

Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor mediating the toxicity and tumor-promoting properties of dioxin. AHR has been reported to be overexpressed and constitutively active in a variety of solid tumors, but few data are currently available concerning its role in thyroid cancer. In this study we quantitatively explored a series of 51 paired-normal and papillary thyroid carcinoma (PTC) tissues for AHR-related genes. We identified an increased AHR expression/activity in PTC, independently from its nuclear dimerization partner and repressor but strictly related to a constitutive active MAPK/ERK pathway. The AHR up-regulation followed by an increased expression of AHR target genes was confirmed by a meta-analysis of published microarray data, suggesting a ligand-independent active AHR pathway in PTC. In-vitro studies using a PTC-derived cell line (BCPAP) and HEK293 cells showed that BRAFV600E may directly modulate AHR localization, induce AHR expression and activity in an exogenous ligand-independent manner. The AHR pathway might represent a potential novel therapeutic target for PTC in the clinical practice.
Keywords: papillary thyroid cancer, aryl hydrocarbon receptor, BRAF, gene expression, meta-analysis
INTRODUCTION
Thyroid cancer is considered the most common endocrine malignancy and the incidence of papillary thyroid carcinoma (PTC), its most frequent histologic subtype - representing approximately 80% of all thyroid cancers - [1], is rapidly increasing [2].
About 36%-69% of PTCs harbor activating mutations in the BRAF gene (v-raf murine sarcoma viral oncogene homolog B1), which encodes a member of the raf/mil family of serine/threonine kinases. BRAF functions to regulate the MAPK/ERK pathway transducing extracellular stimuli to the nucleus [3]. Active BRAF phosphorylates and activates MEK1/2 beginning a kinase cascade that, through ERK1/2, signals for ligand- and cell-specific responses. More than 90% of the BRAF mutated PTCs are characterized by a T1799A transversion that results in the V600E aminoacidic substitution (BRAFV600E) within the activating domain of the protein [4]. Disrupting hydrophobic interactions, BRAFV600E enables the protein to fold into a catalytically active form with a nearly 500-fold increased kinase activity [5]. This event leads to the activation of the downstream signaling cascade in the absence of extracellular stimuli, allowing the cell to become self-sufficient in growth signals within this pathway [6]. In transgenic mice, BRAFV600E induces the development of thyroid cancer with high penetrance and short latency, thus suggesting that BRAF mutations may function as the initial transforming event during thyroid tumor development [7]. All these data support the central role of BRAFV600E and MAPK signaling pathway in transformation in PTC, however the mechanism of concomitant activation of different signaling pathways by BRAFV600E and their effects in thyroid cancer are not fully elucidated.
Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that mediates the effects of many environmental pollutants, including polycyclic aromatic hydrocarbons (PAH) and 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), through the induction of several phase I (e.g. CYP1A1, CYP1B1) and II biotransforming enzymes (e.g. UDP-glucoronosyl transferase UGT1A6, NADPH-quinone-oxidoreductase, NQO1) [8]. In its ligand-free inactive form, the AHR is a cytosolic protein complexed to two HSP90 molecules, the HSP90-interacting protein p23 and the AHR-interacting protein AIP [9]. Upon ligand binding, AHR translocates to the nucleus where it heterodimerizes with the aryl hydrocarbon nuclear translocator (ARNT). This active complex binds to xenobiotic-response elements (XRE) located in the enhancer/promoter regions, thus regulating the expression of target genes [8].
AHR has long been studied for its role in mediating TCDD toxicity [10]. However, its involvement in many other biological processes, including development, immunity and cancer biology, is strongly emerging [8, 11, 12]. AHR is overexpressed and constitutively active in a variety of tumor types, in cancer cell lines and in tumors from animal models, where it mostly shows a pro-oncogenic role [12]. A recent study on multiple cell lines from the Cancer Cell Line Encyclopedia reported relative high level of AHR mRNA in some cellular models of solid tumor (e.g. pancreatic, liver and chondrosarcoma derived cell lines); low levels were instead detected in many leukemia subtypes [13]. More recently, investigating the role of genetic events responsible for the onset of the thyroid cancer in acromegaly, we have shown that AHR is selectively overexpressed in a small cohort of PTC compared to the normal surrounding tissue. We found, in addition, that such increase was more marked in PTC samples harboring BRAFV600E, irrespective of acromegaly status [14]. Given the proven cross-talk between AHR and MAPK pathway [15] and the role of AHR in modulating growth and migration of cancer cells [12], in the present work we aim to confirm previous associations between the BRAFV600E and AHR overexpression in a large, independent cohort of patients with PTC. In addition, cellular models of PTC are used to systematically analyze the expression levels of other components of the AHR signaling pathway, and to better elucidate the molecular link between BRAFV600E and AHR in thyroid tumor.
RESULTS
AHR, AHRR and ARNT expression in tumor and paired normal tissue
To ascertain the possible role of the components of the AHR pathway in the pathogenesis of PTC, the steady-state levels of AHR, AHRR and ARNT mRNAs in tumoral specimens and paired normal tissues were evaluated by quantitative Real-time PCR (qPCR). The mean expression levels of AHRR (2.27 ± 1.80 vs 1.81 ± 2.06, p = 0.065) and ARNT (1.68 ± 0.70 vs 1.62 ± 0.89, p = 0.843) were similar in tumoral and paired normal tissue from 51 PTC, while a significant increase of AHR (3.36 ± 2.83 vs 2.08 ± 1.84, p < 0.0001, Figure 1A) was observed.
Figure 1. AHR expression in PTC.
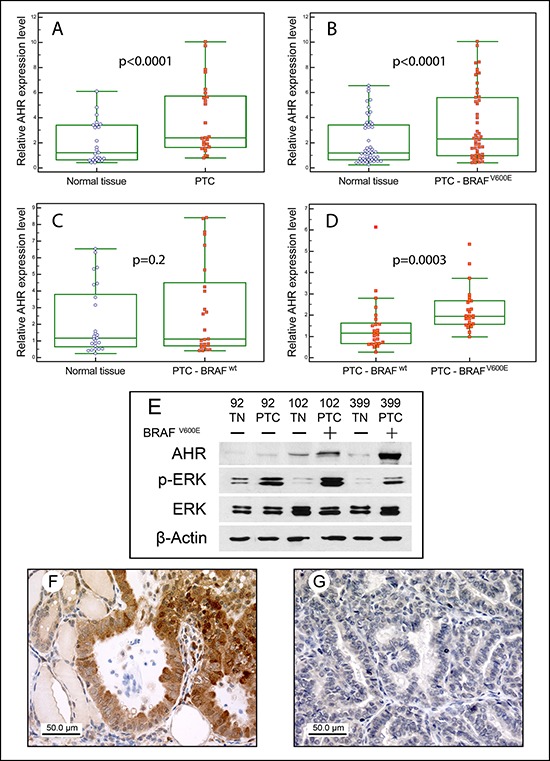
Box plots of relative qPCR gene expression measurements of AHR in either all PTCs A. only the BRAFV600E B. or BRAFwt C. PTCs and the relative paired normal tissues. Each value was referred to a pool of normal thyroid tissues that was set to 1. In D. the AHR expression of BRAFV600E vs BRAFwt PTCs is shown. For each sample the reported value represents the fold increase in tumoral specimen compared to its normal counterpart, which was set to 1. Boxes indicate the range from lower to upper quartile values, with the line inside the box representing the median. The vertical lines mark the highest and lowest value observed within a distance of 1.5 times the inter-quartile range from the bottom and the top of the boxes, respectively. Each dot represents a single sample. E. Representative Western blot analysis of normal/tumoral match-pair samples for the expression of AHR, phospho-ERK and total ERK. Samples were corrected for protein loading by β-Actin and the BRAF mutational status was reported on the top. AHR immunostaining in BRAFV600E F. and in BRAFwt G. PTC samples. Normal thyroid areas surrounding cancer cells are also visible. Original magnification x40.
PTCs were then grouped according to the BRAF mutational status, being mutated in 49% (25/51) of cases, and possible differences in term of AHR expression were investigated. As shown in Figure 1 (Panels B and C), significant differences between tumoral and normal tissues could be observed in the BRAF mutated PTCs (mean of differences 1.88, 95% CI: 1.16–2.59, p < 0.0001), but not in those carrying the wild-type form (0.69, 95% CI: 0.17–1.55, p = 0.20). Moreover, as expected, BRAFV600E cases expressed AHR at a higher level than the BRAFwt (2.27 ± 1.01 vs 1.44 ± 1.21, p = 0.0003, Figure 1D). Conversely, we could not find any correlation when samples were grouped according to RAS genes mutational status.
Parallel western blot experiments testing AHR expression in normal/tumoral match-pair samples showed a good positive correlation between AHR mRNA and protein expression. This confirms that the difference in AHR expression between PTC and its normal paired tissue was significantly higher in tumors carrying the BRAFV600E compared to BRAFwt PTC (Figure 1E).
AHR protein expression was then evaluated in tumoral specimens by immunohistochemistry. As shown in Figure 1F and in accordance with a previous report [14], a weak cytoplasmic and nuclear AHR immunostaining was observed in normal thyroid areas surrounding the neoplastic tissue. In BRAFV600E PTC cases a strong and homogeneous cytoplasmic AHR staining has been detected. Conversely, BRAFwt PTC samples displayed lower cytoplasmic or absent AHR staining (Figure 1G).
To further validate the AHR expression trend observed in our series, and to establish if this might be associated to an increased AHR signaling, a large-scale meta-analysis of microarray data in the public domain has been performed. Gene expression data from five previously published microarray datasets were retrieved from GEO and integrated with a custom methodology. Patients’ tissue samples included normal thyroid (NT, n = 73), follicular thyroid carcinoma (FTC, n = 14), poorly differentiated thyroid carcinoma (PDTC, n = 4), PTC (n = 128) and anaplastic thyroid carcinoma (ATC, n = 22). Expression data showed an upregulation of AHR in PTC compared to normal thyroid tissues (p < 0.0001, Supplemental Figure 1A), while ARNT did not show significant differences between the two groups. We then evaluated the expression of phase I (e.g. CYP1A1 and CYP1B1) and phase II (e.g. NQO1) biotransforming enzymes. A four-fold increase of CYP1B1 was observed in PTC compared to NT (Supplemental Figure 1B), while only a slight 1.5-fold increase was observed for NQO1 (p < 0.0001) (Supplemental Figure 1C). In contrast, CYP1A1 did not significantly change between the two groups (Supplemental Figure 1D). Clinical meta data associated to expression profiles were further analyzed to assess if AHR transcription expression may be a function of thyroid disease progression or histotype. In spite of the different groups’ size and the lack of a statistical significance, in FTC, AHR expression was lower than NT, while ATC showed expression levels higher than NT but similar to PTC (Supplemental Figure 2).
To correlate clinical phenotype with AHR expression, the 51 PTC patients in our series were divided into 2 groups (AHR high- or low-expressing PTCs) according to the receptor median expression values. The clinico-pathological features of the 51 PTC patients were summarized in supplemental Table 1. At the end of a median follow-up of 65 months, 80% of patients were cured, 16% showed a persistent disease, while 4% deceased during the follow-up. Although PTC patients with high AHR expression levels showed preferentially low (I and II) than high (III and IV) PTC stages (68% vs 32%) – low AHR expressing cases are instead equally shared between the two groups – no significant correlation between AHR expression and patients’ age, tumor size, lymph node and distant metastases, and poor outcome could be established.
AHR expression in thyroid cell lines and effect of BRAFV600E on AHR expression
AHR expression levels were then assessed in four thyroid cell lines, all but one (TT) carrying the BRAFV600E in either a homozygous (BCPAP) or heterozygous state (K1 and 8505C). As shown in Figure 2, 8505C showed the highest expression both at mRNA and at protein level. AHR was instead detected at lower but similar protein levels in BCPAP and K1, while apparently no expression was found in TT cell line.
Figure 2. AHR expression in thyroid tumors derived cell lines.
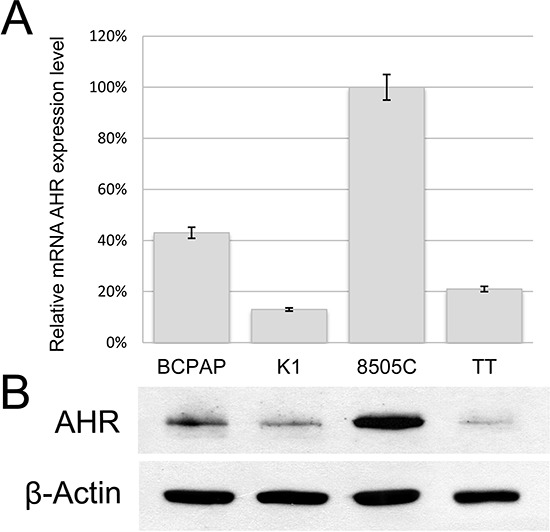
The PTC-derived BCPAP and K1, the medullary thyroid cancer cell line TT, and the 8505C established from an undifferentiated thyroid carcinoma were evaluated for AHR expression at both mRNA A. and protein level B. Samples were corrected for protein loading by β-Actin. Error bars represent standard deviations.
To exclude that the association between BRAFV600E and AHR overexpression was fortuitous and to ascertain if an active BRAF protein up-regulates AHR, the BRAFwt HEK293 cells, were co-transfected with a human BRAFV600E and a reporter plasmid in which luciferase expression is driven by AHR, through XRE elements. The constitutively active BRAFV600E induces a significant increase in luciferase activity compared to mock transfected cells (44% ± 13%, p < 0.01, Figure 3A). Interestingly, this positive stimulus could be reversed by the kinase inhibitor SB590885 acting on both the exogenous BRAFV600E and/or the endogenous BRAF, in a dose dependent manner (Figure 3A). To establish the reason for such increase, quantitative and qualitative evaluations of BRAFV600E effect on AHR expression and/or localization were performed. BRAFV600E apparently does not induce AHR expression (Figure 3B), but rather a tendency to increase nuclear translocation (Figure 3C).
Figure 3. Effect of BRAFV600E on AHR expression/activity in HEK293 cells.
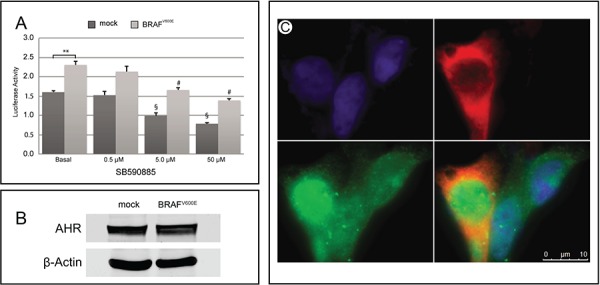
A. HEK293 cells were co-transfected with the XRE-luc reporter plasmid and either the human Flag-tagged BRAFV600E plasmid or the empty vector pcDNA3.1 and treated with increasing concentration of the BRAF inhibitor SB590885. The relative activity was adjusted for transfection efficiency using pRL-TK. Error bars represent standard deviation. ** reflects p < 0.01 value for luciferase activity in mock versus BRAFV600E transfected cells at basal level; # § reflect p < 0.01 values for treated versus untreated cells. B. Representative Western blot analysis of AHR in HEK293 cells transfected with either BRAFV600E or pcDNA3.1. β-Actin was used to normalize for loading variations. C. Immunofluorescence staining of Hoechst (blue), Flag (red) and AHR (green) in BRAFV600E transfected HEK293. Only one of three cells in the picture is positive for Flag-tagged BRAFV600E, for which an increased in nuclear AHR could be observed. All experiments were performed at least in triplicate.
To further study the effect of BRAF inhibitors on AHR expression in thyroid-originating tumor cells, the BRAFV600E carrying cell lines were treated with RAF265 or SB590885, which exert their inhibitory activity with more potency towards BRAF active conformation than the inactive one [4]. As expected p-ERK was strongly reduced in all cell lines after both treatments. In the BCPAP the inhibited BRAF activity is followed by a significant decrease of AHR expression (Figure 4A) and activity (Figure 4B), while no effect could instead be observed in 8505C and K1 (Figure 4A).
Figure 4. Effect of BRAF inhibitors on AHR expression and activity in thyroid cell lines.
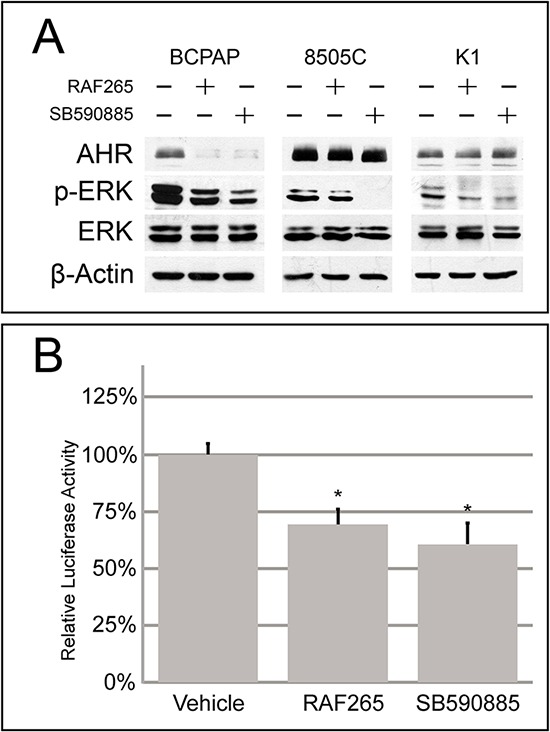
A. Representative Western blot analysis of BCPAP, 8505C and K1 cells treated with IC50 doses of RAF265 and SB590885 for the expression of AHR, phospho-ERK and total ERK. Samples were corrected for protein loading by β-Actin. B. Relative luciferase activity of BCPAP cells transfected with XRE-luc and treated with RAF265 and SB590885 with the same concentration as above. Error bars represent standard deviation *p < 0.05, compared with a group given no drug treatment (one-way ANOVA). All experiments were performed at least in triplicate.
DISCUSSION
AHR has been considered for years as a major regulator of xenobiotic-induced carcinogenesis. Now it is becoming widely recognized that AHR and its abnormal expression play an important role in multiple stages of tumor development and progression. Several recently published works, demonstrated that even in the absence of exogenous ligands, AHR is overexpressed and constitutively active in a variety of human and/or rodent tumors including breast cancer, lung adenocarcinoma, pancreatic and prostate cancer [12, 16, 17].
In the present work, we clearly demonstrated in a large series of PTC – and further confirmed by data emerging from a large-scale meta-analysis of microarray studies in thyroid tumors – an increased expression/activity of AHR, independently from its nuclear dimerization partner ARNT and its repressor AHRR, but strictly related to a constitutive active MAPK/ERK pathway.
A strong body of evidences supports the mutual interaction between the AHR and MAPK/ERK pathways [15, 18]. Our finding that BRAFV600E mutation modulates AHR levels, localization and activity in PTC in an exogenous ligand-independent manner further reinforces this concept. Previous reports demonstrated that typical AHR activators (e.g. TCDD, benzo[a]pyrene) trigger MAP kinases at different levels and in different cellular systems [15, 18]. On the other hand, MAPK are key players in regulating AHR function and stability [19–21]. Changes in the AHR levels and the enhanced TCDD-initiated transactivation potential of the receptor have been indeed observed in cell overexpressing constitutively active ERK1 or MEK1 [20, 22]. Moreover, AHR is significantly expressed in a large subset of N-RAS mutated cell lines whose sensitivity to the MEK inhibitor PD0325901 positively correlates with the receptor expression [13]. The direct or indirect involvement of serine/threonine kinases in AHR function regulation is further strengthened by the observation that phosphorylation of AHR co-chaperons HSP90 and of AHR-associated ancillary proteins, including ARNT, modulates the formation of a functional cytosolic AHR multicomponent complex [23] and/or potentiates the transcriptional activity of AHR/ARNT complexes [20].
In most tumoral specimens in our series, AHR was abundantly expressed in the cytoplasm of papillary thyroid cancer cells and was instead absent in the normal adjacent tissue. Although our data do not permit to distinguish whether the BRAFV600E either induces an increased expression/stability or reduces AHR degradation, by analogy with ERK phosphorylation, we suggest that BRAFV600E might stabilize the AHR, increase its nuclear uptake and protect it from proteasome digestion [24]. As expected, cytosolic AHR overexpression may be associated to the consequent enhancement of AHR-regulated downstream gene expression induction [25]. Accordingly, by a large-scale meta-analysis of published microarray data we observed an increased phase I (CYP1B1) and phase II (NQO1) biotransforming enzymes, supporting the idea that in PTC cells AHR can potentially heterodimerize with ARNT in the absence of ligands.
Three different aspects emerging from our cellular studies further support the inductive role of BRAFV600E on AHR expression/activity. 1- AHR is expressed in 8505C, BCPAP and at lower level in K1 all of them carrying the BRAFV600E, while it is barely detectable in the BRAFwt TT cells. Moreover, in HEK293 with a constitutively active MAPK pathway, the AHR activity was significantly increased. 2- The positive stimulus of BRAFV600E on AHR can be significantly reverted by kinases inhibitors in both BCPAP and HEK293. By specifically blocking BRAF, RAF265 and SB590885 therefore not only reduce cell proliferation and promote apoptosis in BCPAP as we previously demonstrated [4], but also inhibit AHR expression/activity pathway. Additional data could be provided by the use of selective MEK inhibitors. However, the possible concomitant effects of these drugs on AHR [26, 27], prevents their use for studying the effects of blocking the MAPK/ERK pathway downstream of BRAF on AHR activity. In K1 and 8505C cell lines, however, the inhibition of ERK phosphorylation is not accompanied by a decrease in AHR expression. The reason of this uncoupling is unclear, however it is reasonable to hypothesize that other molecular pathways may sustain AHR expression. Further studies would help clarifying this point. 3- The introduction of a constitutive active BRAFV600E in HEK293 induces AHR activity. This effect seems not to reflect an increase in AHR expression as observed in PTC tumoral specimens, but more likely an increased nuclear shuttling. This may reflects the tissue-specific regulation of AHR localization [28].
Both AHR and CYP1B1 represent potential targets for chemoprevention in several cases. Some AHR agonists inhibit the growth of pancreatic cancer cells expressing AHR at high levels [29]. Many flavonoids may prevent or improve tumorigenic outcomes by reducing AHR and/or CYP1A1/CYP1B1 activity, or by preventing PAH-induced genotoxicity [30]. CYP1B1 represents a therapeutic target also for potential anticancer drugs that can be metabolically activated by this biotransforming enzyme [29]. For instance 3,4-Methylenedioxy-3′,4′,5′-trimethoxy chalcone, has shown promise in the treatment and prevention of gastrointestinal tumors in mouse models [31]. Our data raise hence the possibility to consider AHR and CYP1B as possible novel therapeutic targets also for PTC. Anyway, the lack of any link between AHR expression and poor prognostic factors in our PTC series, together with the lack of any correlation with more de-differentiated histotypes from meta data analysis, prompt us to consider AHR as one of initial mediators of thyroid cancer development endorsed by BRAF, rather than a driver of the disease progression process.
In conclusion, this study supports end extends our previous findings concerning the relationship between BRAFV600E and AHR expression in human PTC [14]. Indeed, it provides the first evidence that BRAFV600E activating the MAPK/ERK signaling is able to upregulate the AHR pathway in these tumors, suggesting that targeting the AHR pathway might have potential therapeutic benefit in their clinical management. It raises, however also novel relevant issues we aim to clarify in the next future, including the role of the proteasome in the high AHR expression in mutated PTCs and the nature of the interaction between BRAF and AHR (i.e. direct or mediated through other proteins). Finally, as BRAF somatic mutations characterize other human malignancies including colon cancer and melanoma, future research should clarify if the BRAF-mediated AHR activation is peculiar of PTC or a more general mechanism.
MATERIALS AND METHODS
Patients, DNA extraction and mutation analysis
From 2010 to 2013, we collected a series of paired-normal and PTC tissues from 51 patients: 17 males and 34 females with a mean age 46 years (range 5–69) with a median follow-up of 65 months (range 15–79). All patients were treated with total thyroidectomy for PTC, and therapeutic neck dissection was performed in patients with standard indications. According to the 6th TNM classification 29 were stage I, 2 were stage II, 14 were stage III and 6 were stage IV. Follow-up or survival time was defined as the time from the initial surgical treatment to patient's death due to PTC or to the most recent clinic visit. Patients gave written informed consent for their thyroid tissues to be used for research purposes. The local ethical committee approved the study.
DNA was extracted from frozen tissues after surgery using the DNeasy Blood and Tissue kit (Qiagen, Italy), according to the manufacturer's protocol. Mutation analyses were performed for BRAF (NM_004333.4), N-RAS (NM_002524.3; exons 2 and 3), K-RAS (NM_033360.2; exons 2 and 3), and H-RAS (NM_005343.2; exons 2 and 3) by direct sequencing, as described [14, 32].
Cell lines and treatments
Four human thyroid cell lines that have been recently authenticated to be unique thyroid cancer cell lines [33, 34], were used: the PTC-derived BCPAP (Leibniz Institute-DSMZ, Germany) and K1, the medullary thyroid cancer cell line TT, and the 8505C established from an undifferentiated thyroid carcinoma (ECACC, Sigma-Aldrich, Italy). In addition, the human embryonic kidney HEK293 cell line (American Type Culture Collection, VA) was used for transfection experiments. All cell lines but HEK293, which was maintained in DMEM, were cultured in RPMI 1640 (Gibco, Italy) supplemented with 10% FBS (Gibco), L-glutamine (2 mM) and penicillin/streptomycin (100 IU/mL/100 μg/mL, respectively). Adherent monolayer cultures were maintained in T75 culture flasks and incubated at 37°C with 5% CO2 until they achieved 85% confluency. Cells were detached using 0.25% trypsin (Sigma-Aldrich) and plated into T75 flasks at a density of 2 × 106 cells.
Novartis International (Basel, Switzerland) kindly provided RAF265, while SB590885 were purchased from Selleckchem (Houston, TX), and dissolved in DMSO following the manufacturer's instructions. Cells were incubated with the drugs for 72 hours at the IC50 doses we recently established [4]. IC50 for RAF265 was 0.12 μM, 0.55 μM and 1.32 μM in 8505C, BCPAP and K1 cells, respectively. IC50 for SB590885 was 5.2 μM in BCPAP and K1 cells and 6.2 μM in 8505C cells.
Cell transfection and dual-luciferase assay
Twenty-four hours before the experiment, BCPAP (2.5 × 105 cells/well) or HEK293 cells (2 × 105 cells/well) were seeded in 12-well plates. Cells were transiently transfected as reported elsewhere [35] using 2 μl of Lipofectamine 2000 (Invitrogen, Italy) and 1.5 μg of total DNA consisting of the Flag-tagged BRAFV600E/pcDNA3.1 plasmids, and/or XRE-luc, and pRL-TK (Promega, Italy). After 6 hours incubation the medium was removed, proteins harvested in passive lysis buffer and the relative luciferase activity measured with the Dual Luciferase Reporter Assay System and a GloMax 20/20 luminometer (Promega), according to the manufacturer's instructions.
Immunofluorescence microscopy
HEK293 cells were seeded in cover-glass-bottom microwell dishes and transfected with either the Flag- tagged BRAFV600E plasmid or the empty vector pcDNA3.1, as described above. After 24 h, cells were washed twice with PBS, fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, and permeabilized incubating with 0.3% Triton X-100 and BSA 3% in PBS for 30 min. Cells were stained for endogenous AHR and exogenous BRAF, by incubating with a 1:300 rabbit anti-FLAG polyclonal antibody (F7425, Sigma-Aldrich) and a mouse anti-AHR monoclonal antibody (clone 3B12, Novus Biologicals, 1:100) for 1 h at RT. The cells were then washed three times in PBS and incubated with AlexaFluor™ 488 or 594-labeled secondary antibodies (1:250 dilution) (Life Technologies, Italy) for 1 hour at RT. The cells were subsequently washed three times in PBS and nuclei counterstained with 1.5 μg/mL Hoechst 33258 (Sigma-Aldrich) and mounted with Fluorescent Mounting Medium (Dako, Cat.No. S3023).
The preparations were examined with a Leica DMI6000CS fluorescence microscope (Leica Microsystems CMS) using a 100 × /1.40 oil-immersion objective. Images were acquired by means of a DFC365FX camera and analyzed with Leica LAS-AF 3.1.0 software.
Immunohistochemistry
In a subgroup of nineteen PTC cases (ten BRAFwt and nine carrying the BRAFV600E) for which enough material was available, immunohistochemistry was performed on formalin-fixed, paraffin-embedded 4–6 μm thick tissue sections, using a polyclonal rabbit anti-AHR antibody (sc-5579, Santa Cruz Biotechnology, USA, 1:50). Appropriate positive and negative controls were run concurrently. Immunostaining for AHR was semi-quantitatively scored as previously reported [14]. Two different pathologists assessed the analysis in blind. The intensity of the staining was indicated as weak, moderate or strong. The subcellular localization of staining was also considered (cytoplasmic, nuclear or both).
Meta-analysis of microarray expression data
Raw microarray gene expression profiles were obtained from the NCBI Gene Expression Omnibus (GEO) [36]. We considered five series (GSE53157 [37], GSE33630 [38], GSE29265, GSE3678 and GSE27155 [39]), which comprise 294 samples. Based on tissue type, 241 samples were selected, organized in a proper database, using the open source web application A-MADMAN [40] and considered for further analysis. For details, see Supplemental Materials and Methods.
Protein extraction and western blotting
Proteins were extracted from the different cell lines (6 × 105 cells into 60 mm cell culture dishes) possibly incubated with RAF265 and SB590885 as described elsewhere [41]. Briefly, cells were PBS-rinsed, lysed in RIPA buffer supplemented with proteases inhibitors and clarified by centrifugation. Available tumoral specimens and normal tissue pairs were re-suspended in lyses buffer and homogenized with Tissue Lyser (Qiagen). Protein concentrations were determined using the Bio-Rad DC protein assay kit (BioRad, Italy) following the manufacturer's instructions. For each sample, 20 μg were resolved by SDS-PAGE on a 10% NuPAGE gel (Invitrogen) and transferred onto nitrocellulose membrane by Trans-Blot Turbo transfer system (BioRad). Membranes were blocked for 2 h with 5% non-fat dry milk, incubated overnight at 4°C with a primary antibody [1:1000 anti-AHR, (clone 3B12, Novus Biological), 1:1000 anti-Erk1/2 and anti-phospho-Erk1/2 (Thr202/Tyr204) (Cell Signaling, Euroclone, Italy)] followed by a 1 h incubation with an HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, US). Expression was corrected for differences in protein loading by probing blots with mouse anti-β-actin antibody (1:5000, clone AC-15, Sigma-Aldrich). Blots were developed using Pierce ECL Substrate and exposed to CL-XPosure Film (Thermo Scientific, Rockford, US). Band intensity was quantified with Image J software 1.44p.
RNA isolation, reverse transcription and qPCR
Total RNA from frozen tissue after surgery, as well as from each thyroid cell line was extracted using TRIzol (Invitrogen) as previously reported [42]. RNA yield was determined on a spectrophotometer (NanoDrop Technologies, Wilmington, USA). One μg of DNase-treated RNA was reverse-transcribed using M-MuLV Reverse Transcriptase RNase H- (Euroclone) according to the manufacturer's recommendations. qPCR experiments were performed according to the MIQE guidelines [43]. For details, see Supplemental Materials and Methods.
Statistical analysis
We calculated proportions and rates for categorical variables, means ± standard deviations, or medians and ranges for parametric or non-parametric variables. In qPCR experiments, groups were compared with the Mann-Whitney test or Wilcoxon test for quantitative variables not normally distributed. The MedCalc version 14.7 (MedCalc software, Ostand, Belgium) was used to manage patients’ dataset and for the statistical analyses. The significance level was set at p < 0.05 for all tests.
SUPPLEMENTARY DATA
Acknowledgments
The authors are grateful to Prof. Detour and Dr. Tarabichi (IRIBHM - Université Libre de Bruxelles), for sharing clinical information concerning the GSE33630 dataset, Dr. Walter Kolch, Systems Biology Ireland and The Conway Institute, University College Dublin) for the Flag-tagged BRAFV600E plasmids and Dr Xavier Coumoul (Université Paris Descartes, Sorbonne Paris Cité, Paris, France) for XRE-luc plasmid.
Footnotes
CONFLICTS OF INTEREST
The authors have nothing to disclose.
GRANT SUPPORT
This work was supported by the Italian Ministry for the University and Research (PRIN Project Grant 2010TYCL9B_007 and 2010BX2SNA).
REFERENCES
- 1.Gloeckler Ries LA, Reichman ME, Lewis DR, Hankey BF, Edwards BK. Cancer survival and incidence from the Surveillance, Epidemiology, and End Results (SEER) program. The oncologist. 2003;8:541–552. doi: 10.1634/theoncologist.8-6-541. [DOI] [PubMed] [Google Scholar]
- 2.Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocrine reviews. 2007;28:742–762. doi: 10.1210/er.2007-0007. [DOI] [PubMed] [Google Scholar]
- 3.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Current opinion in cell biology. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 4.Barollo S, Bertazza L, Baldini E, Ulisse S, Cavedon E, Boscaro M, Pezzani R, Mian C. The combination of RAF265, SB590885, ZSTK474 on thyroid cancer cell lines deeply impact on proliferation and MAPK and PI3K/Akt signaling pathways. Investigational new drugs. 2014;32:626–635. doi: 10.1007/s10637-014-0108-3. [DOI] [PubMed] [Google Scholar]
- 5.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 6.Caronia LM, Phay JE, Shah MH. Role of BRAF in thyroid oncogenesis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:7511–7517. doi: 10.1158/1078-0432.CCR-11-1155. [DOI] [PubMed] [Google Scholar]
- 7.Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, Refetoff S, Nikiforov YE, Fagin JA. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer research. 2005;65:4238–4245. doi: 10.1158/0008-5472.CAN-05-0047. [DOI] [PubMed] [Google Scholar]
- 8.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicological sciences: an official journal of the Society of Toxicology. 2011;124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel J, Haarmann-Stemmann T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biological chemistry. 2010;391:1235–1248. doi: 10.1515/BC.2010.128. [DOI] [PubMed] [Google Scholar]
- 10.Van den Berg M, Birnbaum LS, Denison M, De Vito M, Farland W, Feeley M, Fiedler H, Hakansson H, Hanberg A, Haws L, Rose M, Safe S, Schrenk D, et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicological sciences: an official journal of the Society of Toxicology. 2006;93:223–241. doi: 10.1093/toxsci/kfl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Julliard W, Fechner JH, Mezrich JD. The aryl hydrocarbon receptor meets immunology: friend or foe? A little of both. Frontiers in immunology. 2014;5:458. doi: 10.3389/fimmu.2014.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safe S, Lee SO, Jin UH. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicological sciences: an official journal of the Society of Toxicology. 2013;135:1–16. doi: 10.1093/toxsci/kft128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mian C, Ceccato F, Barollo S, Watutantrige-Fernando S, Albiger N, Regazzo D, de Lazzari P, Pennelli G, Rotondi S, Nacamulli D, Pelizzo MR, Jaffrain-Rea ML, Grimaldi F, et al. AHR over-expression in papillary thyroid carcinoma: clinical and molecular assessments in a series of Italian acromegalic patients with a long-term follow-up. PloS one. 2014;9:e101560. doi: 10.1371/journal.pone.0101560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henklova P, Vrzal R, Ulrichova J, Dvorak Z. Role of mitogen-activated protein kinases in aryl hydrocarbon receptor signaling. Chemico-biological interactions. 2008;172:93–104. doi: 10.1016/j.cbi.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Feng S, Cao Z, Wang X. Role of aryl hydrocarbon receptor in cancer. Biochimica et biophysica acta. 2013;1836:197–210. doi: 10.1016/j.bbcan.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nature reviews. Cancer. 2014;14:801–814. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puga A, Ma C, Marlowe JL. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochemical pharmacology. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Z, Chang X, Puga A, Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochemical pharmacology. 2002;64:771–780. doi: 10.1016/s0006-2952(02)01138-3. [DOI] [PubMed] [Google Scholar]
- 20.Tan Z, Huang M, Puga A, Xia Y. A critical role for MAP kinases in the control of Ah receptor complex activity. Toxicological sciences: an official journal of the Society of Toxicology. 2004;82:80–87. doi: 10.1093/toxsci/kfh228. [DOI] [PubMed] [Google Scholar]
- 21.Hoffer A, Chang CY, Puga A. Dioxin induces transcription of fos and jun genes by Ah receptor-dependent and -independent pathways. Toxicology and applied pharmacology. 1996;141:238–247. doi: 10.1006/taap.1996.0280. [DOI] [PubMed] [Google Scholar]
- 22.Chen S, Operana T, Bonzo J, Nguyen N, Tukey RH. ERK kinase inhibition stabilizes the aryl hydrocarbon receptor: implications for transcriptional activation and protein degradation. The Journal of biological chemistry. 2005;280:4350–4359. doi: 10.1074/jbc.M411554200. [DOI] [PubMed] [Google Scholar]
- 23.Ogiso H, Kagi N, Matsumoto E, Nishimoto M, Arai R, Shirouzu M, Mimura J, Fujii-Kuriyama Y, Yokoyama S. Phosphorylation analysis of 90 kDa heat shock protein within the cytosolic arylhydrocarbon receptor complex. Biochemistry. 2004;43:15510–15519. doi: 10.1021/bi048736m. [DOI] [PubMed] [Google Scholar]
- 24.Yim S, Oh M, Choi SM, Park H. Inhibition of the MEK-1/p42 MAP kinase reduces aryl hydrocarbon receptor-DNA interactions. Biochemical and biophysical research communications. 2004;322:9–16. doi: 10.1016/j.bbrc.2004.07.072. [DOI] [PubMed] [Google Scholar]
- 25.Spivack SD, Hurteau GJ, Reilly AA, Aldous KM, Ding X, Kaminsky LS. CYP1B1 expression in human lung. Drug metabolism and disposition: the biological fate of chemicals. 2001;29:916–922. [PubMed] [Google Scholar]
- 26.Reiners JJ, Jr, Lee JY, Clift RE, Dudley DT, Myrand SP. PD98059 is an equipotent antagonist of the aryl hydrocarbon receptor and inhibitor of mitogen-activated protein kinase kinase. Molecular pharmacology. 1998;53:438–445. doi: 10.1124/mol.53.3.438. [DOI] [PubMed] [Google Scholar]
- 27.Bachleda P, Dvorak Z. Pharmacological inhibitors of JNK and ERK kinases SP600125 and U0126 are not appropriate tools for studies of drug metabolism because they activate aryl hydrocarbon receptor. General physiology and biophysics. 2008;27:143–145. [PubMed] [Google Scholar]
- 28.Petrulis JR, Hord NG, Perdew GH. Subcellular localization of the aryl hydrocarbon receptor is modulated by the immunophilin homolog hepatitis B virus X-associated protein 2. The Journal of biological chemistry. 2000;275:37448–37453. doi: 10.1074/jbc.M006873200. [DOI] [PubMed] [Google Scholar]
- 29.Koliopanos A, Kleeff J, Xiao Y, Safe S, Zimmermann A, Buchler MW, Friess H. Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene. 2002;21:6059–6070. doi: 10.1038/sj.onc.1205633. [DOI] [PubMed] [Google Scholar]
- 30.Nishiumi S, Miyamoto S, Kawabata K, Ohnishi K, Mukai R, Murakami A, Ashida H, Terao J. Dietary flavonoids as cancer-preventive and therapeutic biofactors. Frontiers in bioscience (Scholar edition) 2011;3:1332–1362. doi: 10.2741/229. [DOI] [PubMed] [Google Scholar]
- 31.Sale S, Tunstall RG, Ruparelia KC, Butler PC, Potter GA, Steward WP, Gescher AJ. Effects of the potential chemopreventive agent DMU-135 on adenoma development in the ApcMin+ mouse. Investigational new drugs. 2006;24:459–464. doi: 10.1007/s10637-006-5947-0. [DOI] [PubMed] [Google Scholar]
- 32.Barollo S, Pezzani R, Cristiani A, Redaelli M, Zambonin L, Rubin B, Bertazza L, Zane M, Mucignat-Caretta C, Bulfone A, Pennelli G, Casal Ide E, Pelizzo MR, et al. Prevalence, tumorigenic role, and biochemical implications of rare BRAF alterations. Thyroid: official journal of the American Thyroid Association. 2014;24:809–819. doi: 10.1089/thy.2013.0403. [DOI] [PubMed] [Google Scholar]
- 33.Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, Fagin JA, Marlow LA, Copland JA, Smallridge RC, Haugen BR. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. The Journal of clinical endocrinology and metabolism. 2008;93:4331–4341. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dadon T, Ball D, Nelkin BD. Misidentification of putative medullary thyroid cancer cell lines RO-H85-1 and RO-D81-1. The Journal of clinical endocrinology and metabolism. 2013;98:954–955. doi: 10.1210/jc.2012-4181. [DOI] [PubMed] [Google Scholar]
- 35.Occhi G, Losa M, Albiger N, Trivellin G, Regazzo D, Scanarini M, Monteserin-Garcia JL, Frohlich B, Ferasin S, Terreni MR, Fassina A, Vitiello L, Stalla G, et al. The glucose-dependent insulinotropic polypeptide receptor is overexpressed amongst GNAS1 mutation-negative somatotropinomas and drives growth hormone (GH)-promoter activity in GH3 cells. Journal of neuroendocrinology. 2011;23:641–649. doi: 10.1111/j.1365-2826.2011.02155.x. [DOI] [PubMed] [Google Scholar]
- 36.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, et al. NCBI, GEO: archive for functional genomics data sets—update. Nucleic acids research. 2013;41:D991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pita JM, Banito A, Cavaco BM, Leite V. Gene expression profiling associated with the progression to poorly differentiated thyroid carcinomas. British journal of cancer. 2009;101:1782–1791. doi: 10.1038/sj.bjc.6605340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomas G, Tarabichi M, Gacquer D, Hebrant A, Dom G, Dumont JE, Keutgen X, Fahey TJ, 3rd, Maenhaut C, Detours V. A general method to derive robust organ-specific gene expression-based differentiation indices: application to thyroid cancer diagnostic. Oncogene. 2012;31:4490–4498. doi: 10.1038/onc.2011.626. [DOI] [PubMed] [Google Scholar]
- 39.Giordano TJ, Au AY, Kuick R, Thomas DG, Rhodes DR, Wilhelm KG, Jr, Vinco M, Misek DE, Sanders D, Zhu Z, Ciampi R, Hanash S, Chinnaiyan A, et al. Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:1983–1993. doi: 10.1158/1078-0432.CCR-05-2039. [DOI] [PubMed] [Google Scholar]
- 40.Bisognin A, Coppe A, Ferrari F, Risso D, Romualdi C, Bicciato S, Bortoluzzi S. A-MADMAN: annotation-based microarray data meta-analysis tool. BMC bioinformatics. 2009;10 doi: 10.1186/1471-2105-10-201. 201-2105-10-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, Ferasin S, Cetani F, Pardi E, Korbonits M, Pellegata NS, Sidarovich V, Quattrone A, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS genetics. 2013;9:e1003350. doi: 10.1371/journal.pgen.1003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Occhi G, Albiger N, Berlucchi S, Gardiman M, Scanarini M, Scienza R, Fassina A, Mantero F, Scaroni C. Peroxisome proliferator-activated receptor gamma in the human pituitary gland: expression and splicing pattern in adenomas versus normal pituitary. Journal of neuroendocrinology. 2007;19:552–559. doi: 10.1111/j.1365-2826.2007.01562.x. [DOI] [PubMed] [Google Scholar]
- 43.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.