

Abstract
Through comprehensive comparison study, we found that ibrutinib, a clinically approved covalent BTK kinase inhibitor, was highly active against EGFR (L858R, del19) mutant driven NSCLC cells, but moderately active to the T790M ‘gatekeeper’ mutant cells and not active to wild-type EGFR NSCLC cells. Ibrutinib strongly affected EGFR mediated signaling pathways and induced apoptosis and cell cycle arrest (G0/G1) in mutant EGFR but not wt EGFR cells. However, ibrutinib only slowed down tumor progression in PC-9 and H1975 xenograft models. MEK kinase inhibitor, GSK1120212, could potentiate ibrutinib's effect against the EGFR (L858R/T790M) mutation in vitro but not in vivo. These results suggest that special drug administration might be required to achieve best clinical response in the ongoing phase I/II clinical trial with ibrutinib for NSCLC.
Keywords: ibrutinib, NSCLC, EGFR mutation, drug resistance, drug combination
INTRODUCTION
Ibrutinib (PCI-32765), an irreversible BTK kinase inhibitor, has been extensively studied in a variety of hematopoietic malignancies, such as mantle cell lymphoma (MCL), chronic lymphatic lymphoma (CLL), diffuse large B-cell lymphoma (DLBCL), multiple myeloma (MM), and acute myeloid leukemia (AML). Recently it was approved for the clinical treatment of MCL and CLL. [1–7] Ibrutinib has also been studied in preclinical inflammation models. [8–10] Besides its potent activity against BTK kinase, previous studies have shown that ibrutinib may also potently inhibit BMX, BLK, EGFR, and Her2 kianses. [10]
EGFR is a receptor tyrosine kinase that regulates downstream signaling cascades such as PI3K/Akt/mTOR and Ras/RAF/MEK/ERK etc. [11] Gain-of-function mutations of EGFR such as L858R, exon 19 deletion (Del 19), which account for around 90% of all EGFR mutations, can lead to tumorigenesis. [12] First generation EGFR inhibitors, such as Gefitinib and Erlotinib, have exhibited great anti-tumor activity against the EGFR (L858R) and EGFR (del 19) mutation- driven NSCLC. [13] However, the efficacy of chronic treatment with Gefitinib and Erlotinib is at best transient due to signaling bypass resistance mechanisms such as Her2 and MET amplification, or acquired EGFR gatekeeper T790M mutation. [14, 15] This has encouraged the development of second-generation EGFR inhibitors such as Apatinib (BIBW2992), Neratinib and Dacomitinib, which work through formation of a covalent bond with Cys797. [16–18] However, due to the lack of selectivity between the wt EGFR and mutant EGFR, these drugs showed dose-limiting toxicities, which led to develop third generation EGFR (T790M) inhibitors, such as WZ4002, CO-1686 and AZD9291. [19–22]
Recently ibrutinib has been reported to exhibit anti-tumor activities in EGFR mutant NSCLC and currently under phase I/II clinical trial for the previously treated NSCLC (NCT02321540). [23] Combining the previously reported biochemical activity of ibrutinib against wt EGFR and the fact that similar reactive cysteine residues are located at the same positions in EGFR and BTK, we performed a comprehensive comparison study of ibrutinib against NSCLC in vitro and in vivo. We found that ibrutinib exhibited selective anti-proliferation activity against EGFR primary mutants (L858R, del19)-expressing cancer cells but moderately active against drug induced secondary gate-keeper T790M mutation and not active against wt EGFR-expressing cells. [10] In addition, ibrutinib exhibited superior selectivity between mutant and wide type EGFR in comparison with clinical stage EGFR inhibitors such as BIBW2992, CO1686 and AZD9291. Ibrutinib could be effectively combined with MEK kinase inhibitor GSK1120212 against the EGFR secondary mutant (L858/T790M). However, these potent anti-proliferation activities could not directly transform into PC-9 and H1975 xenografts models. These results indicated that ibrutinib might be a potential drug candidate for the EGFR (L858R, del19) mutant driven NSCLC and a useful candidate for combinatorial therapy aimed at overcoming (L858R/T790M) resistance in NSCLC. But considering the discrepancies observed between the in vitro and in vivo, special administration design might be required to achieve the best drug response in the solid NSCLC tumor.
RESULTS
Ibrutinib selectively inhibits the proliferation of EGFR mutant NSCLC cancer cell lines
We first screened a panel of NSCLC cancer cell lines and found that only mutant EGFR-expressing cell lines, such as H3255 (EGFR L858R, GI50: 0.11 μM), PC-9 (EGFR Del 19, GI50: 0.05 μM), and HCC827 (EGFR Del 19, GI50: 0.063 μM), are sensitive to ibrutinib treatment. (Table 1) A similar trend was observed with other EGFR inhibitors, including BIBW2992, WZ4002, CO-1686, AZD9291 and Gefitinib. Interestingly, the H1975 cell line (EGFR L858R/T790M) was only moderately sensitive to ibrutinib (GI50: 1.2 μM) compared with other EGFR T790M mutation-targeting drugs. Unlike the other agents, ibrutinib did not exhibit any inhibitory effect against wt EGFR-expressing cancer cell lines, which suggests a superior selectivity window (Table 1). Intriguingly, a reversible version of ibrutinib (PCI-R, structure shown in Fig. S1), in which the acrylamide is replaced with a propionamide group, lost most of the activity against these otherwise sensitive cell lines. To further confirm the activity of ibrutinib against mutant EGFR-expressing cell lines, we conducted colony formation assays. The results demonstrated that for the EGFR L858R/T790M mutant-driven H1975 cell line, ibrutinib showed moderate anti-colony formation activity (EC50: 604.7 nM), which is similar to that of CO-1686 (EC50: 605.8 nM). (Table S1 and Fig. S2A) WZ4002 showed the strongest inhibitory activity (EC50: 12 nM) and AZD9291 exhibited less activity (EC50: 118 nM). AZD9291 exhibited an EC50 of 2.5 nM against the PC-9 cell line. Ibrutinib and WZ4002 showed similar efficacy (EC50: 16 nM and 11 nM respectively), while CO-1686 exhibited an EC50 of 56 nM. (Fig. S2B) AZD9291 demonstrated the strongest activity against the HCC827 cell line (EC50: 1.2 nM). WZ4002 and CO-1686 showed similar activities (EC50: 10 nM and 11 nM respectively), while ibrutinib was relatively less potent (EC50: 155 nM) (Fig. S2C) For H3255 cell line, AZD9291 displayed the strongest anti-colony formation activity (EC50: 5.5 nM). Ibrutinib and WZ4002 were moderately active (EC50: 23.4 nM and 89 nM), whereas CO1686 was relatively less active (EC50: 210.8 nM). (Fig. S2D)
Table 1. ibrutinib anti-proliferation efficacy against EGFR wt/mutant NSCLC cell lines.
| Cell LineGI50(μM) | EGFR status | BIBW-2992 | WZ-4002 | ibrutinib | PCI-R | CO-1686 | AZD-9291 | Gefitinib |
|---|---|---|---|---|---|---|---|---|
| H3255 | EGFRL858R | 0.001 | 0.12 | 0.11 | >10 | 0.25 | 0.031 | 0.0078 |
| H1975 | EGFRL858R/T790M | 0.33 | 0.3–1 | 1.2 | >10 | 0.44 | 0.037 | >10 |
| PC-9 | EGFR Del19 | 1–3nM | 0.044 | 0.05 | >10 | 0.13 | 0.003 | 0.02 |
| HCC827 | EGFR Del19 | <0.3nM | 0.019 | 0.063 | >10 | 0.097 | 0.004 | 0.0047 |
| H23 | EGFR wt | 4.4 | >10 | >10 | >10 | 1.4 | 4.2 | >10 |
| H460 | EGFR wt | 3.8 | 4.8 | > 10 | > 10 | 2.5 | 4.0 | > 10 |
| A549 | EGFR wt | 7.5 | >10 | >10 | >10 | 0.83 | 5.0 | >10 |
| H358 | EGFR wt | 2.0 | >10 | >10 | >10 | 4.8 | 9.4 | >10 |
| A431 | EGFR wt | 0.3–1 | 1.0 | >10 | >10 | 1.2 | 0.096 | 0.6 |
| H2122 | EGFR wt | 1.8 | 4.5 | >10 | >10 | 1.4 | 7.6 | >10 |
Ibrutinib displays distinct inhibitory activity against EGFR kinase biochemically and in cell
We then studied the inhibitory activity of ibrutinib against the constructed kinase domain of wt EGFR and mutant EGFR with Promega's ADP-Glo™ assay. The result showed that ibrutinib was most potent against EGFR (L858R/T790M) (IC50: 9 nM), moderately potent against EGFR (T790M) (IC50: 50 nM), however slightly less potent against wt EGFR (IC50: 96 nM). (Table 2 and Fig. S3) A similar activity trend was observed for WZ4002, CO-1686 and AZD9291. (Fig. S3) Mass spectrum study with EGFR(T790M) protein revealed that ibrutinib did inhibit EGFR kinase through formation of a covalent bond with Cys797 amino acid and further confirmed the inhibitory activity loss with PCI-R compound observed in Table 1. (Fig. S4) For auto-phosphorylation of EGFR Y1068 in intact cell lines, ibrutinib exhibited an EC50 of 23–58 nM in EGFR primary mutant cell lines (PC-9, HCC827, and H3255) and relatively weaker potency in EGFR L858R/T790M mutant cells (H1975: EC50: 145 nM). (Table 3 and Fig. S5) Interestingly, in the wt EGFR-expressing A431 cell line, the auto-phosphorylation of EGFR Y1068 was significantly inhibited by ibrutinib (EC50: 49 nM), while WZ4002 (EC50: 1436 nM), CO1686 (EC50: > 3000 nM) and AZD9291(EC50: 818 nM) were much less potent. AZD9291 displayed the strongest inhibitory activities against both the primary (L858R, Del19) and secondary drug resistant mutations (L858R/T790M) EGFR Y1068 phosphorylation, and the best selectivity between the drug resistant mutant and wt (EC50: 6 nM versus 818 nM). CO1686 was relatively less potent than AZD9291 and WZ4002, however still exhibited 100-fold selectivity between drug-resistant mutant and wt EGFR Y1068 phosphorylation. These results of inhibitory activities for EGFR Y1068 phosphorylation are in contrast to the results observed in the anti-proliferation assay performed with the A431 cell line (GI50: ibrutinib > 10 μM), which suggests that there may be off-targets effects with WZ4002 (GI50: 1.0 μM), CO1686 (GI50: 1.2 μM) and AZD9291 (GI50: 0.096 μM) that contributed to the growth inhibitory activity other than by inhibition of EGFR in A431 cell line which require further more detailed study. (Table 1) In addition, we found that auto-phosphorylation of Y1173 is in general much more sensitive than Y1068 to all of the drugs which is in accordance with the fact the phosphorylation of Y1068 phosphorylation is a prerequisite for phosphorylation of Y1173. [24]
Table 2. ADP-Glo biochemical assay of ibrutinib against purified EGFR proteins.
| Drug/Proteins (IC50: nM) | EGFR wt | EGFR (T790M) | EGFR (L858R/T790M) |
|---|---|---|---|
| ibrutinib | 96 | 51 | 3 |
| WZ4002 | 44 | 10 | 2 |
| CO-1686 | 114 | 45 | 16 |
| AZD9291 | 134 | 22 | 5 |
Table 3. ibrutinib inhibitory efficacy for EGFR Y1068/Y1173 auto-phosphorylation in intact cells.
| Drug (EC50: nM) Y1068/Y1173 | H1975 | PC-9 | HCC827 | H3255 | A431 |
|---|---|---|---|---|---|
| ibrutinib | 145/63 | 46/15 | 58/13 | 23/20 | 49/18 |
| WZ4002 | 7/4 | 77/19 | 50/36 | 42/38 | 1436/375 |
| CO-1686 | 29/7 | 123/10 | 72/36 | 250/63 | > 3000/1200 |
| AZD9291 | 6/1 | 25/24 | 22/19 | 17/5 | 818/108 |
Ibrutinib selectively affects mutant EGFR mediated signaling pathways
We then investigated the effect of ibrutinib on EGFR-mediated signaling in both ibrutinib- sensitive and ibrutinib-insensitive NSCLC cell lines. The results demonstrated that ibrutinib potently inhibited both EGFR wt/mutant auto-phosphorylation at Y1068. (Fig. 1A and Fig. S6) In sensitive cell lines (H1975, PC-9, H3255, HCC827) it also potently inhibited targets downstream of ERK. (Fig. 1A and Fig. S7A) Interestingly, it significantly affected the phosphorylation of Akt Thr308 and Ser473 in H1975 cells, however not the other three mutant EGFR-expressing cell lines. (Fig. 1A, Fig. S7B, S7C) Although it did not have apparent enzymatic activity against PI3K kinase. (Fig. S8) This is different from other reported third generation EGFR inhibitors (WZ4002, CO1686 and AZD9291) that can significantly affect the phosphorylation of AktS473 in L858R and del 19 mutant cancer cell lines. In addition, in wt EGFR-expressing NSCLC cell lines, such as H460, A549 and A431, none of the downstream mediators were affected. (Fig. S6)
Figure 1. Effect of ibrutinib on EGFR wt/mutant NSCLCs.
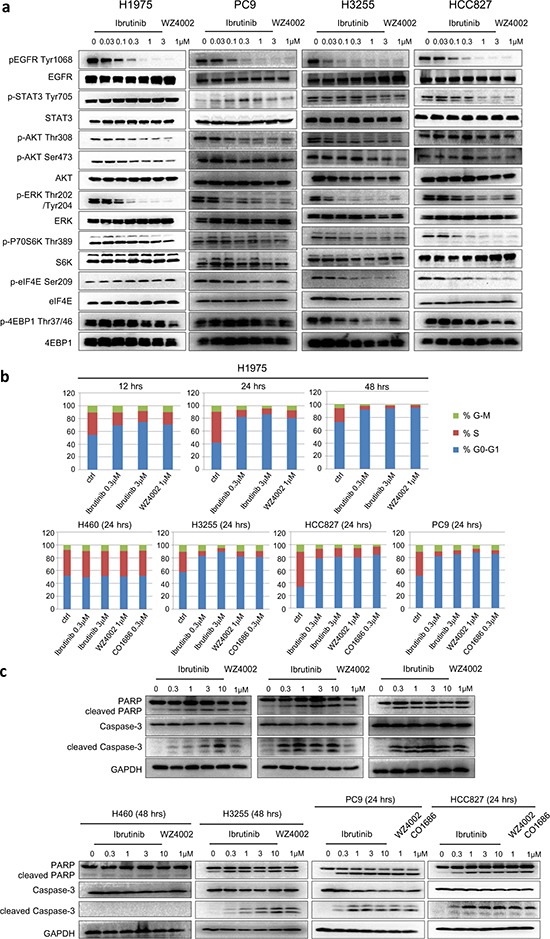
a. Ibrutinib effects on wt EGFR and mutant EGFR- mediated signaling pathways; b. Ibrutinib selectively arrests mutant EGFR-expressing NSCLC cell lines in G0/G1 phase. c. Ibrutinib selectively induces apoptosis in EGFR mutant-expressing NSCLC cell lines.
Ibrutinb selectively inhibits EGFR mutant cells lines cell cycle progression and induces apoptosis
We next investigated the effect of ibrutinib on cell cycle progression. The FACS results showed that ibrutinib could arrest H1975 cells in G0/G1 phase in a concentration- and time-dependent manner, which mimics the efficacy of WZ4002. (Fig. 1B) A similar effect was observed in PC-9, HCC827 and H3255 cell lines. (Fig. 1B and Fig. S9) However, neither ibrutinib nor WZ4002 or CO-1686 could arrest cell cycle progression in wt EGFR-expressing H460 cells. In addition, ibrutinib also demonstrated a concentration- and time-dependent induction of apoptosis in H1975 cells as evidenced by PARP and Caspase-3 cleavage. This apoptotic effect was also observed in PC-9 and H3255 cell lines, however not in the H460 cell line. (Fig. 1C) These results again validate that ibrutinib is effective against mutant EGFR-expressing cells through induction of cell cycle arrest and apoptosis, however not wt EGFR-expressing cells.
Ibrutinib slows down EGFR mutant driven NSCLC tumors progression
We next determined the anti-tumor activity of ibrutinib in subcutaneous inoculated PC-9 and H1975 xenograft mouse models. In the PC-9 model, continuous ibrutinib treatment for 22 days showed a tumor progression slow down efficacy, though there was no significant difference between the different dosages (25 mg/Kg, versus 50 mg/Kg and 100 mg/Kg BID). (Fig. 2A) However, Gefitinib and WZ4002, which have exhibited similar anti-proliferation effect for PC-9 cells in vitro, almost completely suppressed the tumor growth. In the H1975 xenograft mouse model, 23-day continuous (50 mg/kg BID) drug treatment also slowed down tumor progression. Not surprisingly, Gefitinib completely lost the anti-tumor efficacy and WZ4002 almost completely suppressed the tumor growth. Again, a higher dosage of ibrutinib (100 mg/Kg BID) did not improve efficacy. (Fig. 2B)
Figure 2. Ibrutinib anti-tumor efficacy in PC-9 and H1975 xenograft mouse models.
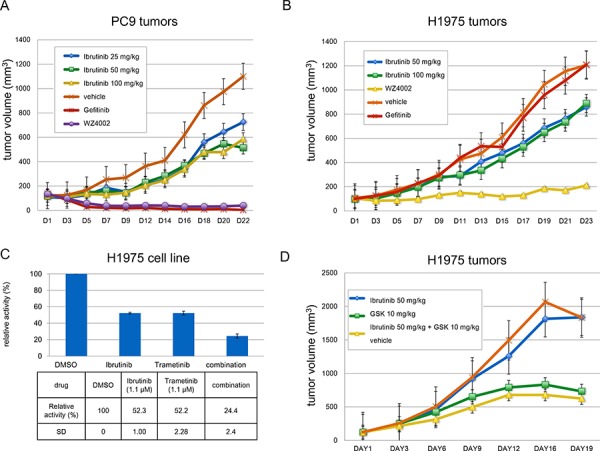
A. Ibrutinib slows down tumor progression in a PC-9 xenograft model at 25 mg/Kg, 50 mg/Kg, 100 mg/Kg BID; B. Ibrutinb slightly slowed down tumor progression in an H1975 xenograft model at 50 mg/Kg, 100 mg/Kg BID; C. Combination study with Ibrutinib and GSK1120212 showed combinatorial anti-proliferation effect against H1975 cells; D. Combination of ibrutinib (50 mg/Kg) and GSK1120212(10 mg/Kg) in H1975 xenograft model.
Ibrutinib and the MEK inhibitor GSK1120212 combinatorially suppress EGFR (L858R/T790M) mutant H1975 cells proliferation
Considering ibrutinib only exhibited moderate activity against drug acquired resistant mutant EGFR (L858R/T790M)-driven H1975 cells, we sought to explore a drug combination strategy to potentiate the growth inhibitory efficacy of ibrutinib. The EGFR gatekeeper mutant T790M causes drug resistance primarily due to the increased binding affinity to ATP but not the canonical steric hindrance. [25] Several different combination strategies have been explored, such as EGFR monoclonal antibody Cetuximab combination with EGFR inhibitor Afatinib or BIBW2992, allele-specific DNAzyme cDzT with EGFR inhibitor BIBW-2992, and MAPK signaling pathway MEK inhibitor GSK1120212 combination with EGFR inhibitor CO1686. [26–28] Given the fact that both in biochemical assays and cellular colony formation assays ibrutinib and CO-1686 have similar potencies against the EGFR T790M mutation, we then tried to combine MEK kinase inhibitor GSK1120212 with ibrutinib to see if it could potentiate the anti-proliferation efficacy of ibrutinib. The results demonstrated that the combination of Trametinib (used at around the GI50 of 1.1μM) with 1.1μM ibrutinib is able to effectively block the growth of the cells. (Fig. 2C and Table S2) However, the combination with ibrutinib (50 mg/Kg, BID) and Trametinib (10 mg/Kg, BID) did not demonstrate apparent combinatorial effect in H1975 tumor xenograft model. (Fig. 2D)
DISCUSSION
Interestingly, despite the preferred selectivity between wt EGFR and EGFR L858R/T790M shown in the in vitro biochemical assays, this trend did not translate into the in-cell auto-phosphorylation assay by looking at Y1068 site. This might be due to assay format differences, since the biochemical assay examines the inhibitory activity against downstream target phosphorylation (here is a peptide substrate), while the auto-phosphorylation assay examines the inhibitory ability to phosphorylate the target protein (EGFR) itself. The other possibility is that in biochemical assay the protein only comprises the kinase domain while in the intact cells the EGFR is full-length. The regulatory domain might play some roles in the protein conformation and activities.
Although ibrutinib exhibited highly potent anti-proliferation efficacy against PC-9 cells, it did not completely suppress the PC-9 cell inoculated tumor progression but only slowed down it. Furthermore, it did not exhibit the dose-dependent efficacy among different dosages in both PC-9 and H1975 cells mediated tumors. However, with the similar formulation, Gefintib and WZ4002 demonstrated much more superior anti-tumor activities against PC-9 and/or H1975 tumor models. One possible reason of this discrepancy between the in vitro and in vivo might be due to the formulation problems since we did not have access to the clinically used formulation of the drug. However, the other reason might be the unfavorable PK property of ibrutinib against solid tumors since it has been primarily developed against the leukemia (CLL) and lymphoma (MCL) cancers. If, then, an alternative drug formulation, such as nanomaterial-mediated controlled release, might be helpful to improve the anti-tumor efficacy, which requires further detailed study.
In summary, the anti-tumor progression efficacy combined with the already clinically validated safety profile during clinical testing as a BTK kinase inhibitor makes ibrutinib a potentially useful drug candidate for first line treatment of EGFR primary mutation- driven NSCLC. The combination of ibrutinib with other drugs as second line treatment option could also be explored to overcome the EGFR T790M induced drug resistance.
MATERIALS AND METHODS
Inhibitors
Ibrutinib, BIBW-2992, W4002, CO-1686, AZD9291, Gefinib and GSK1120212 were purchased from Haoyuan Chemexpress Inc. PCI-R was synthesized in the lab based on the procedure reported previously. [29]
Cell lines and cell culture
The human cancer cell lines A549, A431, H3255, H1975, PC-9, HCC827, H23, H460, A549, H358 and H2122 cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). H1975, PC-9, HCC827, H23, H460, H358, H2122 and EGFR mutant isogenic BaF3 cells lines were cultured in RPMI 1640 media (Corning, USA) with 10% fetal bovine serum (FBS) and supplemented with 2% L-glutamine and 1% penicillin/streptomycin. A549 and A431 were cultured in DMEM media (Corning, USA) with 10% FBS and supplemented with 2% L-glutamine and 1% pen/strep. H3255 was cultured in BEGM media (LONZA, USA) with 10% FBS and supplemented with 2% L-glutamine and 1% pen/strep.
Antibodies and immunoblotting
The following antibodies were purchased from Cell Signaling Technology (Danvers, MA): Akt (pan) (C67E7) Rabbit mAb (#4691), Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb (#4060), Phospho-Akt (Thr308) (D25E6) XP® Rabbit mAb (#13038), p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb (#4695), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb (#4370), GAPDH (D16H11) XP® Rabbit mAb, 4E-BP1 (#9644), 4E-BP1 (53H11) Rabbit mAb (#9644), eIF4E (C46H6) Rabbit mAb (#2067), Phospho-eIF4E (Ser209) (#9741), EGF Receptor (D38B1) XP® Rabbit mAb (#4267), Phospho-EGF Receptor (Tyr1068) (D7A5) XP® Rabbit mAb (#3777), Stat3 (#9132), Phospho-Stat3 (Tyr705) (D3A7) XP® Rabbit mAb (#9145), Src (36D10) Rabbit mAb (#2109), Phospho-Src Family (Tyr416) (D49G4) Rabbit mAb (#6943). Antibodies were used at 1:1000.
EGFR proteins purification for biochemical assay
A construct encoding EGFR residues 696–1022 with a GST tag was cloned into baculovirus expression vector pAcG2T. The protein was expressed by infecting SF9 cells with high titer viral stocks for 48 h. Cells were harvested and lysed in 25 mM Tris (pH 7.9), 150 mM NaCl, and 1 mM DTT. The supernatant was incubated with glutathione Sepharose beads (Genscript). After washing with wash buffer (40 mM Tris pH 7.9, 500 mM NaCl, 1% Glycerol, 1 mM DTT), the beads were incubated overnight with 5ml wash buffer containing 5ul of 5 mg/ml alpha-thrombin to remove GST tag. The eluted EGFR protein was loaded on desalt column PD-10 (GE) to change the buffer to 25 mM Tris pH7.5, 50 mM NaCl, 20 mM MgCl2, and 1 mM DTT. The protein was concentrated to 1 mg/ml and aliquots were frozen and stored at −80C.
ADP-Glo biochemical assay
The ADP-Glo™ kinase assay (Promega, Madison, WI) was used to screen Ibrutinib, WZ4002, CO1686, and AZD9291 for its EGFR inhibition effects. The kinase reaction system contains 4.95 μl EGFR (WT) (1.5 ng/μl), EGFR (T790M) (6 ng/μl) or EGFR (T790M/L858R) (1.5 ng/μl), 0.55 μl of serially diluted drug, and 5.5 μl EGFR substrate Poly (4:1 Glu, Tyr) peptide (0.2 μg/μl) (Promega, Madison, WI) with 40 μM ATP (Promega, Madison, WI). The reaction in each tube was started immediately by adding ATP and kept going for an hour under 37°C. After the tube cooled for 5 minutes at room temperature, 5 μl solvent reactions were carried out in a 384-well plate. Then 5 μl of ADP-Glo™ reagent was added into each well to stop the reaction and consume the remaining ADP within 40 minutes. At the end, 10 μl of kinase detection reagent was added into the well and incubated for 30 minutes to produce a luminescence signal. Luminescence signal was measured with an automated plate reader (Perkin-Elmer Envision) and each measurement was performed in triplicate.
Proliferation studies
Cells were grown in 96-well culture plates (2500–3000/well). For adherent cell lines, compounds of various concentrations were added into the plates after cells were cultured for 12 hours. Cell proliferation was determined after treatment with compounds for 72 hours. Cell viability was measured using the CellTiter–Glo assay (Promega, USA), according to the manufacturer's instructions, and luminescence was measured in a multi-label reader (Envision, PerkinElmer, USA). Data were normalized to control groups (DMSO) and represented by the mean of three independent measurements with standard error <20%. GI50 values were calculated using Prism 5.0 (GraphPad Software, San Diego, CA).
Cell cycle analysis
H1975 cells were treated with DMSO, PCI32765 (0.3 μM, 3 μM), WZ4002 (0.3 μM) for 12, 24, 48 hours before cells were harvested by trypsin and washed with cold PBS. HCC827, PC9, H460 and H3255 cells were treated with DMSO, PCI32765 (0.3 μM, 3 μM), WZ4002 (0.3 μM), CO1686 (0.3 μM) for 24 hours before cells were harvested by trypsin and washed with cold PBS. The cells were fixed in 70% cold ethanol and incubated at −20°C overnight then stained with PI/RNase staining buffer (BD Pharmingen). Flow cytometry was performed using a FACS Calibur (BD), and results were analyzed by ModFit software.
Signaling pathway effect examination
H1975, HCC827, PC9 and H3255 cells were treated with serially diluted PCI32765, WZ4002 (1 μM) for 4 hours. Cells were then washed with PBS and lysed in cell lysis buffer. Phospho-EGF Receptor, Phospho-EGF Receptor (Tyr1068), Stat3, Phospho-Stat3 (Tyr705), AKT, Phospho-AKT (Ser473), Phospho-AKT (Thr308), p44/42 MAPK (Erk1/2), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), p70 S6 Kinase, Phospho-p70 S6 Kinase (Thr389) Src, Phospho-Src Family (Tyr416), eIF4E, Phospho-eIF4E (Ser209), 4E-BP1, Phospho-4E-BP1 (Thr37/46) antibody (Cell signaling Technology) were used for immunoblotting.
Apoptosis effect examination
H1975 cells were treated with serially diluted PCI32765, WZ4002 (1 μM) for 8, 18, 24 hours. H460 cells were treated with serially diluted PCI32765, WZ4002 (1 μM) for 48 hours. PC9, HCC827, H3255 were treated with serially diluted PCI32765, WZ4002 (1 μM) for 24 hours. Cells were then washed in PBS and lysed in cell lysis buffer. PARP, Caspase-3, GAPDH antibody (Cell signaling Technology) were used for immunoblotting.
Mass spectrum experiment
The purified EGFR protein (30 μg protein in 40 μL buffer of 50 mM Tris, 50 mM NaCl and 20 mM MgCl2) was incubated with DMSO or Ibrutinib (50 μM) for 60 min at room temperature. The samples were then subjected to in-solution trypsin digestion. Briefly, the samples were denatured in 8 M urea (160 μl of 10 M urea in PBS was added), reduced by 10 mM of dithiothreitol for 30 min at room temperature (10 μl of 200 mM stock in water was added) and alkylated by 20 mM iodoacetamide for 30 min at room temperature in dark (20 μl of 100 mM stock in water was added). The samples were diluted with ammonium bicarbonate (25 mM, 570 μl) to 2 M urea and subjected to trypsin digestion (Promega; 4 μl of 0.5 μg/μl) overnight at 37°C in the presence of 2 mM CaCl2. Digested peptide samples were desalted and re-solubilized in 20uL Buffer A (95% water, 5% acetonitrile, 0.1% formic acid). 2 uL of each sample was analyzed by LC-MS/MS on a Velos Pro Orbitrap Elite mass spectrometer (Thermo Scientific) coupled to an Easy NanoLC. Peptides were eluted from the C18 column using a 30 min gradient of 2–75% Buffer B (100% acetonitrile, 0.1% formic acid). The flow rate through the column was 225 nL/min and the spray voltage was 2.2 kV. The LTQ-Orbitrap was operated in data-dependent scanning mode, with one full MS scan (375–1600 m/z) in the orbitrap followed by MS/MS scans of the 5 most abundant ions using the linear ion trap with dynamic exclusion enabled. The MS data was analyzed by Mascot v2.3.02 using a Uniprot Human sequence database with two differential modifications: Carbamidomethyl modification of 57.02146 on Cysteine and ibrutinib modification of 440.1961 on Cysteine. For the T790M mutant only, an additional “Thr to Met” modification on Threonine was included. For each of the wild-type and mutant samples, the LC-MS/MS analysis was repeated twice with consistent results.
Colony formation assay
The clonogenic activity of the cells was determined by colony forming assays. Every six conditions of drugs were analyzed (10 μM, 1 μM, 0.1 μM, 0.01 μM, 0.001 μM, DMSO). The cells (H1975, PC-9, HCC827, H3255) were seeded at 600 cells/well in 2 ml medium in 6-well plates. Cells were maintained in a humidified 5% CO2 incubator at 37°C for 10 days without changing medium. After 10 days, the assays were stopped and the cells were fixed with 3.5% formaldehyde and 70% ethanol and subsequently stained with 0.02% crystal violet. The total number of colonies exceeding 50 cells per colony was counted and data were given as percentage with respect to the controls.
Xenograft experiment
Xenograft models. 5 × 106 (PC-9) or 2 × 106 (NCI-H1975) cells (Per 100ul in PBS) were injected subcutaneously into the right flanks of six-week-old female Nude (Nu/Nu) mice (Charles River Laboratories). Animals with 100 mm3 tumors were then randomized into groups of 8~10 animals, and dosed twice daily (BID) with Ibrutinib (25, 50, or 100 mg/kg/day) or administrated once daily (QD) with Gefitinib (25 mg/kg/day) or WZ4002 (25 mg/kg/day) by oral gavage for about 3 weeks. Tumor diameter was measured 3 times weekly. Tumor volumes (V) were calculated by the equation (V = length × width × width/2). ALL experiments and procedures followed the guidelines of the Institutional Animal Care and Use Committees of National Institue of Biological Sciences, Beijing.
Supplemental information
Supplemental Information includes Supplemental Experimental Procedures, nine figures, and two tables and can be found with this article are online available.
SUPPLEMENTARY MATERIALS
Acknowledgments
J. Liu, Q. Liu, W. Wang and L. Cheng are supported by the grant of “Cross-disciplinary Collaborative Teams Program for Science, Technology and Innovation (2014–2016)” from Chinese Academy of Sciences. N. Gray is supported by NIH grant P01 CA154303-03. We want to thank China “Thousand Talents Program” support for Q. Liu and C. Wang; “Hundred Talents Program” of The Chinese Academy of Sciences support for J. Liu, and W. Wang. Z. Zhao is supported by Anhui Province Natural Science Foundation Annual Key Program (grant number: 1301023011), C.-H.Y. is funded by the National Science Foundation of China (No. 31270769) and the National Basic Research Program of China (973 Program, No. 2012CB917202). Q. Liu is also supported by Scientific Research Grant of Hefei Science Center of CAS (SRG-HSC # 2015SRG-HSC022). The authors thank the mass spectrometry facility of National Center for Protein Sciences at Peking University for assistance with data collection and Dr. Rong Meng for technical support.
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Herrera AF, Jacobsen ED. Ibrutinib for the treatment of mantle cell lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:5365–5371. doi: 10.1158/1078-0432.CCR-14-0010. [DOI] [PubMed] [Google Scholar]
- 2.Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA, Smith LL, Dubovsky JA, Towns WH, MacMurray J, Harrington BK, Davis ME, Gobessi S, Laurenti L, Chang BY, Buggy JJ, et al. Bruton's tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL) Blood. 2014;123:1207–1213. doi: 10.1182/blood-2013-07-515361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, Hsi ED. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. British journal of haematology. 2015;168:765–8. doi: 10.1111/bjh.13149. [DOI] [PubMed] [Google Scholar]
- 4.Rushworth SA, Murray MY, Zaitseva L, Bowles KM, MacEwan DJ. Identification of Bruton's tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood. 2014;123:1229–1238. doi: 10.1182/blood-2013-06-511154. [DOI] [PubMed] [Google Scholar]
- 5.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. The New England journal of medicine. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. The New England journal of medicine. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cellular signalling. 2013;25:106–112. doi: 10.1016/j.cellsig.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Hsu J, Gu Y, Tan SL, Narula S, DeMartino JA, Liao C. Bruton's Tyrosine Kinase mediates platelet receptor-induced generation of microparticles: a potential mechanism for amplification of inflammatory responses in rheumatoid arthritis synovial joints. Immunology letters. 2013;150:97–104. doi: 10.1016/j.imlet.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, Robinson WH, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis research & therapy. 2011;13:R115. doi: 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocrine-related cancer. 2001;8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 12.Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap) Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2013;24:2371–2376. doi: 10.1093/annonc/mdt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giaccone G. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell lung cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:3235–3242. doi: 10.1200/JCO.2005.08.409. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 15.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smaill JB, Rewcastle GW, Loo JA, Greis KD, Chan OH, Reyner EL, Lipka E, Showalter HD, Vincent PW, Elliott WL, Denny WA. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido [3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. Journal of medicinal chemistry. 2000;43:1380–1397. doi: 10.1021/jm990482t. [DOI] [PubMed] [Google Scholar]
- 18.Ou SH. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Critical reviews in oncology/hematology. 2012;83:407–421. doi: 10.1016/j.critrevonc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer discovery. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer discovery. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yap TA, Popat S. Toward precision medicine with next-generation EGFR inhibitors in non-small-cell lung cancer. Pharmacogenomics and personalized medicine. 2014;7:285–295. doi: 10.2147/PGPM.S55339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao W, Wang M, Wang L, Lu H, Wu S, Dai B, Ou Z, Zhang L, Heymach JV, Gold KA, Minna J, Roth JA, Hofstetter WL, Swisher SG, Fang B. Selective antitumor activity of ibrutinib in EGFR-mutant non-small cell lung cancer cells. Journal of the National Cancer Institute. 2014;106 doi: 10.1093/jnci/dju204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends in cell biology. 2006;16:649–656. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, Koutcher JA, Spassova M, Ouerfelli O, Mellinghoff IK, Zakowski MF, Politi KA, Pao W. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. The Journal of clinical investigation. 2009;119:3000–3010. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson ML, Riely GJ, Rizvi NA, Azzoli CG, Kris MG, Sima CS, Ginsberg MS, Pao W, Miller VA. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2011;6:1128–1131. doi: 10.1097/JTO.0b013e3182161508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai WY, Chen CY, Yang SC, Wu JY, Chang CJ, Yang PC, Peck K. Overcoming EGFR T790M-based Tyrosine Kinase Inhibitor Resistance with an Allele-specific DNAzyme. Molecular therapy Nucleic acids. 2014;3:e150. doi: 10.1038/mtna.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.