

Abstract
Stromal fibrosis and tissue necrosis are major histological sequelae of hypoxia. The hypoxia-to-fibrosis sequence is well-documented in pancreatic ductal adenocarcinoma (PDAC). However, hypoxic and necrotic PDAC phenotypes are insufficiently characterized. Recently, reduction of tuberous sclerosis expression in mice together with oncogenic Kras demonstrated a rapidly metastasizing phenotype with histologically eccentric necrosis, transitional hypoxia and devascularisation. We established cell lines from these tumors and transplanted them orthotopically into wild-type mice to test their abilities to recapitulate the histological features of the primary lesions. Notably, the necrotic phenotype was reproduced by only a subset of cell lines while others gave rise to dedifferentiated tumors with significantly reduced necrosis. In vitro analysis of the necrotic tumor-inducing cell lines revealed that these cells released a significant amount of vascular endothelial growth factor A (VEGFA). However, its release was not further increased under hypoxic conditions. Defective hypoxia-induced VEGFA secretion was not due to impaired VEGFA transcription or hypoxia-inducible factor 1-alpha activation, but rather a result of hypoxia-induced endoplasmic reticulum (ER) stress. We thus identified hypoxia-induced ER stress as an important pathway in PDACs with tissue necrosis and rapid metastasis.
Keywords: ER stress, VEGFA, hypoxia, necrosis, pancreatic cancer
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is notoriously aggressive and hypoxic [1, 2]. As the extent of hypoxia has been reported to be associated with the prognosis of PDAC patients [3–5], it constitutes an important aspect of its lethal biology. Indeed, hypoxia fosters a variety of malignant features of PDAC by 1) promoting the metastatic spread [6]; 2) facilitating the acquisition of stem cell-like properties [7, 8], and 3) limiting the delivery of conventional chemotherapeutic agents [9]. Thus, targeting tumor hypoxia by using hypoxia-activated substances (or bioreductive prodrugs, e.g. TH-302) has emerged as a promising strategy to improve the outcome of PDAC patients [10].
Stromal fibrosis and tissue necrosis are major histological sequelae of hypoxia in PDAC [11–13], probably reflecting the biological consequences of dissociated tumor expansion and insufficient neo-angiogenesis. We have previously provided in vitro evidence suggesting that an interaction between cancer cells and pancreatic stellate cells (PSCs) initiates and perpetuates a fibrotic, hypoxic and tumor-supportive microenvironment [11, 14]. This notion has recently been challenged by experimental data generated in vivo [15, 16]. Despite the fact that different transgenic mouse models and different approaches for depleting fibrosis were used, two independent studies reached the same conclusion that the fibrotic component of stroma restrained tumor growth rather than supporting it. However, the role of fibrosis in angiogenesis and hypoxia is largely inconsistent: disruption of fibrosis promoted angiogenesis in one study [16] while it inhibited angiogenesis and aggravated tissue hypoxia in another one [15]. Therefore, fibrosis, hypoxia and impaired angiogenesis play complex and not always concerted roles in pancreatic carcinogenesis.
The less well characterized hypoxic/necrotic phenotypes of PDAC add further complexity. Previous descriptive studies in human PDAC have demonstrated that micro- and macro-necrosis can be observed in the majority of cases and that the presence of tumor necrosis is associated with unfavourable prognosis, implying a potential tumor-promoting role associated with the hypoxic/necrotic phenotype [12, 13]. Indeed, we previously characterized a murine model of highly metastatic PDAC driven by oncogenic Kras/Mek-mTOR (mechanistic target of rapamycin) signalling in which the observed massive tumor necrosis was associated with metastatic spread [17]. In the current study, we aimed at uncovering potential molecular mechanisms underlying the hypoxic/necrotic phenotype.
RESULTS
Identification of a hypoxic/necrotic tumor phenotype
Histological examination of PDAC tissue resulting from oncogenic Kras/Mek-mTOR signalling in a mouse model revealed large and eccentric necrosis surrounded by avascular areas [17] (Figure 1A). To further characterize tumor histology, we labelled vessels using CD31 antibodies. This analysis revealed that the necrotic region was surrounded by devascularized tumor areas (Figure 1B) in which cancer cells were specifically carbonic anhydrase IX- (Caix; a hypoxia marker) and cleaved-Caspase 3-positive (Figure 1C, 1D). In addition, other hypoxia-induced molecular changes were also observed: membrane expression of E-Cadherin was lost in cancer cells close to devascularized areas. Furthermore, acquisition of a fibroblastic characteristic (resembling features of epithelial-to-mesenchymal transition (EMT)) and increased expression of Hk2, a surrogate marker of Hif1α activation, was seen (Figure 1E, 1F). Collectively, these results demonstrated a tumor histology in this mouse model closely resembling the human hypoxic/necrotic PDAC phenotype [12, 13].
Figure 1. Identification of a hypoxic/necrotic tumor phenotype.
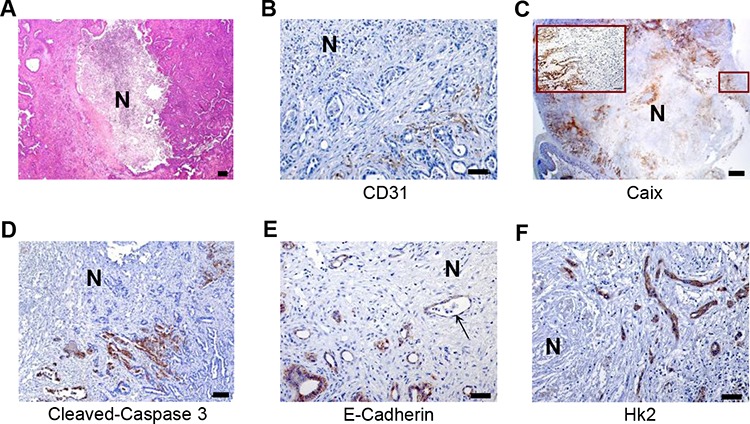
A. Representative H&E-stained sections of necrotic PDACs; scale bar: 100 μm, B–D. IHC studies of CD31, Caix and cleaved-caspase 3 as surrogate markers for vessel localization (scale bar: 50 μm), hypoxic regions (scale bar: 500 μm) and apoptotic cells (scale bar: 100 μm) in metastatic PDACs. E–F. IHC studies for E-Cadherin and Hk2 show loss of membrane staining of E-Cadherin (arrow) and increased expression of Hk2 in cancer cells in the vicinity of tissue necrosis (also hypoxic regions) in metastatic PDACs, scale bar: 50 μm, N: necrosis.
In vivo screening of PDAC cells carrying the hypoxic/necrotic phenotype
To test whether this particular phenotype was preserved in isolated PDAC cell lines, we transplanted a number of these cell lines (n = 6, established from above described mouse models) orthotopically into wild type (WT) mice to test their ability to recapitulate the histology of the primary tumor. Five cell lines (399, 403, 445Li, 907 and 908Li) formed tumors at a 100% (35/35) penetrance while only 3 out of 7 (43%) of the 897 cell-transplanted mice developed tumors. Clear differences in tumor histology were observed: the 399, 403 and 445Li cells gave rise to PDAC characterized by large areas of central necrosis (Figure 2A); the 907, 908Li and 897 cells developed into solid tumors (Figure 2B). The solid tumors contained a higher proportion of dedifferentiated/sarcomatoid-like components than the necrotic ones (Figure 2C; as exemplified by E-Cadherin Immunohistochemistry (IHC)). The necrotic tumors recapitulated the core histological features of the hypoxic/necrotic phenotype including devascularized tumor areas, hypoxia and apoptosis (Figure 2D, 2E and 2F, upper panel). No such features were observed in the histology of solid tumors (Figure 2D, 2E and 2F, lower panel). Thus, the cell lines giving rise to the necrotic tumors with features of hypoxia were labelled “hypoxic/necrotic” whereas the cell lines forming solid dedifferentiated tumors without pronounced features of hypoxia and significantly less necrosis were labelled “non-hypoxic/solid”. In addition, Kras sequencing of these cell lines revealed that the hypoxic/necrotic cells were heterozygous for KrasG12D while the non-hypoxic/solid contained only one mutated allele, showing loss of heterozygosity (LOH) at the Kras locus. PCR analysis confirmed loss of the WT Kras allele in these cells (Supplementary Figure 1A), comparable to previous reports [18]. Western-blot analyses and Ras pull-down assays revealed that neither Kras expression nor the steady-state/serum-induced Kras activity (GTP-Kras) were affected by KrasG12D-LOH (Supplementary Figure 1B).
Figure 2. In vivo screening of PDAC cells with an in vivo preserved hypoxic/necrotic phenotype.
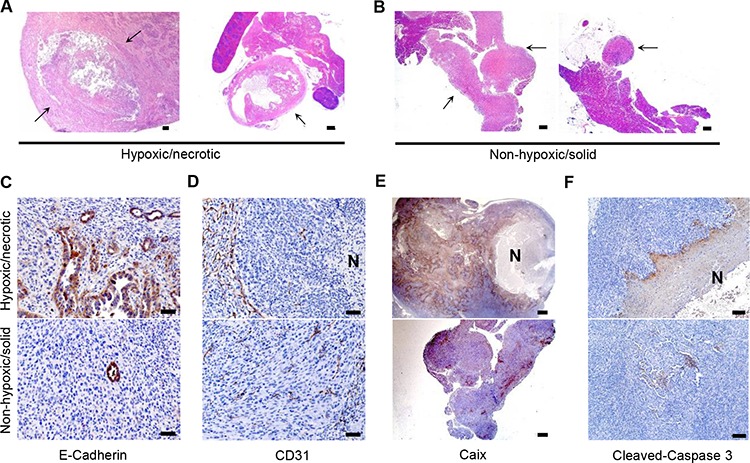
A–B. H&E–stained sections show centrally-necrotic tumors formed after orthotopic implantation of the hypoxic/necrotic cells and more solid forms of tumors formed by dedifferentiated cells, scale bar: 500 μm; arrow: tumor tissues; C. IHC with anti-E-Cadherin demonstrates a high proportion of anaplastic components in tumors developing from the hypoxic/necrotic cells, but not in tumors developing from the dedifferentiated cells, scale bar: 50 μm; D–F. IHC of anti-CD31 (scale bar: 50 μm), anti-Caix (scale bar: 500 μm) and anti-cleaved-caspase 3 (scale bar: 100 μm) show devascularized tumor regions, hypoxic zones and apoptosis in the necrotic tumors forming after transplantation of the hypoxic/necrotic cells, but not in the anaplastic tumors forming after transplantation of the dedifferentiated cells; N: necrosis.
Compromised hypoxia-driven VEGFA secretion in PDAC cells with a hypoxic/necrotic phenotype
As hypoxia-driven angiogenesis relies on the secretion of a plethora of hypoxia-induced angiogenic factors such as most importantly VEGFA, we measured the release of VEGFA following a 24 hour-exposure to hypoxia in hypoxic/necrotic cells (399 and 403 cells). The non-hypoxic/solid cells (907 and 897 cells) and so-called cystic cell lines established from previously described cystic tumors (926 and 928 cells [17]: “cystic”) were used as internal and external controls, respectively. Notably, the hypoxic/necrotic cells secreted high levels of VEGFA but did not increase VEGFA secretion after exposure to hypoxia; however, both the non-hypoxic/solid and cystic cell lines had no such “defect” (Figure 3A). Delineation of the underlying mechanisms demonstrated that hypoxia-induced VEGFA transcription and Hif1alpha activation were significantly increased in all cell lines including the hypoxic/necrotic cells (Figure 3B, 3C). In addition, the hypoxia-induced responses such as dephosphorylation of S6 (p-S6Ser235/236), phosphorylation of Ampk (p-AmpkThr172) and expression of Bnip3 (BCL2/adenovirus E1B interacting protein 3) or of REDD1 were not altered in hypoxic/necrotic cells (Figure 3C). Taken together, these data demonstrate that hypoxia-driven VEGFA secretion is compromised in PDAC cells with the hypoxic/necrotic phenotype; additionally, such defects are not due to impaired VEGFA transcription or Hif1α activation.
Figure 3. Compromised hypoxia-driven VEGFA secretion in PDAC cells with the hypoxic/necrotic phenotype.
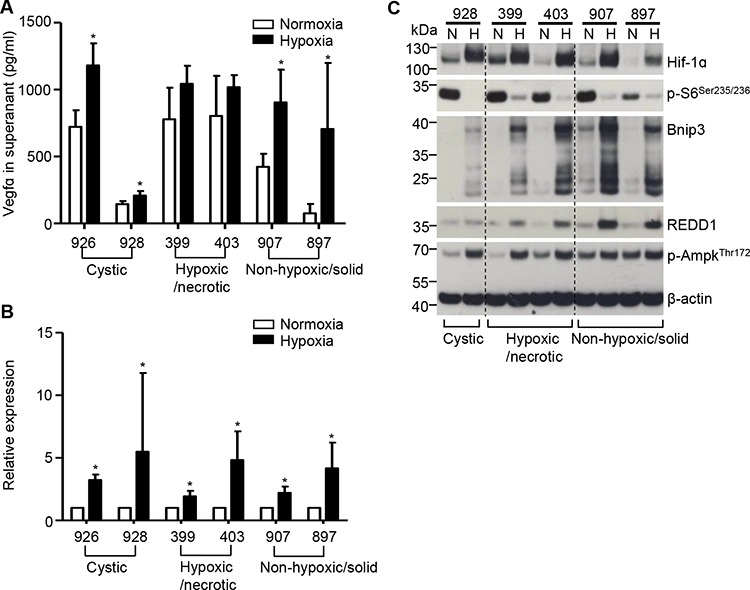
A. ELISA assays show the release of VEGFA into the supernatants of hypoxic/necrotic cells (399 and 403 cells), dedifferentiated cells (907 and 897 cells) and “cystic” cells (926 and 928 cells) after exposure to hypoxia for 24 hours. Values shown are obtained from at least three independent experiments; *p < 0.05; B. VEGFA mRNA levels (measured by QRT-PCR) after induction of hypoxia in all tested cell lines. Data are presented as relative expression (normalized to the median of the respective expression level under normoxia); an unpaired t-test was performed; values shown are obtained from at least three independent experiments; *p < 0.05; C, Western-blot analysis shows expression of Hif1alpha, p-S6Ser235/236, Bnip3, Redd1 and p-AmpkThr172 in hypoxic/necrotic cells (399 and 403 cells), dedifferentiated cells (907 and 897 cells) and “cystic” cells (928 cells) after exposure to hypoxia for 24 hours; N: normoxia, H: hypoxia; one representative blot out of three independent experiments is shown.
Sensitivity of the hypoxic/necrotic cells to hypoxia-induced ER stress
Because secretion and folding of VEGFA rely on the formation of a disulfide bond (an oxidative reaction) in the endoplasmic reticulum (ER) [19, 20], it is likely that the VEGFA secretion in ER is highly vulnerable to oxygen deprivation. Here, Bip (heat shock protein 5), a marker of ER stress, was indeed induced by hypoxia in hypoxic/necrotic cells. The non-hypoxic/solid and cystic cell lines were more resistant to hypoxia-induced ER stress (Figure 4A). Expression of the protein disulfide isomerase (PDI) responsible for disulfide formation remained unchanged under hypoxic conditions (internal control, Figure 4A). In line with these findings, Bip expressing cells were specifically detected in the hypoxic, devascularized regions of primary hypoxic/necrotic PDACs (Figure 4B) and of transplanted hypoxic/necrotic tumors (Figure 4C). No such patterns were observed in the transplanted dedifferentiated tumors (Figure 4B, 4C). Besides, no differences in PDI expression were found comparing these tumor entities (Figure 4B, 4C). Taken together, the hypoxic/necrotic cells were highly sensitive to hypoxia-induced ER stress.
Figure 4. Sensitivity of the hypoxic/necrotic cells to hypoxia-induced ER stress.
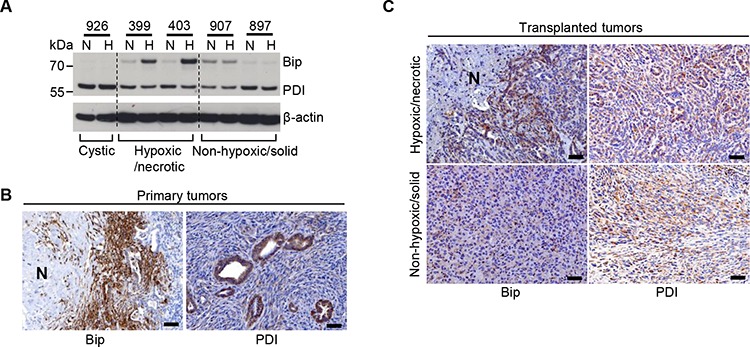
A. Western-blot analysis demonstrates induced expression of Bip and PDI in hypoxic/necrotic cells (399, 403), in dedifferentiated cells (907, 897) and in cystic cells (926) after exposure to hypoxia for 24 hours; N: normoxia, H: hypoxia; one representative blot out of three independent experiments is shown, B. Representative IHC pictures show expression of Bip and PDI in metastatic PDACs, scale bar: 50 μm, C. Representative IHC pictures show expression of Bip and PDI in the transplanted tumor tissues induced by transplantation of hypoxic/necrotic or dedifferentiated cells, scale bar: 50 μm.
DISCUSSION
In general, hypoxia constitutes an important source of ER stress and activates the unfolded protein response (UPR) system to mediate a set of adaptive changes within solid tumors [21, 22]. However, why protein secretion is impaired and how ER stress is induced by hypoxia is not clear. One of the potential reasons is that oxygen provides the ultimate oxidative potential for disulfide formation, which is essential for the folding of secretory proteins. VEGFA is a typical example: the folding and proper function of VEGFA requires both intramolecular and intersubunit disulfide bonds. Therefore, secretion of VEGFA relies on the maintenance of the oxidative potential of the ER [20]. Upon oxygen deprivation, unfolded VEGFA (or other secreted proteins) accumulate in the ER and cause ER stress and UPR. This is especially important for cancer cell lines with high levels of VEGFA secretion. Here, the oxygen supply needs to be constantly maintained to support oxidative folding of high-throughput VEGFA secretion. Subtle changes in oxygen supply may disturb proper folding of VEGFA and cause a severe “traffic jam” along the secretory pathway. This partially explains why cancer cells with high levels of VEGFA secretion are susceptible to hypoxia-induced ER stress.
Interestingly, the secretion of VEGFA is not the only determinant factor: one cystic cell line sustained by the oncogenic PI3K/Akt-mTOR signal secretes (926 cells) a significant amount of VEGFA [17], however, no hypoxic/necrotic phenotype was observed. Importantly, this cell line was resistant to hypoxia-induced ER stress. These data demonstrate that it is not the level of VEGFA secretion that correlates with the hypoxic/necrotic phenotype. Instead, the hypoxia-induced ER stress correlates with the hypoxic/necrotic phenotype. Whether this correlation constitutes a causal link requires further investigations. In particular, the functional relevance of VEGFA secretion in the development of hypoxic/necrotic phenotype seems to be largely context-dependent and complex (see below).
In line with previous reports [23], the hypoxic/necrotic cells sustained by the oncogenic Kras/Mek-mTOR signal produced relatively high levels of VEGFA. It is conceivable that an angiogenic switch can easily be achieved by hypoxic/necrotic cells at the incipient stage of carcinogenesis. However, as the tumor volume increases, hypoxia invariably occurs, which readily triggers ER stress in these cells due to its intrinsic sensitivity. The underlying reasons for this intrinsic sensitivity and the biological relevance of such hypoxia-induced ER stress requires further elucidation. In particular, it is unclear whether it contributes to the aggravation of local hypoxia by compromising VEGFA secretion, whether it is responsible for cell death, or whether it promotes the metastatic spread of PDAC. Interestingly, we observed that a subset of cancer cells appeared to lose the hypoxic/necrotic phenotype with a concomitant acquisition of LOH status at Kras locus due to unknown reasons. To clarify these questions warrants further investigation. Nevertheless, these data suggest that the hypoxia-induced ER stress constitutes a potential target for ameliorating the hypoxic/necrotic phenotype. In this regard, we recently identified a subtype of human PDAC with high ER stress levels which is labelled by a common variant of MIA2 gene (melanoma inhibitory activity 2, MIA2I141M, [24]). Certainly, it would be worthwhile to analyse whether the hypoxic/necrotic phenotype is more dominant in this PDAC subtype.
MATERIALS AND METHODS
Mouse breeding and collection of mouse tissues
Mouse breeding and tissues collection were described previously [17]. Briefly, the metastatic/necrotic PDACs developed in triple transgenic p48Cre/+; LSL-KrasG12D/+; Tsc1flox/+ compound mice while the cystic tumors were derived from triple transgenic p48Cre/+; Ptenflox/flox; Tsc1flox/+ compound mice. Wild-type (WT; C57BL/6J) mice were obtained from Charles River Laboratory (Sulzfeld, Germany). Mouse breeding was performed and husbandry was maintained at the specific pathogen free (SPF) mouse facility at the Technical University of Munich. The compound transgenic mice were maintained on a mixed background. All mouse experiments and procedures were approved by Bavarian Government (No. 55.2–1-54–2532-42–13). All procedures were in accordance with the Office of Laboratory Animal Welfare and the German Federal Animal Protection Laws.
Cell transplantation experiments
Orthotopic transplantations of mouse cells were carried out as described [25]. Briefly, mice were anesthetized, and a left-lateral incision of the abdomen was made to visualize the tail region of the pancreas. 106 cells suspended in 50 μl of PBS were carefully injected into the pancreatic tail. The abdominal wall and skin were closed using running sutures. All mice were sacrificed for histological evaluation after 4 weeks.
Hypoxia assays
Pancreatic cancer cell lines were incubated in a modular chamber saturated with a hypoxic air mixture (89.25% N2 + 10% CO2 + 0.75% O2) for 24 h at 37°C. For measurements of VEGFA, serum-free medium was used. The remaining experiments were performed in medium supplemented with 10% FCS. All experiments were repeated three times.
Further materials and methods
A detailed materials and methods section is provided as a supplement to this manuscript.
Statistical analysis
Either GraphPad Prism 5 Software (GraphPad, San Diego, CA, USA) was used for the statistical analysis. Unless otherwise stated, an unpaired t-test was used for group-wise comparisons. Statistical significance was set at p < 0.05.
SUPPLEMENTARY DATA FIGURE
ACKNOWLEDGMENTS AND FUNDING
We would like to thank Manja Thorwirth and Nadja Maeritz for their excellent technical support.
This project was supported in part by a grant from the TU Munich commission for clinical research (KKF C21-11 to BK), the Deutsche Forschungsgemeinschaft (MI 1173/5-1 to CWM, BK and JK) and the European Union (FP7, PacaNet to CWM and JK).
Abbreviations
- PDAC
pancreatic ductal adenocarcinoma
- ER
endoplasmic reticulum
- VEGFA
vascular endothelial growth factor a
- Tsc
Tuberous sclerosis complex
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositol-4, 5-bisphosphate 3-kinase
- Hif1α
hypoxia-inducible factor 1 alpha
Footnotes
CONFLICTS OF INTEREST
None.
REFERENCES
- 1.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M. Pancreatic tumors show high levels of hypoxia. International journal of radiation oncology, biology, physics. 2000;48:919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Seki S, Sakaguchi H, Sawada T, Hirakawa K, Wakasa K. Clinicopathological significance of hypoxia-inducible factor-1alpha expression in human pancreatic carcinoma. Histopathology. 2003;43:550–555. doi: 10.1111/j.1365-2559.2003.01733.x. [DOI] [PubMed] [Google Scholar]
- 4.Shibaji T, Nagao M, Ikeda N, Kanehiro H, Hisanaga M, Ko S, Fukumoto A, Nakajima Y. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003;23:4721–4727. [PubMed] [Google Scholar]
- 5.Hoffmann AC, Mori R, Vallbohmer D, Brabender J, Klein E, Drebber U, Baldus SE, Cooc J, Azuma M, Metzger R, Hoelscher AH, Danenberg KD, Prenzel KL, Danenberg PV. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia. 2008;10:674–679. doi: 10.1593/neo.08292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niizeki H, Kobayashi M, Horiuchi I, Akakura N, Chen J, Wang J, Hamada JI, Seth P, Katoh H, Watanabe H, Raz A, Hosokawa M. Hypoxia enhances the expression of autocrine motility factor and the motility of human pancreatic cancer cells. Br J Cancer. 2002;86:1914–1919. doi: 10.1038/sj.bjc.6600331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penchev VR, Rasheed ZA, Maitra A, Matsui W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin Cancer Res. 2012;18:4277–4284. doi: 10.1158/1078-0432.CCR-11-3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borad MJ, Reddy SG, Bahary N, Uronis HE, Sigal D, Cohn AL, Schelman WR, Stephenson J, Jr, Chiorean EG, Rosen PJ, Ulrich B, Dragovich T, Del Prete SA, Rarick M, Eng C, Kroll S, et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. J Clin Oncol. 2015;33:1475–1481. doi: 10.1200/JCO.2014.55.7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11:497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiraoka N, Ino Y, Sekine S, Tsuda H, Shimada K, Kosuge T, Zavada J, Yoshida M, Yamada K, Koyama T, Kanai Y. Tumour necrosis is a postoperative prognostic marker for pancreatic cancer patients with a high interobserver reproducibility in histological evaluation. Br J Cancer. 2010;103:1057–1065. doi: 10.1038/sj.bjc.6605854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitsunaga S, Hasebe T, Iwasaki M, Kinoshita T, Ochiai A, Shimizu N. Important prognostic histological parameters for patients with invasive ductal carcinoma of the pancreas. Cancer science. 2005;96:858–865. doi: 10.1111/j.1349-7006.2005.00128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erkan M, Kleeff J, Gorbachevski A, Reiser C, Mitkus T, Esposito I, Giese T, Buchler MW, Giese NA, Friess H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology. 2007;132:1447–1464. doi: 10.1053/j.gastro.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 15.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong B, Wu W, Cheng T, Schlitter AM, Qian C, Bruns P, Jian Z, Jager C, Regel I, Raulefs S, Behler N, Irmler M, Beckers J, Friess H, Erkan M, Siveke JT, et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut. 2015 doi: 10.1136/gutjnl-2014-307616. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, Chen D, Arking DE, Lowy AM, Hruban RH, Remotti HE, Su GH. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–873. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 20.May D, Itin A, Gal O, Kalinski H, Feinstein E, Keshet E. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene. 2005;24:1011–1020. doi: 10.1038/sj.onc.1208325. [DOI] [PubMed] [Google Scholar]
- 21.Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Molecular cancer research: MCR. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]
- 22.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 23.El-Hashemite N, Walker V, Zhang H, Kwiatkowski DJ. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 2003;63:5173–5177. [PubMed] [Google Scholar]
- 24.Kong B, Wu W, Valkovska N, Jager C, Hong X, Nitsche U, Friess H, Esposito I, Erkan M, Kleeff J, Michalski CW. A common genetic variation of melanoma inhibitory activity-2 labels a subtype of pancreatic adenocarcinoma with high endoplasmic reticulum stress levels. Scientific reports. 2015;5:8109. doi: 10.1038/srep08109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.