

Abstract
More than 30 independent single-nucleotide polymorphisms (SNPs) have been associated with Alzheimer's disease (AD) risk by genome-wide association studies (GWAS) in European. We aimed to confirm these SNPs in Chinese Han and investigate the utility of these genetic markers. We randomly divided 459 sporadic AD (SAD) patients and 751 cognitively normal controls into two sets (discovery and testing). Thirty-three SAD risk-associated SNPs were firstly tested in the discovery set. Significant SNPs were used to calculate genetic risk score (GRS) in the testing set. Predictive performance of GRS was evaluated using the area under the receiver operating characteristic curve (AUC). In the discovery set, 6 SNPs were confirmed (P = 7.87 × 10−11~0.048), including rs9349407 in CD2AP, rs11218343 in SORL1, rs17125944 in FERMT2, rs6859 in PVRL2, rs157580 and rs2075650 in TOMM40. The first three SNPs were associated with SAD risk independent of APOE genotypes. GRS based on these three SNPs were significantly associated with SAD risk in the independent testing set (P = 0.002). The AUC for discriminating cases from controls was 0.58 for GRS, 0.60 for APOE, and 0.64 for GRS and APOE. Our data demonstrated that GRS based on AD risk-associated SNPs may supplement APOE for better assessing individual risk for AD in Chinese.
Keywords: Alzheimer's disease, genetic risk score, risk prediction, single nucleotide polymorphism, association, Gerotarget
INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder characterized with progressive deterioration in cognition and behavior. AD is the most common form of dementia in aging population with a worldwide prevalence of 35.6 millions in 2010, and is expected to increase to 115.4 millions by 2050 [1]. In China, the burden of AD increased considerably in recent years due to aging population. The incidence of AD among people aged 60 years or older was 6.25 cases per 1000 person-years in 2010 in China [2]. Because several risk factors have been associated with AD risk [3], the disease may be preventable by reducing these risk factors. It is widely believed that targeted prevention for subjects with higher risk for AD is likely a more effective strategy.
AD is highly heritable and its heritability is estimated up to 76% [4]. Previous work suggested that genetic variants play an important role in the development of the disease. Mutations in APP, PSEN1 and PSEN2 lead to early onset familial AD [5-8]. APOE ε4 allele has been found to be the strongest risk factor for sporadic AD (SAD), the most common form of AD including early- and late-onset SAD [9-12]. However, because about 40-50% of SAD do not carry the APOE ε4 allele [11, 13], additional genetic variants that are related to SAD risk likely exist. Several genome-wide association studies (GWAS) in European descent have identified a number of independent AD risk-associated single nucleotide polymorphisms (SNPs) [14-19]. A recent meta-analysis combined four of these AD GWAS samples identified additional 11 independent AD susceptibility SNPs [20]. To date, 11 of these AD risk-associated SNPs (in 9 genes) have been reported to be significantly associated with AD risk in the Han Chinese population [21-27].
Although these AD risk-associated SNPs have a modest effect size individually (odds ratio [OR] of each individual risk allele is typically < 1.3), it is hypothesized that these SNPs may confer a stronger cumulative effect to AD. A genetic risk score (GRS) that captures the cumulative effect of SNPs has been comprehensively studied to stratify individual risk in several complex diseases [28-31]. In this study, we aimed to first determine which AD risk-associated SNPs reported in European descent are associated with SAD risk in Han Chinese in a discovery set, and then to calculate GRS using these implicated SNPs and asses its discriminative performance in a testing set.
RESULTS
Key demographic and clinical information of study subjects
After quality control analyses, 1210 subjects retained in the study, including 459 SAD patients and 751 control subjects. Key demographic and clinical information for these subjects is presented in Table 1. Because the study was frequency matched for sex, no statistically significant difference in proportion of gender was found (P > 0.05). However, due to the frequency match for age (within 5-years), the mean age at examination was slightly, but statistically significantly younger in cases (71.2 years) than controls (72.7 years), P = 0.004. The age at onset in cases ranged from 45 to 88 years, with a mean of 68.5 years. About 35% SAD cases were early age onset (< 65 years). The mean MMSE score was significantly lower in cases (14.7) than in controls (25.1), P < 0.001. Similarly, APOE ε4 carrier rate was significantly higher in cases (41.4%) than in controls (20.0%), P < 0.001.
Table 1. Characteristics of study subjects in the entire cohort.
| Characteristic | SAD (n=459) | Controls (n=751) | P value |
|---|---|---|---|
| Age at examinationa, mean ± SD, yr | 71.2±9.6 | 72.7 ± 5.9 | 0.004 |
| Age at onset, mean ± SD, yr | 68.5±9.7 | ||
| Age at onset <65, n (%) | 161(35.1) | ||
| Age at onset ≥65, n (%) | 298(64.9) | ||
| Sexa, n (%) | 0.407 | ||
| Male | 228(49.7) | 354(47.1) | |
| Female | 231(50.3) | 397(52.9) | |
| MMSE score, mean±SD | 14.7±6.6 | 25.1±3.5 | <0.001 |
| Missing | 1 | ||
| APOE, n (%) | <0.001 | ||
| APOE ε4 carriers | 190 (41.4) | 150 (20.0) | |
| Non-APOE ε4 carriers | 269 (58.6) | 601 (80.0) |
Abbreviation: SAD, Sporadic Alzheimer's disease; MMSE, Mini-mental state examination; SD, Standard deviation.
Frequency matched
These subjects were randomly assigned to the discovery set (232 cases and 373 controls) and testing set (227 cases and 378 controls). As shown in Table S1, there was no significant difference between the case subjects from the discovery and testing sets (P > 0.05), except that mean age at onset was slightly older in the discovery set (69.4 years) than in the testing set (67.6 years), P = 0.048.
Among 33 SNPs selected in the study, 3 SNPs were excluded due to genotyping failure (rs10838725) and minor allele frequency (MAF) < 0.01 (rs7274581 and rs12989701). None of the SNPs significantly deviated from Hardy-Weinberg equilibrium (HWE) among control subjects at P < 0.001. In the following analysis, 30 SNPs were analyzed.
Association of SAD risk with candidate SNPs in the discovery set
In the discovery set, 6 of the 30 SNPs were significantly associated with SAD risk in Chinese (P < 0.05) after adjustment of sex and age (age at onset for SAD patients and age at examination for control subjects) (Table 2). These 6 SNPs were rs9349407 at 6p12 in CD2AP (P = 1.23 × 10−4), rs11218343 at 11q24 in SORL1 (P = 8.32 × 10−4), rs17125944 at 14q22 in FERMT2 (P = 0.048), rs6859 at 19q13 in PVRL2 (P = 0.001), rs157580 at 19q13 in TOMM40 (P = 0.036) and rs2075650 at 19q13 in TOMM40 (P = 7.87 × 10−11). The direction of association was consistent with that of European descent for all 6 SNPs (Table 2). Four of these SNPs (rs9349407, rs11218343, rs17125944, and rs2075650) remained significant after adjusting for APOE genotype, P < 0.05. Meanwhile, we analyzed LD between SNPs at 19 chromosome and APOE genotype (treating ε3 and ε2 as the same allele and ε4 as another allele), and found the last SNP (rs2075650) was in strong LD with APOE genotype, r2 = 0.48 (Table S2). Associations between SNPs and SAD risk were also tested in the testing set (Table S3).
Table 2. Association of sporadic Alzheimer's disease risk with candidate SNPs reported in European descent among Chinese subjects in the discovery set.
| Chr | SNP | Position | Gene | Reported risk allelea | Allele frequency | Association testb | Association testc | |||
|---|---|---|---|---|---|---|---|---|---|---|
| SAD | Controls | OR (95% CI) | P value | OR (95% CI) | P value | |||||
| 1 | rs6656401 | 207,692,049 | CR1 | A | 0.028 | 0.021 | 1.11(0.51-2.39) | 0.800 | 1.13(0.51-2.52) | 0.769 |
| 1 | rs3818361 | 207,784,968 | CR1 | A | 0.354 | 0.378 | 0.93(0.73-1.19) | 0.561 | 0.92(0.72-1.18) | 0.524 |
| 2 | rs7561528 | 127,889,637 | BIN1 | A | 0.123 | 0.151 | 0.82(0.58-1.16) | 0.258 | 0.87(0.61-1.23) | 0.426 |
| 2 | rs744373 | 127,894,615 | BIN1 | G | 0.366 | 0.376 | 0.97(0.76-1.24) | 0.817 | 0.98(0.76-1.25) | 0.848 |
| 2 | rs35349669 | 234,068,476 | INPP5D | T | 0.013 | 0.011 | 1.18(0.39-3.52) | 0.770 | 1.17(0.37-3.67) | 0.793 |
| 5 | rs190982 | 88,223,420 | MEF2C | A | 0.864 | 0.849 | 1.18(0.84-1.68) | 0.342 | 1.27(0.89-1.81) | 0.196 |
| 6 | rs9271192 | 32,578,530 | HLA-DRB5-HLA-DRB1 | C | 0.144 | 0.112 | 1.30(0.91-1.85) | 0.144 | 1.35(0.94-1.94) | 0.108 |
| 6 | rs9349407 | 47,453,378 | CD2AP | C | 0.202 | 0.118 | 1.95(1.39-2.74) | 1.23 × 10−4 | 2.03(1.43-2.88) | 8.38 × 10−5 |
| 6 | rs11754661 | 151,207,078 | MTHFD1L | A | 0.033 | 0.027 | 1.19(0.58-2.41) | 0.634 | 1.19(0.57-2.51) | 0.647 |
| 7 | rs2718058 | 37,841,534 | NME8 | A | 0.754 | 0.797 | 0.79(0.60-1.05) | 0.103 | 0.80(0.60-1.07) | 0.128 |
| 7 | rs1476679 | 100,004,446 | ZCWPW1 | T | 0.708 | 0.705 | 1.07(0.83-1.39) | 0.589 | 1.04(0.80-1.35) | 0.786 |
| 7 | rs11767557 | 143,109,139 | EPHA1 | T | 0.835 | 0.873 | 0.74(0.53-1.03) | 0.072 | 0.76(0.54-1.06) | 0.109 |
| 7 | rs11771145 | 143,110,762 | EPHA1 | G | 0.507 | 0.463 | 1.12(0.88-1.42) | 0.357 | 1.03(0.81-1.32) | 0.803 |
| 8 | rs28834970 | 27,195,121 | PTK2B | C | 0.276 | 0.302 | 0.88(0.67-1.14) | 0.332 | 0.88(0.67-1.16) | 0.367 |
| 8 | rs11136000 | 27,464,519 | CLU | C | 0.832 | 0.806 | 1.18(0.87-1.60) | 0.284 | 1.17(0.86-1.60) | 0.319 |
| 8 | rs569214 | 27,487,790 | CLU | G | 0.489 | 0.505 | 0.97(0.76-1.24) | 0.803 | 0.98(0.76-1.27) | 0.895 |
| 11 | rs983392 | 59,923,508 | MS4A6A | A | 0.959 | 0.972 | 0.77(0.41-1.43) | 0.404 | 0.68(0.36-1.27) | 0.225 |
| 11 | rs610932 | 59,939,307 | MS4A6A | G | 0.667 | 0.646 | 1.11(0.87-1.43) | 0.398 | 1.09(0.84-1.42) | 0.496 |
| 11 | rs4938933 | 60,034,429 | MS4A4A | T | 0.754 | 0.726 | 1.15(0.88-1.51) | 0.305 | 1.15(0.86-1.52) | 0.345 |
| 11 | rs2373115 | 78,091,150 | GAB2 | C | 0.580 | 0.594 | 0.95(0.75-1.20) | 0.648 | 0.93(0.73-1.19) | 0.581 |
| 11 | rs17817600 | 85,677,471 | PICALM | G | 0.037 | 0.043 | 0.85(0.45-1.59) | 0.607 | 0.67(0.35-1.30) | 0.238 |
| 11 | rs3851179 | 85,868,640 | PICALM | C | 0.635 | 0.598 | 1.17(0.92-1.49) | 0.194 | 1.19(0.93-1.52) | 0.177 |
| 11 | rs11218343 | 121,435,587 | SORL1 | T | 0.750 | 0.669 | 1.60(1.22-2.11) | 8.32 × 10−4 | 1.57(1.18-2.09) | 0.002 |
| 14 | rs17125944 | 53,400,629 | FERMT2 | C | 0.263 | 0.218 | 1.32(1.00-1.75) | 0.048 | 1.35(1.02-1.81) | 0.040 |
| 14 | rs10498633 | 92,926,952 | SLC24A4 | G | 0.884 | 0.884 | 1.04(0.72-1.50) | 0.838 | 1.10(0.76-1.61) | 0.606 |
| 19 | rs3764650 | 1,046,520 | ABCA7 | G | 0.321 | 0.276 | 1.25(0.96-1.61) | 0.097 | 1.26(0.96-1.64) | 0.093 |
| 19 | rs6859 | 45,382,034 | PVRL2 | A | 0.403 | 0.307 | 1.51(1.18-1.94) | 0.001 | 1.22(0.93-1.59) | 0.149 |
| 19 | rs157580 | 45,395,266 | TOMM40 | A | 0.531 | 0.473 | 1.29(1.02-1.65) | 0.036 | 1.04(0.80-1.35) | 0.769 |
| 19 | rs2075650 | 45,395,619 | TOMM40 | G | 0.236 | 0.084 | 3.19(2.25-4.52) | 7.87 × 10−11 | 2.50(1.60-3.91) | 6.16 × 10−5 |
| 19 | rs3865444 | 51,727,962 | CD33 | C | 0.838 | 0.840 | 0.97(0.70-1.36) | 0.878 | 1.03(0.73-1.45) | 0.888 |
Abbreviation: SAD, sporadic Alzheimer's disease; Chr, chromosome; SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval.
Risk allele reported in European population.
Association test was adjusted for sex, age (age at onset for SAD patients and age at examination for control subjects).
Association test was adjusted for sex, age (age at onset for SAD patients and age at examination for control subjects) and APOE ε4 status (0 or 1).
GRS calculation and its discriminative performance analysis in the testing set
To assess the cumulative effect of multiple SAD risk-associated SNPs in predicting SAD risk, we calculated GRS for subjects in the independent testing set based on all 6 implicated SNPs in the discovery set (Table 3). The median GRS was significantly higher in SAD cases than in controls, P < 0.001. AUC of GRS in discriminating SAD cases from controls was 0.63, higher than that of APOE (0.60).
Table 3. Association of genetic risk score and SAD risk in the testing set.
| # of subjects (SAD/Controls) | Mean GRS | Median GRS | |||||
|---|---|---|---|---|---|---|---|
| Sample set | SAD | Controls | SAD | Controls | P value | AUC | |
| GRS based on 6 SNPs | 227/378 | 2.48 | 1.15 | 0.85 | 0.55 | <0.001 | 0.63 |
| APOE ε4 carriers | 93/81 | 4.57 | 2.47 | 2.51 | 1.69 | 0.003 | 0.64 |
| APOE ε4 non-carriers | 134/297 | 1.03 | 0.79 | 0.53 | 0.47 | 0.101 | 0.55 |
| Modified GRS based on 3 non APOE-related SNPs | 227/378 | 1.02 | 0.91 | 0.87 | 0.87 | 0.002 | 0.58 |
| APOE ε4 carriers | 93/81 | 1.01 | 0.92 | 0.87 | 0.87 | 0.187 | 0.56 |
| APOE ε4 non-carriers | 134/297 | 1.02 | 0.91 | 0.87 | 0.87 | 0.006 | 0.58 |
Abbreviation: SAD, sporadic Alzheimer's disease; GRS, genetic risk score
Considering that the effect of GRS may be confounded by APOE, we calculated a modified GRS by removing three SNPs that are related to APOE: two SNPs that were no longer significantly associated with SAD risk after adjusting for APOE genotypes (rs6859 and rs157580) and one SNP that was in strong LD with APOE genotypes (rs2075650). In the independent testing set, the modified GRS was significantly higher in SAD cases than in controls, P = 0.002 (Table 3). The AUC of the modified GRS was 0.58. When this modified GRS was combined with APOE genotypes, the AUC was 0.64, significantly higher than that of APOE alone (0.60), P = 0.003 (Figure 1).
Figure 1. Receiver operating characteristic curves for genetic models among Chinese subjects in the testing set.
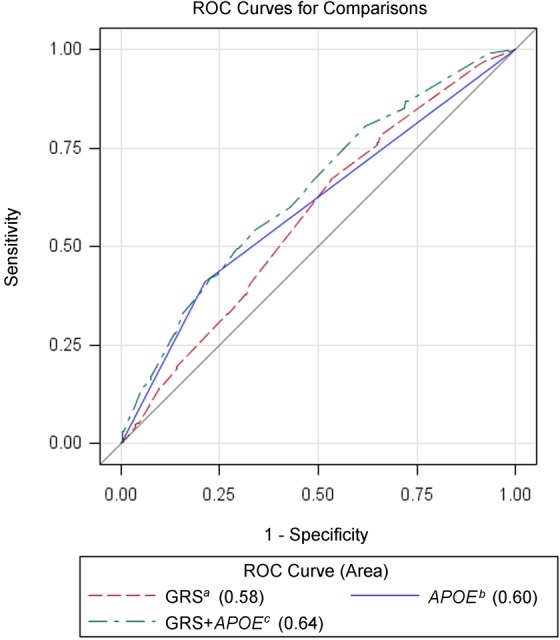
a Modified genetic risk score (GRS) based on 3 SNPs (rs9349407, rs11218343 and rs17125944). b APOE ε4 status (0 or 1). c Combination of modified GRS and APOE.
Association analysis of GRS and SAD risk
When the modified GRS was analyzed in subjects stratified by the APOE ε4 status in the testing set, similar trends were observed in both APOE ε4 carriers and non-carriers, although the association and discriminative performance of modified GRS was slightly stronger in non-carriers (Table 3).
DISCUSSION
The primary purpose of this study was to assess performance of multiple risk-associated SNPs for predicting SAD risk in the Han Chinese population. To achieve this goal, we firstly identified SNPs that were associated with SAD risk among Chinese in a discovery set. We then assessed the cumulative effect of these implicated SNPs, as measured by GRS, on association of SAD risk and ability to discriminate SAD patients from non-dementia controls in the testing set. Furthermore, to assess whether the predictive performance of multiple SAD risk-associated SNPs are independent of APOE genotypes, we calculated a modified GRS that based on three SAD risk-associated SNPs that are independent of APOE genotypes. With this rigorously strategy, we demonstrated that modified GRS was able to discriminate SAD patients from controls, with an AUC of 0.58. When combined the modified GRS with APOE, the AUC increased to 0.64, significantly higher than APOE alone (0.60), P = 0.003.
To our knowledge, this is the first report assessing cumulative effect of multiple AD risk-associated SNPs on association and discrimination of SAD. It is well recognized that effect of individual SNPs on AD risk is modest. However, it is hypothesized that cumulatively they have a stronger effect. As demonstrated in this study, the AUC of modified GRS based on three implicated SAD risk-associated SNPs (0.58) was similar to that of well-established APOE (0.60). This result offers empirical evidence to support this cumulative effect hypothesis and provides basis for additional larger and more comprehensive studies to further test the hypothesis in Chinese and European descent. With more established AD risk-associated SNPs (such as in European descent), it is expected that cumulative effect will be stronger.
The findings that SAD risk-associated SNPs are associated with SAD risk in both APOE ε4 allele carriers and non-carriers and that they add value to APOE in discriminating SAD cases from controls are important. On one hand, these results suggest SAD risk-associated SNPs play similar roles in the etiology of SAD among APOE ε4 allele carriers and non-carriers. On the other hand, it is practically important in assessment of SAD risk. The APOE ε4 allele has been found to be the strongest risk factor for SAD [9-12]. A meta-analysis of clinical and autopsy-based studies demonstrated that individuals with ε4 allele have increased AD risk compared with ε3/ε3 genotype in Caucasian population (OR was 2.6, 3.2, and 14.9 for individuals with ε2/ε4, ε3/ε4, and ε4/ε4, respectively) [32]. However, the AUC of APOE alone in discriminating SAD remains moderate (for example, AUC was 0.62 and 0.60 in the discovery set and testing set of our study, respectively). Furthermore, it is recognized that 40~50% of SAD patients do not carry APOE ε4 allele [11, 13]. This number was even higher in our study where 58.6% SAD patients did not carry APOE ε4 allele (Table 1). Therefore, identifying tool for better risk assessment of AD risk, especially among subjects without APOE ε4 allele is necessary. Better risk assessment may identify subjects at higher risk for SAD for targeted prevention. Subjects with a higher risk for SAD may be more motivated to take action to prevent AD through reducing life-style risk factors associated with AD.
Family history is another well-established risk factor for AD. The relative risk of AD for those with at least one first degree relative with dementia was estimated at 3.2-3.8 [33-35]. Unfortunately, we could not assess the effect of family history on AD risk in this study because we focused on SAD in this study, and none of the AD patients had a positive family history by our inclusion criteria. However, the fact that we demonstrated GRS based multiple inherited risk-associated SNPs is a predictor of SAD risk among patients without a known family history indirectly suggests family history alone is not sufficient to capture inherited risk for AD. In contrast to GRS that is based on individual's own risk-associated SNPs, family history is an indirect measurement of familial risk (inherited and shared household environment) through their relatives, and is therefore influenced by the number, age, and competing mortality of their relatives. This limitation is more prominent for late age onset diseases such as AD. Thus, lack of a known family at the time of examination may not necessarily indicate that individuals are at lower risk for AD. Studies are needed to assess the combined performance of family history, GRS, and APOE in assessing AD risk.
We randomly divided our study subjects into two sets of equal sample size: discovery and testing. The major advantage of this approach is that we can identify SAD risk-associated SNPs in Han Chinese and obtain their OR in the discovery set and then objectively assess the performance of these SNPs in the independent testing set. However, this approach reduced the statistical power to detect association of SNPs with SAD risk. Among the 30 SNPs tested in the discovery set, 19 SNPs had the same direction of association as in the studies of European descent, although only 6 SNPs reached the statistical significance of P < 0.05. Larger sample size may be needed to confirm additional AD risk-associated SNPs in Han Chinese.
Several case-control studies on association of AD risk-associated SNPs reported in GWAS of European descent with AD risk in Chinese population were published in the last several years. However, few SNPs were consistently implicated among these studies. For example, Chen et al. [21] evaluated 7 SNPs (rs3818361 and rs6656401 in CR1; rs11136000, rs2279590, and rs9331888 in CLU; rs3851179 and rs541458 in PICALM) among 462 AD patients and 350 control subjects from southern Chinese population. Of the 7 SNPs, rs6656401 (P = 0.035) and rs3818361 (P = 0.029) in CR1, and rs11136000 in CLU (P = 0.038) were confirmed; rs3851179 in PACALM showed significant association with LOAD only in APOE ε4 non-carriers (P = 0.028). Tan et al. [25] assessed a total of 10 SNPs among 612 sporadic late-onset AD (LOAD) patients and 612 control subjects from northern Han Chinese, including 2 in BIN1 (rs7561528 and rs744373), 2 in ABCA7 (rs3752246 and rs3764650), 3 in the MS4A gene cluster (rs4938933, rs610932, and rs670139), and 1 each in CD2AP (rs9349407), CD33 (rs3865444), and EPHA1 (rs11767557). Based on a multivariate analysis, rs610932 in MS4A6A (P = 0.019) and rs3865444 in CD33 (P = 0.017) were confirmed; rs7561528 in BIN1 was confirmed only in APOE ε4 carriers (P = 0.039). Ma et al. [26] conducted a replication study of rs11754661 and rs2073067 in MTHFD1L among 582 LOAD subjects and 607 healthy controls from northern Han Chinese. The rs11754661 was confirmed (P = 0.016). Liu et al. [23] evaluated and confirmed the association of rs3764650 in ABCA7 with SAD in 350 SAD and 283 non-demented elderly controls from Han Chinese, P = 0.004. Ma et al. [27] investigated the association of three SNP (rs157580, rs2075650 and rs11556505 in TOMM40) with LOAD among 787 LOAD patients and 791 healthy subjects. The rs157580 (P < 0.001) and rs2075650 (P = 0.001) were confirmed. Among these confirmed SNPs (rs3818361, rs6656401, rs7561528, rs11754661, rs11136000, rs610932, rs3851179, rs3764650, rs3865444, rs157580 and rs2075650), 2 SNPs (rs157580 and rs2075650 in TOMM40) were also found to be significantly associated with SAD in our study. Except for 2 SNPs (rs3818361 and rs7561528), the rest SNPs have the same direction of association with previous findings in Chinese. Multiple factors may contribute to these different confirmed SNPs, including small sample size, different criteria for AD patients and controls, and different genetic background between northern and southern Han Chinese.
The three SNPs, independent of APOE genotypes and used in GRS calculating to predict SAD risk in the study, are rs9349407 in CD2AP (intron 1), rs11218343 in SORL1 (intron 21), and rs17125944 in FERMT2 (intron 14). The functions of these 3 genes have been reported to be relevant to the development of AD. Both CD2AP and FERMT2 have been implicated in cell adhesion [36, 37]. In Drosophila model of AD, CD2AP and FERMT2 were identified as modifier of Tau neurotoxicity, which related to neurofibrillary tangle pathology in AD [38]. SORL1 encodes a neuronal sorting protein that binds APP protein and directs it towards the endosome-recycling pathways [39] and variants in SORL1 were significantly associated with cerebrospinal Aβ42 levels [40], which reflect the metabolic process in brain and was used to aid the diagnosis of AD at an early stage of disease.
In summary, results from this well-designed but underpowered study provided preliminary evidence that multiple AD risk-associated SNPs can be used to supplement APOE to better define individual's risk for AD. Larger studies are justified to formally test the hypothesis and assess its predictive performance.
MATERIALS AND METHODS
Study population
Subjects included in this study (515 SAD patients and 770 cognitively normal controls) were Chinese Han, and were recruited during 2008-2013. SAD patients, comprising early- and late-onset SAD (age at onset ranged from 45 to 88 years), were recruited from Huashan Hospital in Shanghai and were diagnosed as probable AD according to DSM-IV-R and NINCDS-ADRDA criteria [41, 42]. All SAD patients reported no family history of AD. The cognitively normal controls were recruited from communities in Shanghai and were carefully evaluated based on mini-mental state examination (MMSE) and years of education. They were frequency matched for SAD cases by gender and age. Two senior neurologists reviewed all data and confirmed the diagnosis. APOE genotype status, measured by method described by Donohoe et al. [43], was available in cases and controls. This study was approved by the ethics committee of Huashan Hospital and written informed consents were completed for all study subjects.
After genotyping, subjects with a missing rate of > 20% were removed from the study (56 SAD patients and 19 control subjects). The retained subjects (459 SAD patients and 751 controls) were randomly divided into discovery set (232 cases and 373 controls) and testing set (227 cases and 378 controls). Association of AD risk-associated SNPs reported in European descent with SAD risk was firstly tested in the discovery set. Significant SNPs were used to calculate GRS in the testing set.
SNP selection
A total of 33 SAD risk-associated SNPs were selected using the following criteria: 1) association with AD risk in European population exceeded the threshold of a genome-wide significance level (P < 5 × 10−8) and published before Jan 2014; 2) if multiple SNPs that are in strong linkage disequilibrium (LD) met the above criterion, defined by pairwise r2 > = 0.2 estimated from the HapMap CHB (Han Chinese in Beijing, China) population, the most commonly cited SNP was selected. The information of all SNPs that be chosen was listed in Table S4.
SNP genotyping and Quality control
Genotyping of selected SNPs was performed using the Sequenom MassArray system (iPLEX; Sequenom, Inc. San Diego, CA) at the Centre for Genomic Translational Medicine and Prevention, School of Public Health, Fudan University. Duplicates from two subjects and two water samples (negative control) were included in each 96-well plate for genotyping quality control. All assays were conducted blinded to case-control status. The overall concordance rate was 100% among the duplicated quality control samples. A quality control was conducted to select the samples (mentioned in “Study Population” section) and SNPs for further analysis. SNPs with a missing rate of > 5%, the minor allele frequency (MAF) of < 0.01 in either cases or controls, or with Hardy-Weinberg equilibrium (HWE) test at P < 0.001 among controls were excluded.
Statistical analysis
Differences between cases and controls were tested using t-test for quantitative variables and Chi-square test for qualitative variables. Associations between SNPs and SAD risk were tested for each SNP using an additive model adjusted for 1) sex, age (age at onset for SAD patients and age at examination for control subjects); 2) sex, age (age at onset for SAD patients and age at examination for control subjects) and APOE ε4 status (0 or 1). The allelic odds ratio (OR) and 95% confidence intervals (CI) were estimated using a logistic regression model.
GRS for each subject was calculated based on SAD risk-associated SNPs established in the discovery set using the method described by Pharoah et al. [44]. Briefly, 1) the allelic OR of each SNP was obtained from the discovery set, 2) the genotypic OR of each SNP was estimated from the allelic OR assuming a multiplicative model, 3) the risk relative to the average risk in the population was calculated for each genotype based on genotypic OR and genotype frequency in the HapMap CHB (Han Chinese in Beijing, China) population, and 4) a GRS was obtained by multiplying the risks relative to the population of all SNPs. Therefore, a GRS of 1.0 indicates an average risk in the general population.
Non-parametric analysis (Wilcoxon Rank Sum Test) was used to test association of GRS and SAD risk. The performance of GRS in discriminating SAD cases from controls was evaluated using the area under the receiver operating characteristic curve (AUC). Difference in AUC between two predictive model was tested using the method described by DeLong and colleagues [45].
All statistical analyses were performed using the PLINK software (version 1.07) [46] and SAS software (version 9.2; SAS Institute, Cary, NC). All statistical tests were two-sided.
Acknowledgments
We sincerely thank all participants who agreed to participate in this study. This work was supported by the grant from the National Natural Science Foundation to Zhi-Ying Wu (81125009, Beijing). This work was also partially supported by the Ellrodt-Schweighauser Family Chair of Cancer Genomic Research of North Shore University HealthSystem to Jianfeng Xu.
Footnotes
Authors' Contributions
Study conception and design: JFX and ZYW. Acquisition, analysis, or interpretation of data: QYX, ZJL, YMS, HLL, HTC and LX. Statistical analysis: QYX, ST, DKJ, BL and CHW. Manuscript drafting: QYX, ST. Manuscript revision: ZYW, JFX, DKJ and SLZ.
CONFLICTS OF INTEREST
All authors report no disclosures relevant to the manuscript and declare no conflicts of interest.
REFERENCES
- 1.Wimo A, Jonsson L, Bond J, Prince M, Winblad B. The worldwide economic impact of dementia 2010. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2013;9:1–11. doi: 10.1016/j.jalz.2012.11.006. e13. [DOI] [PubMed] [Google Scholar]
- 2.Chan KY, Wang W, Wu JJ, Liu L, Theodoratou E, Car J, Middleton L, Russ TC, Deary IJ, Campbell H, Wang W, Rudan I. Epidemiology of Alzheimer's disease and other forms of dementia in China, 1990-2010: a systematic review and analysis. Lancet. 2013;381:2016–2023. doi: 10.1016/S0140-6736(13)60221-4. [DOI] [PubMed] [Google Scholar]
- 3.Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer's disease: an analysis of population-based data. The Lancet Neurology. 2014;13:788–794. doi: 10.1016/S1474-4422(14)70136-X. [DOI] [PubMed] [Google Scholar]
- 4.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 6.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science (New York, NY) 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 7.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 8.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 9.Chartier-Harlin MC, Parfitt M, Legrain S, Perez-Tur J, Brousseau T, Evans A, Berr C, Vidal O, Roques P, Gourlet V, et al. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer's disease: analysis of the 19q13. 2 chromosomal region. Human molecular genetics. 1994;3:569–574. doi: 10.1093/hmg/3.4.569. [DOI] [PubMed] [Google Scholar]
- 10.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science (New York, NY) 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 11.Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I, White LR, Lydersen S, Aasly JO. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer's disease; a case control study from central Norway. BMC neurology. 2008;8:9. doi: 10.1186/1471-2377-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Duijn CM, de Knijff P, Cruts M, Wehnert A, Havekes LM, Hofman A, Van Broeckhoven C. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer's disease. Nature genetics. 1994;7:74–78. doi: 10.1038/ng0594-74. [DOI] [PubMed] [Google Scholar]
- 13.Ashford JW, Mortimer JA. Non-familial Alzheimer's disease is mainly due to genetic factors. Journal of Alzheimer's disease : JAD. 2002;4:169–177. doi: 10.3233/jad-2002-4307. [DOI] [PubMed] [Google Scholar]
- 14.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature genetics. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature genetics. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nature genetics. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 17.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nature genetics. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naj AC, Beecham GW, Martin ER, Gallins PJ, Powell EH, Konidari I, Whitehead PL, Cai G, Haroutunian V, Scott WK, Vance JM, Slifer MA, Gwirtsman HE, et al. Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS genetics. 2010;6:e1001130. doi: 10.1371/journal.pgen.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nature genetics. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen LH, Kao PY, Fan YH, Ho DT, Chan CS, Yik PY, Ha JC, Chu LW, Song YQ. Polymorphisms of CR1, CLU and PICALM confer susceptibility of Alzheimer's disease in a southern Chinese population. Neurobiology of aging. 2012;33:210–e211-217. doi: 10.1016/j.neurobiolaging.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Deng YL, Liu LH, Wang Y, Tang HD, Ren RJ, Xu W, Ma JF, Wang LL, Zhuang JP, Wang G, Chen SD. The prevalence of CD33 and MS4A6A variant in Chinese Han population with Alzheimer's disease. Human genetics. 2012;131:1245–1249. doi: 10.1007/s00439-012-1154-6. [DOI] [PubMed] [Google Scholar]
- 23.Liu LH, Xu J, Deng YL, Tang HD, Wang Y, Ren RJ, Xu W, Ma JF, Wang G, Chen SD. A complex association of ABCA7 genotypes with sporadic Alzheimer disease in Chinese Han population. Alzheimer disease and associated disorders. 2014;28:141–144. doi: 10.1097/WAD.0000000000000000. [DOI] [PubMed] [Google Scholar]
- 24.Ma JF, Liu LH, Zhang Y, Wang Y, Deng YL, Huang Y, Wang G, Xu W, Cui PJ, Fei QZ, Ding JQ, Tang HD, Chen SD. Association study of clusterin polymorphism rs11136000 with late onset Alzheimer's disease in Chinese Han population. American journal of Alzheimer's disease and other dementias. 2011;26:627–630. doi: 10.1177/1533317511432735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan L, Yu JT, Zhang W, Wu ZC, Zhang Q, Liu QY, Wang W, Wang HF, Ma XY, Cui WZ. Association of GWAS-linked loci with late-onset Alzheimer's disease in a northern Han Chinese population. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2013;9:546–553. doi: 10.1016/j.jalz.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Ma XY, Yu JT, Wu ZC, Zhang Q, Liu QY, Wang HF, Wang W, Tan L. Replication of the MTHFD1L gene association with late-onset Alzheimer's disease in a Northern Han Chinese population. Journal of Alzheimer's disease : JAD. 2012;29:521–525. doi: 10.3233/JAD-2011-111847. [DOI] [PubMed] [Google Scholar]
- 27.Ma XY, Yu JT, Wang W, Wang HF, Liu QY, Zhang W, Tan L. Association of TOMM40 polymorphisms with late-onset Alzheimer's disease in a Northern Han Chinese population. Neuromolecular medicine. 2013;15:279–287. doi: 10.1007/s12017-012-8217-7. [DOI] [PubMed] [Google Scholar]
- 28.Kader AK, Sun J, Reck BH, Newcombe PJ, Kim ST, Hsu FC, D'Agostino RB, Jr, Tao S, Zhang Z, Turner AR, Platek GT, Spraggs CF, Whittaker JC, et al. Potential impact of adding genetic markers to clinical parameters in predicting prostate biopsy outcomes in men following an initial negative biopsy: findings from the REDUCE trial. European urology. 2012;62:953–961. doi: 10.1016/j.eururo.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mavaddat N, Pharoah PD, Michailidou K, Tyrer J, Brook MN, Bolla MK, Wang Q, Dennis J, Dunning AM, Shah M, Luben R, Brown J, Bojesen SE, et al. Prediction of breast cancer risk based on profiling with common genetic variants. Journal of the National Cancer Institute. 2015:107. doi: 10.1093/jnci/djv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA, Levy D, D'Agostino RB, Hwang SJ, O'Donnell CJ. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circulation Cardiovascular genetics. 2012;5:113–121. doi: 10.1161/CIRCGENETICS.111.961342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tam CH, Ho JS, Wang Y, Lam VK, Lee HM, Jiang G, Lau ES, Kong AP, Fan X, Woo JL, Tsui SK, Ng MC, So WY, et al. Use of net reclassification improvement (NRI) method confirms the utility of combined genetic risk score to predict type 2 diabetes. PloS one. 2013;8:e83093. doi: 10.1371/journal.pone.0083093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 33.van Duijn CM, Clayton D, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Mortimer JA, Rocca WA, Shalat SL, Soininen H, et al. Familial aggregation of Alzheimer's disease and related disorders: a collaborative re-analysis of case-control studies. International journal of epidemiology. 1991;20(Suppl 2):S13–20. doi: 10.1093/ije/20.supplement_2.s13. [DOI] [PubMed] [Google Scholar]
- 34.Fratiglioni L, Ahlbom A, Viitanen M, Winblad B. Risk factors for late-onset Alzheimer's disease: a population-based, case-control study. Annals of neurology. 1993;33:258–266. doi: 10.1002/ana.410330306. [DOI] [PubMed] [Google Scholar]
- 35.Payami H, Grimslid H, Oken B, Camicioli R, Sexton G, Dame A, Howieson D, Kaye J. A prospective study of cognitive health in the elderly (Oregon Brain Aging Study): effects of family history and apolipoprotein E genotype. American journal of human genetics. 1997;60:948–956. [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf G, Stahl RA. CD2-associated protein and glomerular disease. Lancet. 2003;362:1746–1748. doi: 10.1016/S0140-6736(03)14856-8. [DOI] [PubMed] [Google Scholar]
- 37.Lai-Cheong JE, Parsons M, McGrath JA. The role of kindlins in cell biology and relevance to human disease. The international journal of biochemistry & cell biology. 2010;42:595–603. doi: 10.1016/j.biocel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, Hu Y, Devenport D, Chipendo P, Chibnik LB, Diamond A, Perrimon N, Brown NH, De Jager PL, Feany MB. Functional screening in Drosophila identifies Alzheimer's disease susceptibility genes and implicates Tau-mediated mechanisms. Human molecular genetics. 2014;23:870–877. doi: 10.1093/hmg/ddt478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, Thomas-Anterion C, Michon A, Croisile B, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Molecular psychiatry. 2012;17:875–879. doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- 40.Alexopoulos P, Guo LH, Kratzer M, Westerteicher C, Kurz A, Perneczky R. Impact of SORL1 single nucleotide polymorphisms on Alzheimer's disease cerebrospinal fluid markers. Dementia and geriatric cognitive disorders. 2011;32:164–170. doi: 10.1159/000332017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, Nordberg A, Backman L, Albert M, Almkvist O, Arai H, Basun H, Blennow K, et al. Mild cognitive impairment—beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. Journal of internal medicine. 2004;256:240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- 42.Mittal VA, Walker EF. Diagnostic and statistical manual of mental disorders. Psychiatry research. 2011;189:158–159. doi: 10.1016/j.psychres.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donohoe GG, Salomaki A, Lehtimaki T, Pulkki K, Kairisto V. Rapid identification of apolipoprotein E genotypes by multiplex amplification refractory mutation system PCR and capillary gel electrophoresis. Clinical chemistry. 1999;45:143–146. [PubMed] [Google Scholar]
- 44.Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. The New England journal of medicine. 2008;358:2796–2803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- 45.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 46.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]