

Abstract
Chemo-resistance is a clinical barrier to more effective anti-cancer therapy. In this context, cancer stem-like cells (CSCs) are thought to be chemo-resistant, resulting in tumor recurrence and distant metastasis. Our hypothesis is that chemo-resistance in CSCs is driven, in part, by enhanced mitochondrial function. Here, we used breast cell lines and metastatic breast cancer patient samples to begin to dissect the role of mitochondrial metabolism in conferring the CSC phenotype. More specifically, we employed fluorescent staining with MitoTracker (MT) to metabolically fractionate these cell lines into mito-high and mito-low sub-populations, by flow-cytometry. Interestingly, cells with high mitochondrial mass (mito-high) were specifically enriched in a number of known CSC markers, such as aldehyde dehydrogenase (ALDH) activity, and they were ESA+/CD24-/low and formed mammospheres with higher efficiency. Large cell size is another independent characteristic of the stem cell phenotype; here, we observed a >2-fold increase in mitochondrial mass in large cells (>12-μm), relative to the smaller cell population (4–8-μm). Moreover, the mito-high cell population showed a 2.4-fold enrichment in tumor-initiating cell activity, based on limiting dilution assays in murine xenografts. Importantly, primary human breast CSCs isolated from patients with metastatic breast cancer or a patient derived xenograft (PDX) also showed the co-enrichment of ALDH activity and mitochondrial mass. Most significantly, our investigations demonstrated that mito-high cells were resistant to paclitaxel, resulting in little or no DNA damage, as measured using the comet assay. In summary, increased mitochondrial mass in a sub-population of breast cancer cells confers a stem-like phenotype and chemo-resistance. As such, our current findings have important clinical implications for over-coming drug resistance, by therapeutically targeting the mito-high CSC population.
Keywords: mitochondria, MitoTracker, cancer stem cells, tumor metabolism
INTRODUCTION
Cancer stem-like cells (CSCs) or tumor-initiating cells (TICs) are thought to be responsible for driving patient relapse (tumor recurrence and metastasis), as a consequence of their chemo-resistance and/or radio-resistance [1–6]. However, the cellular mechanism(s) driving this drug-resistant phenotype remain largely unknown. Elucidation of the mechanisms underpinning drug-resistance would have broad clinical implications, for the prevention of treatment failure in a variety of different cancer types.
One hypothesis is that mitochondria are involved in conferring drug-resistance, as they control both i) energy metabolism and ii) susceptibility towards apoptotic cell death. Previous studies have shown that catabolic cancer-associated fibroblasts (CAFs) produce mitochondrial fuels (such as L-lactate, ketone bodies and L-glutamine), which are sufficient to confer a drug-resistant phenotype in MCF7 breast cancer cells in culture [7, 8]. In this context, treatment with metformin (a mitochondrial inhibitor of Complex I) was sufficient to revert this drug-resistance phenotype. Similar results were also obtained in a variety of distinct patient cohorts followed at the MD Anderson and Dana-Farber Cancer Centers, showing that increased mitochondrial integrity (in stained tumor-tissue sections) was associated with drug-resistance and treatment failure in a number of different cancer types, including multiple myeloma, acute myelogenous and lymphoblastic leukemia, as well as ovarian cancers [9, 10].
Our group has previously shown that breast CSCs are chemo- and radio-resistant and that stem cell regulatory pathways, such as Notch, Wnt, FAK and CXCR1/2, may play a role in their maintenance and resistance mechanisms [11–17]. However, there is a limited understanding of the metabolic activities within CSC-enriched and CSC-depleted populations, although mechanistic differences are undoubtedly present [18–20]. Metabolic investigations in glioma stem cells (GSCs) showed that radio-resistant GSCs were less glycolytic, with a higher mitochondrial reserve capacity [18], indicating that specific inhibition of OXPHOS may target the GSCs. Similarly CSCs isolated from ovarian cancer patients also showed evidence of a metabolic profile dominated by OXPHOS, even in the absence of glucose. The authors suggest that these findings could be instrumental in helping CSCs to escape damage in hypo-oxygenated tumor areas [21].
To determine if breast CSC characteristics were associated with a specific metabolic activity, we used MitoTracker (MT) as an investigational tool to live-stain and metabolically fractionate cancer cell lines and patient samples into mito-high and mito-low cell populations. Our study demonstrates that cells with high mitochondrial mass showed many of the characteristics of CSCs, including increased ALDH activity, mammosphere formation activity and tumor initiation in vivo. In addition, we show increased survival and decreased DNA damage within the mito-high cells after paclitaxel treatment, suggesting that mitochondrial mass confers a chemo-resistance phenotype, as predicted.
Thus, our current findings provide a new mitochondrially-based model for understanding the relationship between CSCs and chemo-resistance. As this approach is technically simple, it could be used to rapidly isolate chemo-resistant CSC populations from possibly any tumor type, facilitating the development of new classes of drugs to target chemo-resistance.
RESULTS
High mitochondrial mass directly correlates with ALDH activity, the ESA+CD24-/low CSC population and larger cell size
Studies show CSC and non-CSC populations may use different metabolic pathways and this may contribute to their increased survival and resistance to chemotherapy [18, 22, 23]. To determine if mitochondrial mass is increased within the CSC population, we fluorescently-labeled mitochondria in MCF7 and MDA MB 231 breast cancer cells with MitoTracker Deep-Red. These cells were co-stained with ALDEFLUOR or CSC cell surface markers ESA/CD24, to identify the CSC population. Both ALDH+ and ESA+/CD24− cell populations have been shown to be enriched in mammosphere- and tumor-initiating cells, key characteristics of CSCs [2, 24–28].
Figure 1A, 1B, 1C shows that MitoTracker staining is specifically enriched in the ALDH+ cell population of MCF7 and MDA MB 231 cells (P < 0.05). A similar fold increase in MitoTracker mean fluorescence intensity was also observed in the ESA+CD24-/low CSC population of the MDA MB 231 cell line (Figure 1D, P < 0.01). These findings suggest that CSCs contain a higher mitochondrial mass than the non-CSC population.
Figure 1. Mitochondrial mass directly correlates with ALDH activity and the ESA+CD24-/low CSC population.
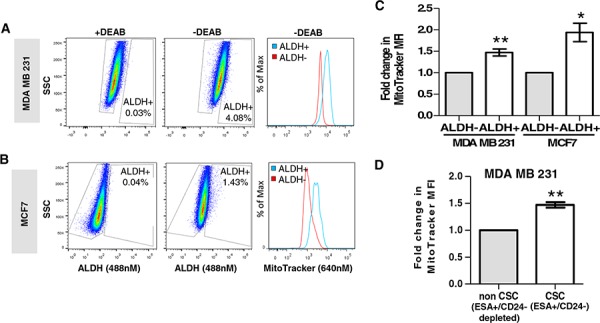
Representative dot plots of ALDH activity in MDA MB 231 A. and MCF7 B. cells, showing ALDH+ and ALDH− cells in the absence of DEAB. Histograms represent typical staining intensity of MitoTracker in ALDH+ and ALDH− populations in both cell lines. C. Graph showing fold change in mean fluorescence intensity (MFI) of MitoTracker (Deep Red; 640 nM), within ALDH+ and ALDH− populations of MCF7 and MDA MB 231 cell lines (n = 4 independent experiments). D. Graph showing fold change in mean fluorescence intensity of MitoTracker (Deep Red; 640 nM) within the ESA+CD24− (cancer stem-like cell, CSC) population and ESA+/CD24− depleted (non-CSC) populations of MDA MB 231 cells (n = 4 independent experiments). Bar graphs are shown as the mean ± SEM, t-test, two-tailed test, *P ≤ 0.05, **P ≤ 0.01.
As an alternative approach to enrich CSCs, we used cell size. Previous studies have shown that cells with mammary stem cell activity tend to be larger than 10 μm [29]. As a consequence, we used forward scatter (FSC) to isolate three different cell populations, based solely on size: 4–8 μm, 9–12 μm and >12 μm (Figure 2A). Quantitative analysis of MitoTracker staining demonstrated that larger cells were associated with significantly higher mitochondrial mass, up to 2.5-fold, consistent with an anabolic CSC phenotype (Figure 2B and 2C, P < 0.001).
Figure 2. Mitochondrial mass directly correlates with the enriched breast CSC population, identified using large cell size.
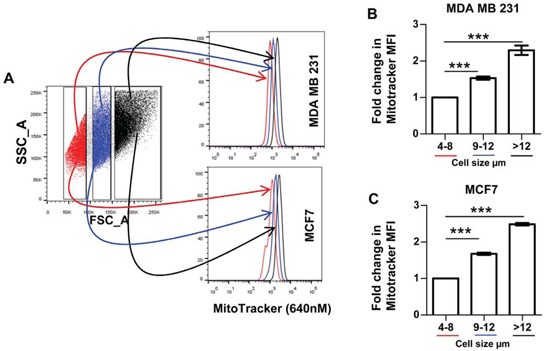
A. Typical dot plot showing side scatter (SSC) and forward scatter (FSC) of live breast cancer cells, gates represent cell size (RED- 4–8 μm; BLUE- 9–12 μm; BLACK > 12 μm; [29]). Histograms show MitoTracker mean fluorescence intensity within the 3 cell size groups of MDA MB 231 and MCF7 cells. Graphs showing fold change in the mean fluorescence intensity (MFI) of MDA MB 231 B. and MCF7s C. within the 9–12 μm and > 12 μm cell size compared to the smallest cells (4–8 μm), n = 3 independent experiments, 2 technical replicates. Bar graphs are shown as the mean ± SEM, t-test, two-tailed test, ***P < 0.001.
These data indicate that high mitochondrial mass, as determined by MitoTracker staining, is associated with breast CSC populations enriched via three independent CSC markers, namely ALDH activity, ESA/CD24 cell surface levels or cell size.
High mitochondrial mass directly correlates with ALDH activity in primary breast cancer cells isolated from metastatic disease sites or a patient derived xenograft (PDX)
To validate the possible in vivo relevance of our above findings, we next examined mitochondrial mass in primary CSC populations from metastatic breast cancer patients. For this purpose, we co-labeled breast cancer cells isolated directly from pleural effusions or ascites fluids (n = 4) with ALDEFLUOR and MitoTracker.
Figure 3A, 3B, and 3D supports our breast cancer cell line data, showing that ALDH+ primary metastatic breast CSCs have significantly higher mitochondrial mass than the ALDH− cells (P < 0.05). Notably, although these findings are of a low sample size, our results appear to be independent of estrogen receptor (ER), progesterone (PR) and HER2 status (Figure 3F). In addition, we also show similar results within the ALDH+ population of human breast cancer cells isolated from a patient derived xenograft (BB3RC50*) (Figure 3C and 3E). These data suggest that high mitochondrial mass is associated with CSC populations from freshly isolated metastatic breast cancer cells.
Figure 3. Mitochondrial mass correlates with ALDH activity in primary breast cancer cells isolated from metastatic breast cancer samples and a patient derived xenograft.
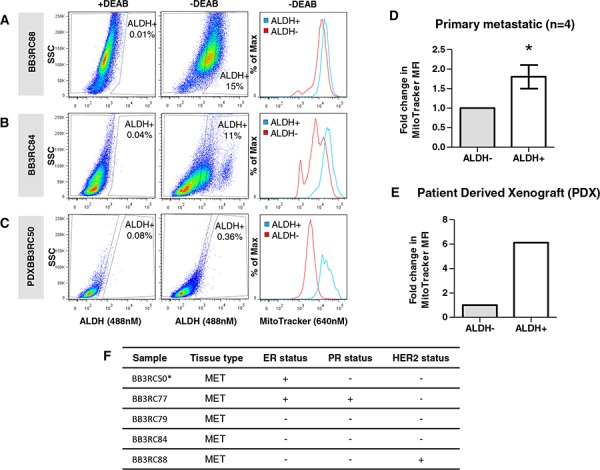
Representative dot plots of ALDH activity in primary metastatic breast cancer samples BB3RC88 A. BB3RC84 B. and patient derived xenograft sample (BB3RC50) C. cells showing ALDH+ and ALDH− cells in the absence of DEAB. Histograms represent typical staining intensity of MitoTracker (MT) in ALDH+ and ALDH− populations. D. Graph showing fold change in mean fluorescence intensity (MFI) of MitoTracker (Deep Red; 640 nM) within ALDH+ and ALDH− populations of primary metastatic breast cancer samples (n = 4), BB3RC77, BB3RC79, BB3RC88, BB3RC84. E. Graph showing fold change in mean fluorescence intensity (MFI) of MitoTracker (Deep Red; 640 nM) within ALDH+ and ALDH− populations of a patient derived xenograft sample (BB3RC50). F. Table showing the original (invasive) breast cancer characteristics of the metastatic (MET) breast cancer samples. Estrogen receptor (ER) and progesterone receptor (PR) status, + = > 10% positive; HER2 status, + = 3+ or 2+ & amplified; BB3RC50* denotes the PDX sample. Bar graphs are shown as the mean ± SEM, t-test, two-tailed test, *P ≤ 0.05.
High mitochondrial mass enriches for mammosphere-forming activity in vitro and tumor-initiating activity in vivo
Although we correlated high mitochondrial mass with a number of CSC markers, it was important to validate our findings with more functional parameters, such as mammosphere-forming activity and tumor initiation, that have been traditionally associated with “stemness” [30–32]. MCF7 and MDA MD 231 cells were sorted into mito-high (top 5% of cells with high MitoTracker staining) and mito-low (bottom 5% of cells with low MitoTracker staining) cell populations using flow-cytometry. The mito-high and mito-low cell populations were then seeded under non-adherent culture conditions and the number of mammospheres >60 μm were counted after 5 days. Both MCF7 and MDA MB 231 cells showed a significant 2- to -3 fold increase in mammosphere forming efficiency within the mito-high population, compared to mito-low cells (Figure 4A, P < 0.01).
Figure 4. High mitochondrial mass is specifically associated with mammosphere formation and tumor-initiating activity.
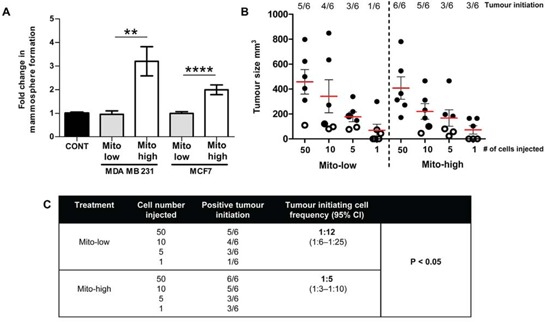
MDA MB 231 and MCF7 cells were subjected to flow-cytometry to isolate different populations of MitoTracker (MT) stained cells: mito-low = lowest 5% of MitoTracker stained cells; mito-high = highest 5% of MitoTracker stained cells. Mito-high and low populations were then seeded into mammosphere cultures A. or injected by limiting dilution into NOD scid gamma (NSG) mice B. (A) Graph showing fold change in mammosphere formation in mito-low and mito-high populations of MDA MB 231 and MCF7 cells, n = 4 independent experiments, ≥3 technical replicates. (B) Graph showing tumor size (mm3), 9 weeks after cell injections (sub-cutaneous), with 1, 5, 10 and 50 cells per injection of mito-low and mito-high MDA MB 231 cell populations. Each dot represents a tumor, hollow dots <100 μm = no tumor initiation. C. Extreme limiting dilution analysis (ELDA) was used to calculate the tumor initiation cell frequency within the mito-low and mito-high MDA MB 231 cell populations. Graphs are shown as the mean ± SEM, t-test, two-tailed test, **P ≤ 0.01, **** ≤ 0.0001.
The most stringent test of CSC activity is the capacity for tumor initiation [31, 32]. Serial dilutions of MDA MB 231 mito-high and mito-low cells (1, 5, 10, and 50 cells) were injected into NOD scid gamma (NSG) mice, and tumor initiation and growth were measured over 9 weeks. Our results show high mitochondrial mass was not associated with increased tumor growth/size (Figure 4B). However, using extreme limiting dilution analysis (ELDA), we demonstrate a 2.4-fold increase in tumor initiation cell frequency within the mito-high population of MDA MB 231 cells, when compared to mito-low (Figure 4C, P < 0.05). Although high mitochondrial mass was not associated with increased tumor size (Figure 4B), these data show that high mitochondrial mass is specifically associated with two key functional CSC characteristics, in vitro mammosphere formation and tumor-initiating cell frequency in vivo.
CSCs with high mitochondrial mass preferentially survive paclitaxel treatment and show reduced DNA strand breaks, conferring chemo-resistance
Another key CSC characteristic is resistance to radio- and chemo-therapy [2, 3, 17]. In order to correlate chemo-resistance with mitochondrial mass, MCF7 and MDA MB 231 cells were first separated into mito-high and mito-low cell fractions by flow-cytometry, using MitoTracker. The two cell populations were then seeded into mammosphere culture in the presence or absence of paclitaxel (0.1–0.5 μM) to assess their survival characteristics and susceptibility towards DNA damage.
Figure 5A and 5B shows that cells with high mitochondrial mass are resistant to paclitaxel, demonstrating little or no decrease in mammosphere formation, compared to vehicle alone controls. In contrast, cells with low mitochondrial mass show significant reductions in mammosphere formation, up to 2-fold, at the same concentrations (P < 0.001). Thus, high mitochondrial mass directly correlates with resistance to paclitaxel in CSCs.
Figure 5. CSCs with high mitochondrial mass preferentially survive paclitaxel treatment and show reduced DNA strand breaks.
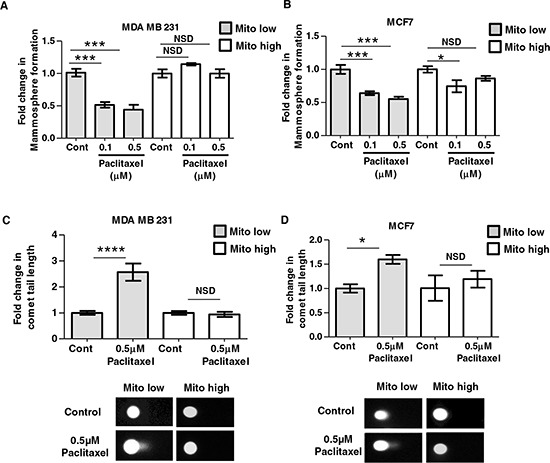
MDA MB 231 and MCF7 cells were subjected to flow-cytometry to isolate different populations of MitoTracker stained cells: mito-low = lowest 5% of MitoTracker stained cells; mito-high = highest 5% of MitoTracker stained cells. Mito-high and low populations were then seeded into mammosphere culture in the presence or absence of paclitaxel (A–B) or treated with paclitaxel or control (DMSO 1:10,000) for 24 h before DNA damage analysis via a comet assay (C–D). A–B. Graphs show fold change in mammosphere formation in mito-low and mito-high cells (MDA MB 231 and MCF7), after treatment with 0.1 μM and 0.5 μM paclitaxel compared to vehicle alone controls, n = 3 independent experiments, ≥3 technical replicates. C–D. Graph shows fold change in comet tail length in mito-low and mito-high cell populations (MDA MB 231 and MCF7), after 24 h of 0.5 μM paclitaxel treatment, the images show representative comet tails under all conditions. Bar graphs are shown as the mean ± SEM, (ANOVA) test with post-hoc dunetts multiple comparisons (A–B), t-test, two-tailed test (C–D), **P ≤ 0.01, **** ≤ 0.0001.
To better mechanistically understand this chemo-resistance phenotype, we used the comet assay to functionally measure the levels of DNA strand breaks after 24 h of paclitaxel treatment [33]. Remarkably, Figure 5C and 5D shows that cells with high mitochondrial mass have significantly reduced DNA damage, whereas cells with low mitochondrial mass are more chemo-sensitive, with increased comet tail length after paclitaxel treatment. These data indicate that high mitochondrial mass allows CSCs to survive chemotherapy, by affording protection or increased repair against DNA damage.
DISCUSSION
Drug resistance is a rate-limiting barrier to successful cancer therapy. Tumor-initiating cells and/or CSCs share a common chemo-resistant phenotype, which ultimately drives tumor recurrence and metastatic disease, resulting in patient relapse [1–6]. In this report, we show that high mitochondrial mass is associated with CSC markers, functional CSC characteristics (mammosphere and tumor initiation) and chemo-resistance. Our findings highlight the need to further understand and target the metabolic pathways in CSCs, to improve clinical outcome in breast cancer patients.
ALDEFLUOR is an investigational tool, which is routinely used to measure ALDH activity in cell lines and primary tumor samples, enriching for CSC activity via flow-cytometry [27, 28]. However, the ALDH gene family is quite large and it still remains controversial which ALDH isoforms contribute towards ALDH activity that is associated with CSC activity [34–37]. As our current report shows that ALDH activity in CSCs is also associated with high levels of mitochondrial mass, in cell lines and patient samples, it is quite possible that this stem-cell associated ALDH activity may also be derived from mitochondria. In fact, at least 7 members of the ALDH family of genes are known to be targeted to mitochondria and are bonafide resident mitochondrial proteins, including ALDH2, ALDH1L2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1 and ALDH18A1. Therefore, this new mitochondrial connection could functionally explain the relationship between ALDH activity and stemness in cancer cells. In support of this idea, it was recently shown that the drug-resistant ALDH-high cell sub-population has significantly increased mitochondrial respiration, leading to increased levels of oxidative stress [38].
We also showed that other CSC markers (ESA/CD24), and large cell size were associated with high mitochondrial mass. This supports other published studies where breast CSCs have been shown to have a higher maximum mitochondrial capacity and mitochondrial proton leak, compared to their differentiated non-CSC progeny [22]. Moreover, we demonstrated that cells with high mitochondrial mass were also functionally more stem-like, showing significant increases in i) mammosphere-forming efficiency and ii) tumor-initiating cell activity in vivo. The mechanism behind the increased mitochondrial mass in our study remains unknown. However, CSC populations are known to have a distinct genomic and proteomic expression profile [39], which can contribute to the stem cell characteristics of CSCs. This differential expression may also influence mitochondrial biogenesis, for example high levels of mTORC1 and its activity are seen in CSC populations of pancreatic and breast cancer cells [40, 41]. Studies have shown mitochondrial activity and biogenesis are controlled by mTORC1 by promotion of the translation of mitochondria-related mRNAs, via inhibition of the eukaryotic translation initiation factor 4E (eIF4E)-binding proteins [42], suggesting the high activity of mTORC1 in CSCs may increase mitochondrial biogenesis. Similarly, the transcription coactivator peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α), a known mediator of mitochondrial biogenesis, has been shown to be overexpressed in circulating tumor cells (with CSC characteristics) from the MMTV-PyMT spontaneous mouse breast cancer model [43]. The over-expression of oncogenes is also linked to increased mitochonidrial mass. Increased expression of Myc maintained the stemness of brain stem cells and glioma CSCs [44, 45] but also stimulated nuclear-encoded mitochondrial genes and mitochondrial biogenesis [46]. These studies suggest the differential expression of proteins and genes within the CSC population may preferentially favor increased mitochondrial biogenesis, compared to the non-CSC population.
The inherent expression of genes that increase mitochondrial biogenesis may, in turn, aid the maintenance of stem cell characteristics, as mitochondrial function has been shown to be important for the self renewal and differentiation capacity of stem cells. Although mitochondrial mass in embryonic stem cells is low, studies in normal adult stem cells and CSCs demonstrate the levels of mitochondrial mass are tissue-specific [47]. In glioblastoma, the depletion of mitochondrial DNA, after 2′-3′-dideoxycytidine treatment, inhibited tumor initiation [48], whereas reduced mitochondrial mass identified lung cancer stem cells [49]. A recent paper also highlighted the asymmetric apportioning of old/aged mitochondria, where the retention of young mitochondria during asymmetric self-renewal was vital to maintaining the stem cell characteristics of normal mammary epithelial cells [50]. Our data suggests that increased mitochondrial mass may increase tumor initiation via increased self renewal. Further investigations on the tumor initiation capacity of breast CSCs after the depletion or inactivation of mitochondria are warranted to validate this hypothesis.
Although high mitochondrial mass increased tumor initiation, especially at low cell numbers, it did not significantly affect overall tumor growth/size. These findings may simply reflect the recent observation that tumor cells have the capacity to “steal” mitochondrial DNA (mt-DNA) from host stromal cells (such as mesenchymal stem cells or endothelial cells), to boost their metabolic capacity towards OXPHOS, depending on the length of time they spend in the host [51]. Interestingly, it has also been reported that the transfer of stromal cell mitochondria to cancer cells increases their respiratory capacity and chemo-resistance [52].
Our findings also demonstrated that cells with high mitochondrial mass were resistant to chemotherapy, effectively protecting, delaying or avoiding DNA strand breaks after 24 h of paclitaxel treatment. Other studies show that inhibiting ALDH activity, which is increased within the mito-high population, can increase chemo-sensitivity [53, 54], although the specific mechanism is currently unknown. Evidence also shows that chemo-resistance solid cancers could be driven by numerous metabolic mechanisms, including reduced reactive oxygen species [55] and increased mitochondrial coupling [56]. Similarly, a recent study has identified that SIRT4, a mitochondria-localized sirtuin, regulates the metabolic responses to DNA damage by repressing mitochondrial glutamine metabolism. This contributes to the control of cell cycle progression and the maintenance of genomic integrity in response to DNA damage [57]. Differential metabolic control of the cell cycle may therefore play a role in reducing DNA damage in our study, as paclitaxel requires cell division to induce microtubule damage. Further exploration to elucidate the exact mechanisms is warranted.
CSCs are known to have increased DNA damage responses and repair mechanisms, characterized by the expression of high levels of DNA repair proteins, e.g. Chk1 and Rad51 [58, 59]. Since our mito-high cell population is enriched in established CSC markers, it is reasonable to presume the reduced DNA damage may be facilitated in part via non-metabolic mechanisms. However, recent studies have described signaling linking the metabolite fumarate to the promotion of DNA repair. Increased fumarate inhibited KDM2B histone demethylase activity, which subsequently increased accumulation of DNA-PK at double strand break regions, driving non-homologous end-joining DNA repair and cell survival [60, 61]. Research to date has only just started to reveal the complex signaling cross-talk linking metabolic processes with the DNA damage response and other biological functions. Further investigations will aid our understanding and deliver new targets to improve cancer treatment.
Interestingly, most markers of stemness that are routinely used to enrich for CSC activity (CD24, ESA, CD133, KRT19) are not generally thought of as therapeutically druggable targets. In contrast, the discovery of mitochondrial mass as a biomarker of CSC activity, as detected using the MitoTracker probe, suggests that we could therapeutically target mitochondria to more effectively eradicate the CSC population. There are many potential targets within mitochondria that could be inhibited to improve cancer treatment. These include targeting mitochondrial control of apoptosis with inhibitors the BCL-2 family [62] or HSP90, which has been shown to be specifically expressed in cancer cell mitochondria, but not their normal counterparts [63, 64]. Specific targeting of upregulated metabolic pathways, such as fatty acid synthesis, glycolysis and OXPHOS have also been shown to have anti-cancer effects (reviewed in Ref [65]). As discussed above, mitochondrial biogenesis can be induced by the hyper-activation of numerous signaling pathways and oncogenes, such as mTOR and Myc, which can be inhibited using rapacycin analogues e.g. everolimus [66] or DCR-MYC, a novel siRNA-based therapeutic, designed to silence the MYC oncogene which is currently in Phase I clinical trials [67].
The “Endo-symbiotic Theory of Mitochondrial Evolution” suggests that mitochondria originally evolved from engulfed aerobic bacteria, over millions of years of adaptation [68, 69]. As such, antibiotics target mitochondria, and therefore this side effect could be exploited to target the increased mitochondrial mass within CSCs. Interestingly, several FDA-approved drugs/antibiotics that target mitochondria have already been shown to eradicate CSC activity, such as OXPHOS inhibitors (metformin and pyrvinium pamoate) [70–73], a potassium ionophore (salinomycin) [74, 75] and known inhibitors of mitochondrial biogenesis and mitochondrial translation (the erythromycins, the tetracyclines and the glycylcyclines) [76, 77]. In fact, doxycycline and azithromycin have already shown significant efficacy in treatment-resistant cancer patients, with MALT lymphoma and non-small cell lung tumors, respectively [78–80].
The discovery that MitoTracker can identify a live population of cancer cells (mito-high) that are enriched for chemo-resistant cells may have a potential role in personalized medicine. For example the isolation of mito-high cells from patient samples could be used to screen for sensitivity to current or new therapies, by measuring cell death, mammosphere formation, or by employing a simple colony survival assay (Figure 6). Mito-high cells could also be isolated for genomic and proteomic analyses. Detailed analysis of the transcriptome and the proteome of mito-high cells, as compared to the mito-low cell populations, could reveal specific pathways that mito-high cells depend on for survival and maintenance. These specific pathways could then be targeted, to eliminate this chemo-resistant cell population. With further development, our new findings could have important clinical relevance for targeting CSCs, by improving sensitivity to chemotherapy and aiding in the discovery of new therapeutics that inhibit mitochondrial biogenesis in CSCs.
Figure 6. Therapeutic targeting of chemo-resistant CSCs: A new systematic approach.
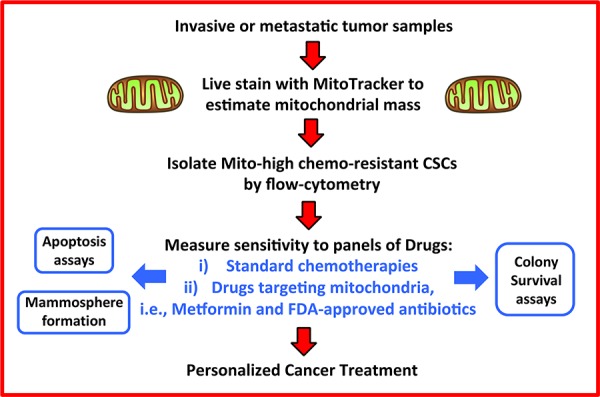
Here, we propose a new clinical strategy for over-coming drug resistance. We suggest that primary or metastatic clinical samples could be used to purify chemo-resistant “mito-high” CSCs by flow-cytometry, after live-staining with MitoTracker, to estimate mitochondrial mass. Then, these chemo-resistant CSCs would be subjected to phenotypic drug screening, with well-defined panels of i) conventional chemotherapies and/or ii) mitochondrially-targeted FDA-approved drugs (e.g., metformin and antibiotics). This would allow us to achieve the goals of personalized cancer treatment, by establishing the chemo-sensitivity profiles of mito-high CSCs in individual patients, leading to the prevention of tumor recurrence and metastasis, in multiple cancer types. Chemo-resistant “mito-high” CSCs could also be expanded by using 3D-spheroid cultures, possibly for biobanking and drug screening.
In summary, metabolic fractionation of cancer cell lines and primary metastatic breast cancer samples, identified a stem-like, mitochondrial-rich sub-population of tumor cells (Figure 7). CSCs with high mitochondrial mass showed an increased capacity for mammosphere formation, tumor initiation and were resistant to DNA-damage induced by paclitaxel. Thus, these results provide a new mitochondrially-based model for understanding the molecular relationship between CSCs and chemo-resistance.
Figure 7. Understanding the relationship between mitochondrial mass, “stemness” and chemo-resistance in cancer cells.
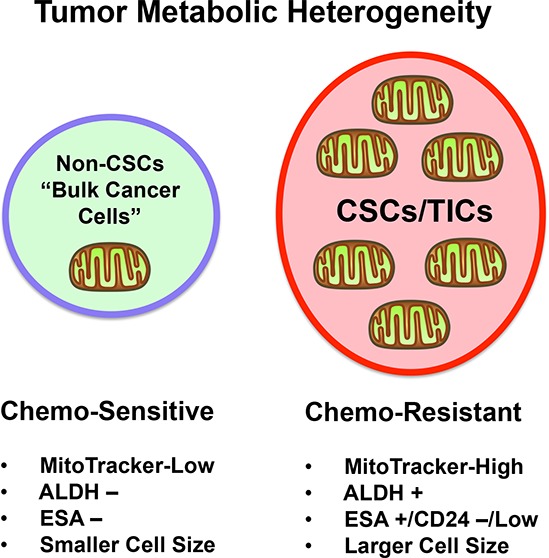
Our report suggests that high mitochondrial mass, quantified using live MitoTracker staining, is associated with a number of stem cell characteristics and biological functions including CSC markers ALDH+, ESA/CD24, large cell size, mammosphere formation in vitro, as well as tumor-initiating activity in vivo. Moreover, cells with high mitochondrial mass were preferentially resistant to paclitaxel during mammosphere formation and the comet assay (a marker DNA damage), when compared to low mitochondrial mass cells. Thus, we conclude that high mitochondrial mass can be used as a biomarker to isolate a sub-population of stem-like cancer cells that are chemo-resistant. This has important implications for better appreciating the role of tumor metabolic heterogeneity in driving chemo-resistance and treatment failure, via recurrence and metastasis.
MATERIALS AND METHODS
Cell lines and media
MCF7 and MDA MB 231 cell lines were purchased from ATCC and maintained in monolayer media—DMEM, supplemented with 10% (v/v) fetal calf serum and 200 mM L-glutamine (Sigma). Mammosphere media consisted of DMEM/F12, phenol-red-free, supplemented with 1 × B27 without vitamin A (Gibco), plus 20 ng/ml human EGF (Milteny Biotech).
Human tumor tissues
Metastatic breast cancer sample were obtained from pleural effusions or ascites. Approval for these samples was granted by the Christie Medical Research and Biobank Ethics Committees. All patients provided written informed consent.
Flow-cytometry analysis and sorting
ALDEFLUOR activity
5 × 105 cells were re-suspended in 1 ml Assay buffer and 5 μl ALDEFLUOR reagent (Stem Cell Technologies) was added and mixed well. 0.5 ml of this cell suspension was transferred into a new tube with 5 μl of DEAB regent (Stem Cell Technologies). Tubes were incubated for 30 mins at 37°C before centrifugation and re-suspension in Assay Buffer (0.5 ml) for FACS analysis (BD, LSR Fortessa). When combined with MitoTracker Deep-Red (Molecular Probes, 0.125 μl/ml), both were added before 30 min incubation. Note the PDX model BB6RC50 was also co-stained with anti-mouse MHC Class I (H-2Kd) antibody conjugated with Pacific Blue (BioLegend, 116616), to exclude mouse cells in the ALDH analysis.
Mitotracker staining
Exponentially dividing MCF7 and MDA MB 231 cells were trypsinized and re-suspended into a 1 × 106 cell/ml solution in PBS. 0.125 μl/ml MitoTracker Deep-Red (Molecular Probes) was added for 30 mins at 37°C before centrifugation and re-suspension in PBS for FACS analysis (BD, LSR Fortessa) or Sorting (BD, Aria III).
CSC cell surface markers
Exponentially dividing MDA MB 231 cells were trypsinized and re-suspended into a 1 × 106 cell/100 μl PBS. MitoTracker (0.125 μl/ml) was added to cell suspension for 30 mins at 37°C before centrifugation and re-suspension in 100 μl PBS and the addition of 10 μl ESA-FITC (DAKO, BerEP4), 10 μl CD24-PE (Pharmingen) for 10 mins at 4°C before centrifugation and re-suspension in PBS for FACS analysis (BD, LSR Fortessa. All tubes were incubated with 1 μl/ml live/dead fixable violet cell stain (Molecular Probes) to exclude dead cells from FACS analysis and sorting.
Comet assay
Alkaline comet assays were modified from [33]. MDA MB 231 and MCF7 cells were FACS sorted for the 5% highest and lowest MitoTracker expressing cells. Cells were seeded into 24 well plates (2 × 104 cells) and treated with 0.1 μM paclitaxel for 24 h. Cell were them trypsinized and resuspended into 250 μl cell media; 750 μl of low melting point agarose (1% w/v in PBS, at 40°C) was added to the cell suspensions before pipetting on to a pre-agarose coated glass slide and a glass cover slip was placed on top. After 15 min at 4°C, slides were placed in lysis buffer (2.5 M NaCl, 1 mM EDTA, 10 mM Tris, 10% DMSO, 1% Triton X-100, pH 10) and left at 4°C overnight. Slides were placed in alkaline solution (0.3 M NaOH, 1 mM EDTA, pH13) for 50 mins at 4°C and electrophoresed at 23 V for 50 mins (4°C). SYBRgold (Molecular Probes) was applied (1:10,000 in TE buffer, 10 mM Tris-Cl, pH7.5, 1 mM EDTA) to stain the DNA. Comets were imaged (Zeiss Axiovert 200 M, x10 lens) and measured using OPENCOMET free software 29 for Image J (v1.3).
Isolation of breast cancer cells from primary samples and patient derived xenografts
Breast cancer cells were isolated from pleural effusions or ascites fluid from metastatic breast cancer patients (n = 4), using the following method. The acquired samples were centrifuged at 4°C for 10 min at 1000 × g, followed by resuspension in cold PBS. Red blood cells were removed using Lymphoprep™ (Axis-Shield) and subsequently leukocytes were removed with CD45-negative magnetic sorting (Miltenyi Biotech), according to the manufacturer's instructions. Metastatic human breast cancer samples were collected from patients at The Christie NHS Foundation Trust. All patients underwent fully informed consent, in accordance with local research ethics committee guidelines (05/Q1403/159 and 05/Q1402/25). The PDX model BB6RC50 was generated from the subcutaneous injection of 1 million metastatic breast cancer cells into NOD scid gamma mice. Passage 2 tumors were removed, dissected into 2–3-mm cubed pieces and then digested for 1–2 hours at 37°C in serum-free Dulbecco's modified Eagle Medium (DMEM;Gibco) containing × 1 collagenase/hyaluronidase (STEMCELL technologies) and penicillin (100 U/mL) – streptomycin (0.1 mg/mL; Sigma). The enzymatically digested tumor was then filtered through a sterile 40-μm mesh/sieve, to obtain a single-cell suspension and washed in cold PBS.
Mammosphere culture of breast cancer cells
Mammospheres were cultured as described [30]. In brief, single cells from MCF7 and MDA MD 231 cell lines or primary metastatic samples were seeded at 500 cells per cm2 on poly-HEMA-coated plates. Mammosphere forming efficiency (MFE) was calculated as follows: [(number of mammospheres formed (≥60 μm) ÷ by the number of cells seeded) × 100].
Tumor initiation and Extreme limiting dilution assay (ELDA)
MDA MB 231 cells were stained with MitoTracker and sorted into two populations; mito-low (lowest 5% mitochondrial mass) and mito-high (highest 5% mitochondrial mass). Cells were then resuspended in 1:1 solution of mammosphere media and growth factor reduced matrigel. NOD scid gamma (NSG) mice were then subcutaneously injected with 1, 5, 10 and 50 cells per flank (n = 3 mice per group) of mito-low and mito-high MDA MB 231 cells. Tumor size was measured bi-weekly (L × W × W/2) with calipers over 9 weeks (<100 μm = no tumor initiation). The tumor initiation rate from each population was then used to calculate tumor initiating cell frequency, using the extreme limiting dilution analysis calculation: http://bioinf.wehi.edu.au/software/elda/ [32].
Statistical analysis
In vitro data is represented as the mean ± standard error of the mean (SEM), taken over ≥3 independent experiments, with ≥3 technical replicates per experiment, unless otherwise stated. Statistical significance was measured using the analysis of variance (ANOVA) test with post-hoc dunetts multiple comparisons or t-test, using Graphpad prism. P ≤ 0.05 was considered significant and all statistical tests were two-sided. All statistics were carried out under the guidance of the Medical Statistics Department, Christie Hospital NHS Trust, UK.
ACKNOWLEDGMENTS AND FUNDING
We thank the University of Manchester for providing start-up funds that contributed to the success of this study. We would like to thank Dr Robert B Clarke, Dr Sacha Howell and Dr Denis Alferez, for aiding in the acquisition of patient samples and the derivation of the PDX model. Gillian Farnie was funded via a Breast Cancer Now Research Fellowship (2008 May SF01). The Sotgia and Lisanti Laboratories were supported, in part, by funding from the European Union (ERC Advanced Grant), Breast Cancer Now, and the Manchester Cancer Research Centre (MCRC).
Abbreviations
- CSCs
cancer stem-like cells
- TICs
tumor-initiating cells
- PDX
patient-derived xenograft
- ALDH
aldehyde dehydrogenase
Footnotes
CONFLICTS OF INTEREST
None.
Author contributions
MPL initiated this collaborative project. GF performed all the experiments, analyzed the data and generated the figures with experimental data. MPL and FS wrote the first draft of the manuscript, which was extensively edited by GF, who also contributed ideas and suggestions for the discussion topics. GF also contributed heavily to the writing of the Material and Methods section and the figure legends. MPL generated the schematic summary diagrams.
REFERENCES
- 1.Zhang M, Rosen JM. Stem cells in the etiology and treatment of cancer. Current opinion in genetics & development. 2006;16:60–64. doi: 10.1016/j.gde.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast cancer research: BCR. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 4.Scopelliti A, Cammareri P, Catalano V, Saladino V, Todaro M, Stassi G. Therapeutic implications of Cancer Initiating Cells. Expert opinion on biological therapy. 2009;9:1005–1016. doi: 10.1517/14712590903066687. [DOI] [PubMed] [Google Scholar]
- 5.Xin H, Kong Y, Jiang X, Wang K, Qin X, Miao ZH, Zhu Y, Tan W. Multi-drug-resistant cells enriched from chronic myeloid leukemia cells by Doxorubicin possess tumor-initiating-cell properties. Journal of pharmacological sciences. 2013;122:299–304. doi: 10.1254/jphs.13025fp. [DOI] [PubMed] [Google Scholar]
- 6.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Molecular cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer biology & therapy. 2011;12:924–938. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Metabolic asymmetry in cancer: a “balancing act” that promotes tumor growth. Cancer cell. 2014;26:5–7. doi: 10.1016/j.ccr.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, Mitsiades CS, Matulonis UA, Drapkin R, Stone R, Deangelo DJ, McConkey DJ, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf DA. Is reliance on mitochondrial respiration a “chink in the armor” of therapy-resistant cancer? Cancer cell. 2014;26:788–795. doi: 10.1016/j.ccell.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ablett MP, O'Brien CS, Sims AH, Farnie G, Clarke RB. A differential role for CXCR4 in the regulation of normal versus malignant breast stem cell activity. Oncotarget. 2013;4 doi: 10.18632/oncotarget.1169. Advanced Online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, Bundred NJ. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst. 2007;99:616–627. doi: 10.1093/jnci/djk133. [DOI] [PubMed] [Google Scholar]
- 13.Farnie G, Willan PM, Clarke RB, Bundred NJ. Combined inhibition of ErbB1/2 and Notch receptors effectively targets breast ductal carcinoma in situ (DCIS) stem/progenitor cell activity regardless of ErbB2 status. PLoS One. 2013;8:e56840. doi: 10.1371/journal.pone.0056840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 16.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams KE, Bundred NJ, Landberg G, Clarke RB, Farnie G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem cells. 2015;33:327–341. doi: 10.1002/stem.1843. [DOI] [PubMed] [Google Scholar]
- 18.Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, Pajonk F. Metabolic state of glioma stem cells and nontumorigenic cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16062–16067. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC. Mitochondrial regulation in pluripotent stem cells. Cell metabolism. 2013;18:325–332. doi: 10.1016/j.cmet.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Burgess RJ, Agathocleous M, Morrison SJ. Metabolic regulation of stem cell function. Journal of internal medicine. 2014;276:12–24. doi: 10.1111/joim.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, Rasola A, Frasson C, Nardo G, Zulato E, Nicoletto MO, Manicone M, Indraccolo S, Amadori A. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget. 2014;5:4305–4319. doi: 10.18632/oncotarget.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, Chan M, Alhiyari Y, Dratver MB, Pajonk F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast cancer research and treatment. 2014;146:525–534. doi: 10.1007/s10549-014-3051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, Cai S, Scheeren FA, Kuo AH, Diehn M. Targeting unique metabolic properties of breast tumor initiating cells. Stem cells. 2014;32:1734–1745. doi: 10.1002/stem.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer research. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 25.Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast cancer research: BCR. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borgna S, Armellin M, di Gennaro A, Maestro R, Santarosa M. Mesenchymal traits are selected along with stem features in breast cancer cells grown as mammospheres. Cell cycle. 2012;11:4242–4251. doi: 10.4161/cc.22543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, Houvenaeghel G, Extra JM, Bertucci F, Jacquemier J, Xerri L, Dontu G, Stassi G, Xiao Y, Barsky SH, Birnbaum D, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Machado HL, Kittrell FS, Edwards D, White AN, Atkinson RL, Rosen JM, Medina D, Lewis MT. Separation by cell size enriches for mammary stem cell repopulation activity. Stem cells translational medicine. 2013;2:199–203. doi: 10.5966/sctm.2012-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G, Clarke RB. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of mammary gland biology and neoplasia. 2012;17:111–117. doi: 10.1007/s10911-012-9255-3. [DOI] [PubMed] [Google Scholar]
- 31.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Tice RR, Andrews PW, Singh NP. The single cell gel assay: a sensitive technique for evaluating intercellular differences in DNA damage and repair. Basic life sciences. 1990;53:291–301. doi: 10.1007/978-1-4613-0637-5_23. [DOI] [PubMed] [Google Scholar]
- 34.Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chemico-biological interactions. 2013;202:2–10. doi: 10.1016/j.cbi.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell cycle. 2011;10:1378–1384. doi: 10.4161/cc.10.9.15486. [DOI] [PubMed] [Google Scholar]
- 36.van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, Hamdy FC, Eaton CL, Thalmann GN, Cecchini MG, Pelger RC, van der Pluijm G. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer research. 2010;70:5163–5173. doi: 10.1158/0008-5472.CAN-09-3806. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Orlicky DJ, Matsumoto A, Singh S, Thompson DC, Vasiliou V. Aldehyde dehydrogenase 1B1 (ALDH1B1) is a potential biomarker for human colon cancer. Biochemical and biophysical research communications. 2011;405:173–179. doi: 10.1016/j.bbrc.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, Wilson C, Merchant M, Settleman J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer research. 2014;74:3579–3590. doi: 10.1158/0008-5472.CAN-13-3456. [DOI] [PubMed] [Google Scholar]
- 39.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, Weaver DT, Padval MV, Pachter JA, Xu Q. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015;75:446–455. doi: 10.1158/0008-5472.CAN-14-1223. [DOI] [PubMed] [Google Scholar]
- 41.Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Scientific reports. 2013;3:3230. doi: 10.1038/srep03230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell metabolism. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 43.LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nature cell biology. 2014;16:992–1003. doi: 10.1038/ncb3039. 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wey A, Knoepfler PS. c-myc and N-myc promote active stem cell metabolism and cycling as architects of the developing brain. Oncotarget. 2010;1:120–130. doi: 10.18632/oncotarget.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769. doi: 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Molecular and cellular biology. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rehman J. Empowering self-renewal and differentiation: the role of mitochondria in stem cells. Journal of molecular medicine. 2010;88:981–986. doi: 10.1007/s00109-010-0678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickinson A, Yeung KY, Donoghue J, Baker MJ, Kelly RD, McKenzie M, Johns TG, St John JC. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell death and differentiation. 2013;20:1644–1653. doi: 10.1038/cdd.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW, Yu SC, Qian GS. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. International journal of cancer Journal international du cancer. 2011;129:820–831. doi: 10.1002/ijc.25944. [DOI] [PubMed] [Google Scholar]
- 50.Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340–343. doi: 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell metabolism. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. Journal of translational medicine. 2013;11:94. doi: 10.1186/1479-5876-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res Treat. 2012;133:75–87. doi: 10.1007/s10549-011-1692-y. [DOI] [PubMed] [Google Scholar]
- 54.Liu P, Kumar IS, Brown S, Kannappan V, Tawari PE, Tang JZ, Jiang W, Armesilla AL, Darling JL, Wang W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. British journal of cancer. 2013;109:1876–1885. doi: 10.1038/bjc.2013.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang CW, Chen YS, Chou SH, Han CL, Chen YJ, Yang CC, Huang CY, Lo JF. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer research. 2014;74:6291–6305. doi: 10.1158/0008-5472.CAN-14-0626. [DOI] [PubMed] [Google Scholar]
- 56.Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR, Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer research. 2008;68:2813–2819. doi: 10.1158/0008-5472.CAN-08-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, Xu X, Li C, Wang RH, Lee J, Csibi A, Cerione R, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer cell. 2013;23:450–463. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu J, Lai G, Wan F, Xiao Z, Zeng L, Wang X, Ye F, Lei T. Knockdown of checkpoint kinase 1 is associated with the increased radiosensitivity of glioblastoma stem-like cells. The Tohoku journal of experimental medicine. 2012;226:267–274. doi: 10.1620/tjem.226.267. [DOI] [PubMed] [Google Scholar]
- 59.Desai A, Webb B, Gerson SL. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother Oncol. 2014;110:538–545. doi: 10.1016/j.radonc.2013.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yogev O, Yogev O, Singer E, Shaulian E, Goldberg M, Fox TD, Pines O. Fumarase: a mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS biology. 2010;8:e1000328. doi: 10.1371/journal.pbio.1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu R, Xia Y, Chen Q, Peng G, Lin SY, Lu Z. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nature cell biology. 2015;17:1158–68. doi: 10.1038/ncb3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kutuk O, Letai A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res. 2008;68:7985–7994. doi: 10.1158/0008-5472.CAN-08-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 64.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 65.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nature reviews Drug discovery. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 66.Fasolo A, Sessa C. Targeting mTOR pathways in human malignancies. Current pharmaceutical design. 2012;18:2766–2777. doi: 10.2174/138161212800626210. [DOI] [PubMed] [Google Scholar]
- 67.Tolcher AW, Papadopoulos KP, Patnaik A, Rasco DW, Martinez D, Wood DL, Fielman B, Sharma M, Janisch LA, Brown BD, Bhargava P, MJ R. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J Clin Oncol. 2015;33 abstr 11006. [Google Scholar]
- 68.Zimorski V, Ku C, Martin WF, Gould SB. Endosymbiotic theory for organelle origins. Current opinion in microbiology. 2014;22:38–48. doi: 10.1016/j.mib.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 69.Degli Esposti M, Chouaia B, Comandatore F, Crotti E, Sassera D, Lievens PM, Daffonchio D, Bandi C. Evolution of mitochondria reconstructed from the energy metabolism of living bacteria. PloS one. 2014;9:e96566. doi: 10.1371/journal.pone.0096566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer research. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee H, Park HJ, Park CS, Oh ET, Choi BH, Williams B, Lee CK, Song CW. Response of breast cancer cells and cancer stem cells to metformin and hyperthermia alone or combined. PloS one. 2014;9:e87979. doi: 10.1371/journal.pone.0087979. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget. 2015;6:4569–84. doi: 10.18632/oncotarget.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu W, Lacerda L, Debeb BG, Atkinson RL, Solley TN, Li L, Orton D, McMurray JS, Hang BI, Lee E, Klopp AH, Ueno NT, Reuben JM, Krishnamurthy S, Woodward WA. The antihelmintic drug pyrvinium pamoate targets aggressive breast cancer. PloS one. 2013;8:e71508. doi: 10.1371/journal.pone.0071508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou S, Wang F, Wong ET, Fonkem E, Hsieh TC, Wu JM, Wu E. Salinomycin: a novel anti-cancer agent with known anti-coccidial activities. Current medicinal chemistry. 2013;20:4095–4101. doi: 10.2174/15672050113109990199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai CK, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge AC, Datti A, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014;5:11029–11037. doi: 10.18632/oncotarget.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ferreri AJ, Ponzoni M, Guidoboni M, De Conciliis C, Resti AG, Mazzi B, Lettini AA, Demeter J, Dell'Oro S, Doglioni C, Villa E, Boiocchi M, Dolcetti R. Regression of ocular adnexal lymphoma after Chlamydia psittaci-eradicating antibiotic therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:5067–5073. doi: 10.1200/JCO.2005.07.083. [DOI] [PubMed] [Google Scholar]
- 79.Ferreri AJ, Ponzoni M, Guidoboni M, Resti AG, Politi LS, Cortelazzo S, Demeter J, Zallio F, Palmas A, Muti G, Dognini GP, Pasini E, Lettini AA, Sacchetti F, De Conciliis C, Doglioni C, et al. Bacteria-eradicating therapy with doxycycline in ocular adnexal MALT lymphoma: a multicenter prospective trial. Journal of the National Cancer Institute. 2006;98:1375–1382. doi: 10.1093/jnci/djj373. [DOI] [PubMed] [Google Scholar]
- 80.Chu DJ, Yao DE, Zhuang YF, Hong Y, Zhu XC, Fang ZR, Yu J, Yu ZY. Azithromycin enhances the favorable results of paclitaxel and cisplatin in patients with advanced non-small cell lung cancer. Genetics and molecular research: GMR. 2014;13:2796–2805. doi: 10.4238/2014.April.14.8. [DOI] [PubMed] [Google Scholar]