.Bonneau J, Dumestre-Perard C, Rinaudo-Gaujous M, Genin C, Sparrow M, Roblin X, Paul S. Systematic review: new serological markers (anti-glycan, anti-GP2, anti-GM-CSF Ab) in the prediction of IBD patient outcomes. Autoimmun Rev. 2015;14:231–245. doi: 10.1016/j.autrev.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 107.Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123–1142. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 108.Suzuki T, Sakagami T, Young LR, Carey BC, Wood RE, Luisetti M, Wert SE, Rubin BK, Kevill K, Chalk C, Whitsett JA, Stevens C, Nogee LM, Campo I, Trapnell BC. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med. 2010;182:1292–1304. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 110.Jouneau S, Kerjouan M, Briens E, Lenormand JP, Meunier C, Letheulle J, Chiforeanu D, Laine-Caroff C, Desrues B, Delaval P. Pulmonary alveolar proteinosis. Rev Mal Respir. 2014;31:975–991. doi: 10.1016/j.rmr.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 111.Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, Abe S, Trapnell C, Carey B, Moritz T, Malik P, Lutzko C, Wood RE, Trapnell BC. Pulmonary macrophage transplantation therapy. Nature. 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garber B, Albores J, Wang T, Neville TH. A plasmapheresis protocol for refractory pulmonary alveolar proteinosis. Lung. 2015;193:209–211. doi: 10.1007/s00408-014-9678-2. [DOI] [PubMed] [Google Scholar]
- 113.Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, Hizawa N, Kasahara Y, Tatsumi K, Hojo M, Ishii H, Yokoba M, Tanaka N, Yamaguchi E, Eda R, Tsuchihashi Y, Morimoto K, Akira M, Terada M, Otsuka J, Ebina M, Kaneko C, Nukiwa T, Krischer JP, Akazawa K, Nakata K. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2010;181:1345–1354. doi: 10.1164/rccm.200906-0978OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reed JA, Ikegami M, Cianciolo ER, Lu W, Cho PS, Hull W, Jobe AH, Whitsett JA. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol. 1999;276:L556–L563. doi: 10.1152/ajplung.1999.276.4.L556. [DOI] [PubMed] [Google Scholar]
- 115.Tazawa R, Hamano E, Arai T, Ohta H, Ishimoto O, Uchida K, Watanabe M, Saito J, Takeshita M, Hirabayashi Y, Ishige I, Eishi Y, Hagiwara K, Ebina M, Inoue Y, Nakata K, Nukiwa T. Granulocyte-macrophage colony-stimulating factor and lung immunity in pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2005;171:1142–1149. doi: 10.1164/rccm.200406-716OC. [DOI] [PubMed] [Google Scholar]