

Abstract
Background and Purpose
Inflammasomes are multimeric complexes that facilitate caspase‐1‐mediated processing of the pro‐inflammatory cytokines IL‐1β and IL‐18. Clinical hypertension is associated with renal inflammation and elevated circulating levels of IL‐1β and IL‐18. Therefore, we investigated whether hypertension in mice is associated with increased expression and/or activation of the inflammasome in the kidney, and if inhibition of inflammasome activity reduces BP, markers of renal inflammation and fibrosis.
Experimental Approach
Wild‐type and inflammasome‐deficient ASC −/− mice were uninephrectomized and received deoxycorticosterone acetate and saline to drink (1K/DOCA/salt). Control mice were uninephrectomized but received a placebo pellet and water. BP was measured by tail cuff; renal expression of inflammasome subunits and inflammatory markers was measured by real‐time PCR and immunoblotting; macrophage and collagen accumulation was assessed by immunohistochemistry.
Key Results
1K/DOCA/salt‐induced hypertension in mice was associated with increased renal mRNA expression of inflammasome subunits NLRP3, ASC and pro‐caspase‐1, and the cytokine, pro‐IL‐1β, as well as protein levels of active caspase‐1 and mature IL‐1β. Following treatment with 1K/DOCA/salt, ASC −/− mice displayed blunted pressor responses and were also protected from increases in renal expression of IL‐6, IL‐17A, CCL2, ICAM‐1 and VCAM‐1, and accumulation of macrophages and collagen. Finally, treatment with a novel inflammasome inhibitor, MCC950, reversed hypertension in 1K/DOCA/salt‐treated mice.
Conclusions and Implications
Renal inflammation, fibrosis and elevated BP induced by 1K/DOCA/salt treatment are dependent on inflammasome activity, highlighting the inflammasome/IL‐1β pathway as a potential therapeutic target in hypertension.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- AIM2
absent in melanoma 2
- ASC
apoptosis‐associated speck‐like protein containing a carboxy‐terminal CARD
- CARD
caspase activation and recruitment domain
- CCL
C‐C motif chemokine ligand
- DAMP
danger‐associated molecular pattern
- DOCA
deoxycorticosterone acetate
- HRP
horseradish peroxidase
- ICAM‐1
intercellular adhesion molecule‐1
- NLRC4 (IPAF)
NOD‐like receptor family CARD domain‐containing protein 4
- NLRP
NOD‐like receptor family pyrin domain‐containing protein
- PAMP
pathogen‐associated molecular pattern
- VCAM‐1
vascular cell adhesion molecule‐1
Tables of Links
| LIGANDS | |||
|---|---|---|---|
| Allopurinol | CCL2 | IFN‐γ | LPS |
| Anakinra | CCL5 | IL‐1β | MCC950 |
| Angiotensin II | Clexane | IL‐6 | Osteopontin |
| ATP | Deoxycorticosterone | IL‐17A | Rilonacept |
| Biotin | ICAM‐1 | IL‐18 | TNF |
| Canakinumab | IL‐23 | VCAM‐1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a, 2013b).
Introduction
Hypertension is now recognized as a chronic, low‐grade inflammatory disease, with the kidneys representing a major site of this inflammation (Harrison et al., 2011; Rodriguez‐Iturbe et al., 2012). Through tight regulation of the pressure–natriuresis relationship, whereby increments in BP are accompanied by a compensatory reduction in sodium reabsorption in the proximal tubules, the kidneys play a major role in BP homeostasis (Rodriguez‐Iturbe et al., 2012). By promoting such processes as the intrarenal accumulation of T‐cells and macrophages (Mattson, 2014; Wei et al., 2014), a loss of the peritubular vascular network (Rodriguez‐Iturbe and Johnson, 2010), up‐regulation of Na+ transport/re‐uptake mechanisms (Meng et al., 2006; Aoi et al., 2007), and interstitial collagen production (Mezzano et al., 2001; Blasi et al., 2003), inflammation may blunt the pressure–natriuresis relationship and thereby contribute to hypertension. Hence, a better understanding of the processes that give rise to renal inflammation, may pave the way for novel anti‐hypertensive therapies.
Inflammasomes are sensors of the innate immune system that play important roles in initiating inflammation in response to acute infections and chronic diseases. Inflammasomes are multimeric complexes that can be activated by a diverse range of pathogen‐ and danger‐associated molecular patterns (‘PAMPs’ and ‘DAMPs’, respectively), including bacterial pore‐forming toxins such as nigericin (Mariathasan et al., 2006), crystalline substances such as uric acid (Martinon et al., 2006) and cholesterol (Duewell et al., 2010), misfolded and/or acute phase proteins such as β‐amyloid (Halle et al., 2008) and serum amyloid A (Niemi et al., 2011), and extracellular ATP (Mariathasan et al., 2006). To date, four inflammasome complexes have been described, each displaying selectivity towards a different suite of PAMPs and DAMPs. Inflammasomes are comprised of one of four distinct pattern recognition receptors – NOD‐like receptor family pyrin domain‐containing protein (NLRP) 1, NLRP3, ICE protease‐activating factor/NOD‐like receptor family CARD domain‐containing protein 4 (NLRC4) or absent in melanoma 2 (AIM2) – and it is this sensor that defines the inflammasome and confers its selectivity (Latz, 2010; Schroder and Tschopp, 2010a). Once activated, the inflammasome recruits pro‐caspase‐1, either via an adapter protein, ASC [apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (CARD)], or homotypically using its own CARD. The clustering of several pro‐caspase‐1 subunits at the inflammasome complex results in auto‐cleavage and formation of active caspase‐1 heterodimers, each consisting of a p10 and p20 subunit. These active caspase‐1 heterodimers convert inactive cytosol‐bound pro‐IL‐1β and pro‐IL‐18 into their mature forms, IL‐1β and IL‐18, respectively, which can be released from the cell to activate pro‐inflammatory signalling pathways in both an autocrine and paracrine manner (Latz, 2010; Schroder and Tschopp, 2010a).
As a safeguard against unwanted inflammatory responses, expression of inflammasome components and of the cytokine precursors, pro‐IL‐1β and pro‐IL‐18, are normally kept at very low levels within cells (Dinarello, 1996; 1999; Bauernfeind et al., 2009). This requires the system to be ‘primed’ before inflammasome activation and IL‐1β/IL‐18 production can commence. Inflammasome priming normally occurs downstream of the transcription factor NF‐κB (Bauernfeind et al., 2009) and essentially involves trans‐activation of the various genes that encode the different inflammasome subunits and their cytokine targets. Stimuli known to lead to inflammasome priming include toll‐like receptor ligands such as LPS, cytokines such as TNF and reactive oxygen species (Kahlenberg et al., 2005; Bauernfeind et al., 2009; 2011).
The NLRP3 inflammasome is the most widely studied of the four inflammasomes identified. Unlike other inflammasomes which are activated by a limited array of specific PAMPs or DAMPs, such as the anthrax lethal toxin and muramyl dipeptide (NLRP1), cytosolic flagellin of various bacteria (NLRC4), or cytosolic double‐stranded DNA (AIM2), the NLRP3 inflammasome can be activated by multiple stimuli including LPS, reactive oxygen species, ATP and microcrystals, and is thus believed to be the most important isoform contributing to inflammation in the setting of chronic diseases (Schroder and Tschopp, 2010a; Schroder et al., 2010b; Wen et al., 2012; Ozaki et al., 2015).
Previous studies have shown that hypertension in humans is associated with increased circulating levels of IL‐1β and IL‐18 (Dalekos et al., 1997; Rabkin, 2009; Krishnan et al., 2014; Barbaro et al., 2015). Hence, in the present study, we investigated whether hypertension induced by uninephrectomy and subsequent treatment with deoxycorticosterone acetate and salt in mice (1K/DOCA/salt) is associated with an increase in inflammasome expression and or activity in the kidneys. Furthermore, we evaluated whether inhibition of inflammasome activity, either due to a genetic deficiency in the adaptor protein, ASC, or as a result of treatment with a novel NLRP3 inhibitor, was effective at reducing renal inflammation, fibrosis and hypertension in response to 1K/DOCA/salt treatment.
Methods
Animals
Male wild‐type and ASC−/− mice (Mariathasan et al., 2004), fully backcrossed onto a C57BL6/J background, aged 10–12 weeks, and weighing 25–30 g were used in this study. Mice were obtained from Monash Animal Services (Clayton, Victoria, Australia) and housed in standard mouse boxes with ad libitum access to normal chow and water. All procedures on mice were performed in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes (8th Edition) and were approved by the Monash University Animal Research Platform Animal Ethics Committee (Ethics number: MARP/2013/043). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
One kidney/deoxycorticosterone acetate/salt model of hypertension
All surgeries were performed under anaesthesia induced by inhalation of 2% isoflurane. While under anaesthesia, hind‐paw withdrawal, blink reflexes and respiratory rate were monitored. Hypertension was induced in wild‐type and ASC−/− mice by removal of their left kidney, implantation of a deoxycorticosterone acetate pellet (DOCA; 2.4 mg·day−1, s.c.; Innovative Research of America, Sarasota, FL, USA) and replacement of their drinking water with 0.9% saline (1K/DOCA/salt) (Manhiani et al., 2009). Normotensive controls for these experiments were mice that were also uninephrectomized but received a placebo pellet (Innovative Research of America) and normal drinking water (1K/placebo).
Angiotensin II‐infusion model of hypertension
In another cohort of wild‐type and ASC−/− mice, hypertension was instead induced by implantation of a micro‐osmotic pump (Model 1004, Alzet, Cupertino, CA, USA) containing angiotensin II (to deliver 0.7 mg·kg−1·day−1, s.c.; Moore et al., 2013). Normotensive controls received the vehicle for angiotensin II (i.e. 0.9% saline; 0.11 μL·h−1, s.c.) via micro‐osmotic pumps.
Administration of MCC950 to mice with 1K/DOCA/salt‐induced hypertension
A separate cohort of mice were treated with MCC950; a novel diarylsulfonylurea‐based compound that acts as a selective inhibitor of NLRP3 inflammasome activity by preventing oligomerization of the complex (Coll et al., 2015). MCC950 was administered as an intervention in mice with established hypertension. Briefly, hypertension was induced in mice by 1K/DOCA/salt treatment as described above. After 10 days (i.e. when BP had increased to a stable plateau), a micro‐osmotic pump was implanted to release either MCC950 (10 mg·kg−1·day−1, s.c.) or vehicle (saline; 0.5 μL·h−1, s.c.) for the remaining 11 days of treatment.
BP measurements
Systolic BP was measured by tail cuff plethysmography (MC4000 Multi Channel Blood Pressure Analysis System, Hatteras Instruments, Cary, NC, USA; Krege et al., 1995). BP was recorded everyday for 3 days prior to surgery in order to acclimatize mice to the procedure. BP was then measured just prior to surgery (day 0) and then re‐measured after 3, 7, 10, 14, 21 and 28 days.
Gene expression in the kidney
At the end of the treatment period, mice were killed by isoflurane overdose and perfused through the left ventricle with PBS containing 0.2% 400 IU of Clexane (Sanofi Aventis, Paris, France). The right kidney was excised, cut in half transversally, snap‐frozen in liquid nitrogen, and stored at −80°C until time of assay. RNA was extracted from one quarter of the kidney using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The yield and purity of RNA was determined using a NanoDrop 1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). RNA was converted to cDNA using a reverse transcription kit (Applied Biosystems, Foster City, CA, USA) as per the manufacturer's instructions. The cDNA was then used as a template in real‐time PCR to measure mRNA expression of NLRP3, ASC, pro‐caspase‐1, pro‐IL‐1β, pro‐IL‐18, IL‐6, IL‐17A, IFN‐γ, TNF, C‐C motif chemokine ligand (CCL) 2, CCL5, intercellular adhesion molecule‐1 (ICAM‐1), vascular cell adhesion molecule‐1 (VCAM‐1), or the house keeping gene, GAPDH (TaqMan Gene Expression Assays, Applied Biosystems). Real‐time PCR was performed in a Bio‐Rad CFX96 Real‐Time PCR Detection System (Bio‐Rad Laboratories, Hercules, CA, USA) and the comparative Ct method was used to calculate the fold‐change in mRNA expression relative to a reference control sample (Schmittgen and Livak, 2008).
Caspase‐1 and IL‐1β protein expression in the kidney
Protein lysates were prepared from the other quarter of the kidney in 1.5× Laemmli buffer (7.5% glycerol, 3.75% β‐mercaptoethanol, 2.25% SDS, 75 mM Tris‐HCl pH 6.8, 0.003% bromophenol blue). One hundred and fifty μg of protein was heated at 80°C and loaded onto a 12.5% polyacrylamide gel, in parallel with a molecular weight marker (Precision Plus Protein Dual Colour Standards; Bio‐Rad Laboratories). Proteins were separated according to molecular weight by electrophoresis (60 V for 30 min followed by 110 V for 2 h) and then transferred onto a 0.45 μm pore Immobilon‐P PVDF membrane (Millipore Corporation, Billercia, MA, USA) using a semi‐dry electroblotting transfer apparatus (25 V for 1 h; Trans‐Blot Semi‐Dry Transfer Cell, Bio‐Rad Laboratories). The membrane was incubated overnight at 4°C with an anti‐caspase‐1 antibody (Santa Cruz Biotechnology, Dallas, TX, USA) at a dilution of 1:1000 or with an anti‐IL‐1β antibody conjugated to biotin (R&D Systems, Minneapolis, MN, USA) at a dilution of 1:250. Secondary antibodies used for detection were a goat anti‐rabbit antibody conjugated to horseradish peroxidase (HRP; 1:10 000; Dako, Glostrup, Denmark) and a streptavidin antibody conjugated to Alexa Fluor 680 (1:2000; Life Technologies) respectively. To control for protein loading, the membrane was probed with a mouse anti‐GAPDH antibody (1:20 000; Abcam, Cambridge, UK), and then with an HRP‐conjugated goat anti‐mouse secondary antibody (1:10 000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Membranes to detect the caspase‐1 p45 and p10 bands were developed using a CP‐1000 X‐ray film processor (Agfa Corporation, Mortsel, Belgium), and quantified using a densitometer (ChemiDoc XRS Universal Hood II, Bio‐Rad Laboratories) and the accompanying 1‐D analysis software (Quantity One 4.6.1, Bio‐Rad Laboratories), whereas the Odyssey Scanner ODY‐1882 (LI‐COR, Lincoln, NE, USA) and ImageJ (National Institutes of Health, Bethesda, MD, USA) was used to quantify the density of IL‐1β p31 and p17 bands.
Immunohistochemistry for detection of macrophage accumulation in the kidneys
Briefly, the kidney was cut in half transversely, fixed in 10% formalin for 3 days and then embedded in paraffin. Kidneys were sectioned (5 μm), deparaffinized, rehydrated and stained with rat anti‐F4/80 (Bio‐Rad Laboratories), followed by a biotinylated anti‐rat IgG secondary antibody (Vector Labs, Burlingame, CA, USA). An avidin‐biotin complex (ABC Elite, Vector Labs) was used for signal amplification and 3,3′‐diaminobenzidine (DAB; Sigma‐Aldrich, St. Louis, MO, USA) substrate was used for detection. Finally, sections were counterstained with hematoxylin and mounted in DePex (VWR International, Radnor, PA, USA). Imaging and analysis was performed in a blinded manner. Six randomly selected fields (viewed under a magnification of ×15) were imaged per slide and fields containing blood vessels or areas with tears in the tissue were excluded. The number of cells positive for F4/80 was counted per field of view.
Picrosirius red staining for detection of renal interstitial collagen
Kidneys were fixed, sectioned and imaged (viewed under a magnification of ×20) as described above. Interstitial collagen content of the renal cortex was quantified using 0.1% Picrosirius red staining solution. Collagen staining was quantified as a percentage of the total area per field of view using ImageJ software (National Institutes of Health).
Statistical analysis
Data are expressed as mean ± SEM. Systolic BP data was analysed by two‐way repeated measures anova followed by Bonferroni post hoc test. Other data were analysed using either Student's unpaired t‐test or one‐way anova followed by Bonferroni's post hoc test. P < 0.05 was considered to be statistically significant. Note: post hoc tests were only performed where the F‐ratio of the anova highlighted a significant difference (P < 0.05). Data were graphed and analysed using GraphPad Prism Software v6.04 (La Jolla, CA, USA).
Results
1K/DOCA/salt‐induced hypertension is associated with increased inflammasome activity in the kidney
1K/DOCA/salt treatment in C57BL/6J mice increased systolic BP by ∼30 mmHg within 7–10 days, and this increase was sustained throughout the remainder of the 21 day treatment period (Figure 1A). By contrast, systolic BP remained largely unchanged over the 21 days in 1K/placebo‐treated mice (Figure 1A).
Figure 1.
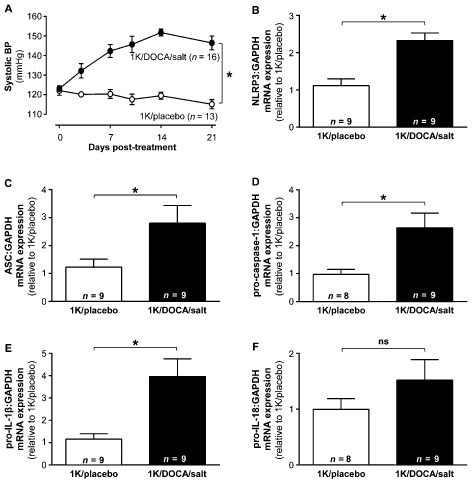
1K/DOCA/salt‐induced hypertension in mice is associated with inflammasome priming. Effects of 1 kidney (1K)/DOCA/salt treatment in mice on systolic BP (A); and renal mRNA expression of the inflammasome‐related genes NLR family, pyrin domain containing 3 (NLRP3; B); apoptosis‐associated speck‐like protein containing a CARD (ASC; C); pro‐caspase‐1 (D), pro‐IL‐1β (E) and pro‐IL‐18 (F). All values are expressed as mean ± SEM. *P < 0.05, ns, not significant for two‐way repeated‐measures anova (A) or Student's unpaired t‐test (B–F).
TaqMan real‐time PCR analysis revealed that there was a significant increase in mRNA expression of inflammasome‐related genes in the kidney of 1K/DOCA/salt‐induced hypertensive mice compared with 1K/placebo‐treated normotensive mice. These genes included NLRP3 (∼2.5‐fold; Figure 1B), ASC (∼3‐fold; Figure 1C), pro‐caspase‐1 (∼2.5‐fold; Figure 1D) and pro‐IL‐1β (∼4‐fold; Figure 1E). There was no significant change in mRNA expression of pro‐IL‐18 (Figure 1F).
Western blotting with an anti‐caspase‐1 antibody that detects both full‐length and the cleaved p10 proteolytically active caspase‐1 revealed that under control conditions (i.e. 1K/placebo), wild‐type mice expressed both a 45 kDa protein and a ∼10 kDa protein that were absent in kidney samples from caspase‐1−/− mice and thus likely to represent the pro‐ and active forms of the enzyme (Figure 2A). 1K/DOCA/salt treatment was associated with an increase in renal protein expression of the pro‐caspase‐1 p45 subunit (i.e. by ∼5‐fold; Figure 2B). Levels of the active caspase‐1 p10 subunit were also elevated in 1K/DOCA/salt mice (∼1.5‐fold; Figure 2C), albeit to a lesser extent than the p45 subunit. Thus, there was an overall reduction in the caspase‐1 p10/45 ratio (∼4‐fold; Figure 2D). Levels of the pro‐IL‐1β p31 subunit (∼1.5 fold; Figure 2E), active IL‐1β p17 subunit (∼2‐fold; Figure 2F) and the ratio of IL‐1β p17/p31 (∼1.5‐fold; Figure 2G) were all elevated in the kidneys of 1K/DOCA/salt‐treated mice compared with 1K/placebo‐treated mice indicative not only of increased IL‐1β expression but also enhanced IL‐1β processing. It is important to note, however, that despite detecting IL‐1β maturation in the kidneys, IL‐1β could not be detected in the plasma of 1K/DOCA/salt‐ or 1K/placebo‐treated mice by cytometric bead array (data not shown).
Figure 2.
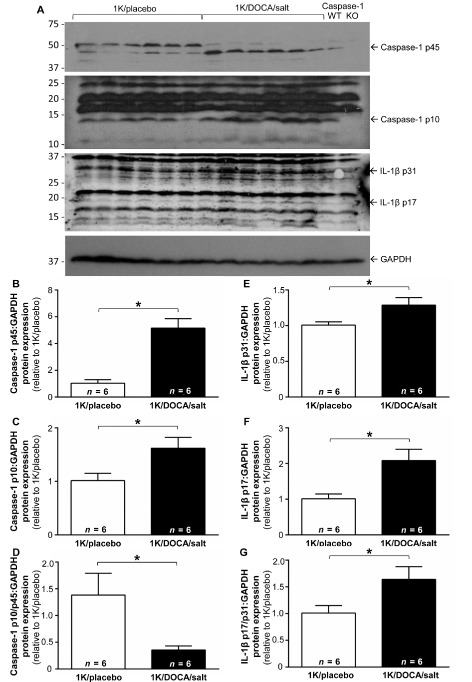
Effect of 1K/DOCA/salt treatment on caspase‐1 and IL‐1β protein expression in the kidney. A shows original Western blots in which membranes were probed with antibodies against either caspase‐1 (two upper blots), IL‐1β (third blot) or the housekeeping gene, GAPDH (lower blot). Note, lanes 1–6 were loaded with protein samples from normotensive (1K/placebo) mice; lanes 7–12 were samples from hypertensive [1 kidney (1K)/DOCA/salt] mice; while lanes 11 and 12 were protein samples from kidneys from littermate caspase‐1+/+ and caspase‐1−/− mice, respectively, the latter serving as a negative control. Also shown are the quantified densitometric data for the uncleaved pro‐caspase‐1 p45 subunit (B); the active caspase‐1 p10 subunit (C); the uncleaved pro‐IL‐1β p31 subunit (E); and the mature IL‐1β p17 subunit (F), all normalized to GAPDH. (D) and (G) show the ratios of caspase‐1 p10:p45 and IL‐1β p17:p31 respectively. All values are expressed as mean ± SEM. *P < 0.05, Student's unpaired t‐test.
ASC −/− mice are protected from 1K/DOCA/salt‐ and angiotensin II‐induced increases in systolic BP
In a separate cohort of animals, 1K/DOCA/salt treatment increased systolic BP in wild‐type mice by ∼40 mmHg (Figure 3A). In ASC−/− mice, the pressor effect of 1K/DOCA/salt treatment was significantly blunted by approximately 40% compared with wild‐type mice (Figure 3A). Furthermore, ASC−/− mice were also resistant to the chronic pressor effects of another hypertensive stimulus, angiotensin II (Figure 3B), suggesting that the crucial role of the inflammasome is not only restricted to hypertension induced by 1K/DOCA/salt.
Figure 3.
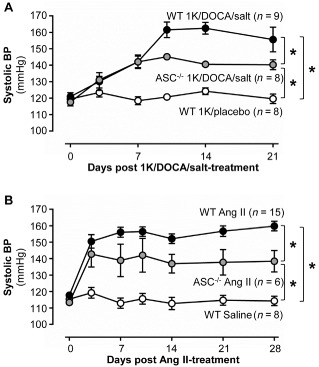
ASC gene deficiency protects mice from the chronic pressor effects of 1K/DOCA/salt and angiotensin II. Effects of 1 kidney (1K)/DOCA/salt (A) and angiotensin (Ang) II (B) on systolic BP in wild‐type and ASC −/− mice. Values are expressed as mean ± SEM. *P < 0.05 for two‐way repeated measures anova.
Effect of ASC deficiency on 1K/DOCA/salt‐induced priming of the inflammasome
In this series of experiments, 1K/DOCA/salt treatment was again found to increase renal expression levels of the inflammasome subunit NLRP3 in wild‐type mice by approximately 2.5‐fold (Figure 4A). Expression of NLRP3 was also elevated (compared to the normotensive controls) in ASC−/− mice that were treated with 1K/DOCA/salt, although to a lesser extent (i.e. by ∼1.7‐fold) than that in wild‐type mice (Figure 4A). Expression of pro‐caspase‐1 (Figure 4B) and pro‐IL‐1β (Figure 4C) were increased to a similar extent in kidneys from both genotypes by treatment with 1K/DOCA/salt (Figure 4B and C). ASC expression was also up‐regulated in kidneys from 1K/DOCA/salt‐treated wild‐type mice, but was barely detectable at the mRNA level in 1K/DOCA/salt‐treated ASC−/− mice (Figure 4D), confirming that these transgenic animals were truly ASC‐deficient. Again, 1K/DOCA/salt treatment appeared to have no effect on renal expression of pro‐IL‐18 (Figure 4E). Taken together, these results suggest that the absence of ASC only has a minor effect on inflammasome priming in the kidney during hypertension.
Figure 4.
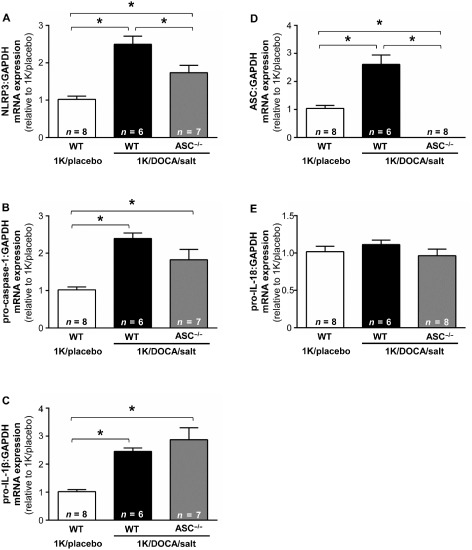
Inflammasome priming in wild‐type and ASC −/− mice following treatment with 1K/DOCA/salt. Effects of 1 kidney (1K)/DOCA/salt treatment on renal expression levels of NLR family, pyrin domain containing 3 (NLRP3); (A), pro‐caspase‐1 (B), pro‐IL‐1β (C), apoptosis‐associated speck‐like protein containing a CARD (ASC; D) and pro‐IL‐18 (E) in wild‐type and ASC −/− mice. Values are expressed as mean ± SEM. *P < 0.05 for one‐way anova followed by Bonferonni's correction for three comparisons.
ASC‐deficiency inhibits 1K/DOCA/salt‐induced increases in expression of pro‐inflammatory cytokines and macrophage infiltration of the kidney
Mature IL‐1β can induce the production of the pro‐inflammatory cytokines IL‐6 and IL‐17A (Cahill and Rogers, 2008; Sutton et al., 2009; Mills et al., 2013). Indeed, we showed that 1K/DOCA/salt‐induced hypertension in wild‐type mice was associated with significant increases in renal mRNA expression of both IL‐6 and IL‐17A (Figure 5A and B). In 1K/DOCA/salt‐treated ASC−/− mice, expression of IL‐6 and IL‐17A was elevated relative to normotensive controls, but was nonetheless 40–60% lower than the levels observed in 1K/DOCA/salt‐treated wild‐type mice (Figure 5A and B). In contrast to the above, expression of IFN‐γ, which lies downstream of mature IL‐18, remained unchanged by 1K/DOCA/salt treatment in both wild‐type and ASC−/− mice (Figure 5C).
Figure 5.
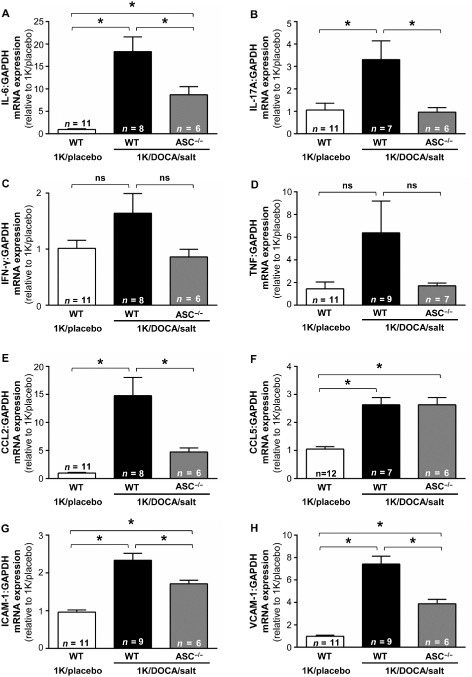
ASC gene deficiency limits 1K/DOCA/salt‐induced increases in multiple renal inflammatory markers. Effects of 1 kidney (1K)/DOCA/salt treatment on renal expression levels of IL‐6 (A), IL‐17A (B), TNF (C), chemokine CCL2 (D), CCL5 (E), ICAM‐1 (F) and VCAM‐1 (G) in wild‐type and ASC −/− mice. Values are expressed as mean ± SEM. *P < 0.05, ns, not significant for one‐way anova followed by Bonferonni's correction for three comparisons.
Renal expression of several additional markers of inflammation including the chemokines, CCL2 (Figure 5E) and CCL5 (Figure 5F), and the adhesion molecules, ICAM‐1 (Figure 5G) and VCAM‐1 (Figure 5H) were also up‐regulated in 1K/DOCA/salt‐ versus 1K/placebo‐treated wild‐type mice. Expression levels of all of these genes, except CCL5, were lower in ASC−/− mice than in wild‐type mice following 1K/DOCA/salt treatment (Figure 5). The pro‐inflammatory cytokine, TNF, also appeared to be upregulated in wild‐type but not in ASC−/− mice following 1K/DOCA/salt treatment; however, these differences failed to reach statistical significance (Figure 5D).
1K/DOCA/salt treatment in wild‐type mice was also associated with a threefold increase in macrophage numbers in the kidney relative to 1K/placebo‐treated mice (Figure 6B). Similar to its effects on the various markers of renal inflammation, ASC deficiency attenuated this increase by ∼30% (Figure 6B). Collectively, these findings indicate that inflammasome activity is a major contributor to the renal inflammation that occurs during hypertension.
Figure 6.
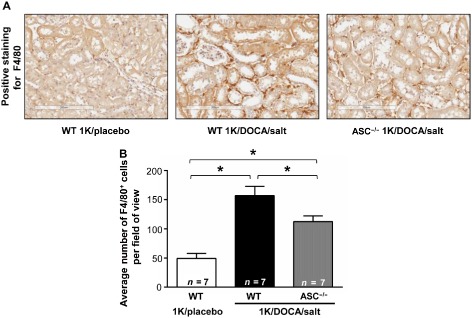
ASC gene deficiency prevents 1K/DOCA/salt‐induced increases in macrophage infiltration of the kidney. F4/80 stained kidney sections from normotensive wild‐type mice (1K/placebo), and from wild‐type and ASC −/− mice following 1 kidney (1K)/DOCA/salt treatment. Upper panels are representative images (×15 magnification; scale bar = 200 μm) while lower panel shows the quantified group data. Values are expressed as mean ± SEM *P < 0.05 for one‐way anova followed by Bonferonni's correction for three comparisons.
ASC −/− mice are protected from renal fibrosis associated with 1K/DOCA/salt‐dependent hypertension
Picrosirius red staining revealed that wild‐type mice treated with 1K/DOCA/salt had significantly elevated levels of renal interstitial collagen compared with the 1K/placebo‐treated control mice (Figure 7). Collagen content in kidneys from 1K/DOCA/salt‐treated ASC−/− mice was markedly lower than that in similarly treated wild‐type mice (Figure 7).
Figure 7.
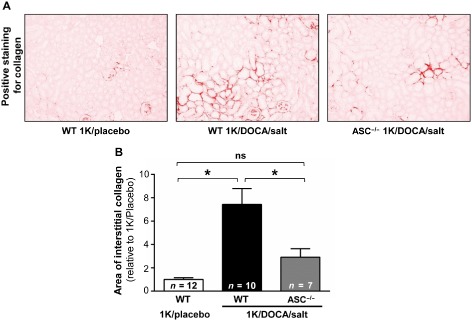
ASC gene deficiency protects mice against 1K/DOCA/salt‐dependent renal fibrosis. Picrosirius red‐stained kidney sections from normotensive wild‐type mice (1K/placebo), and from wild‐type and ASC −/− mice following 1 kidney (1K)/DOCA/salt treatment. Upper panels are representative images (×20 magnification) while lower panel shows the quantified group data. Values are expressed as mean ± SEM. *P < 0.05; ns, not significant for one‐way anova followed by Bonferonni's correction for three comparisons.
A specific NLRP3 inhibitor reverses 1K/DOCA/salt‐induced increases in systolic BP and reduces renal pro‐inflammatory cytokine expression
We evaluated the effects of intervention with a novel, specific inhibitor of NLRP3 inflammasome activity, MCC950, on BP in wild‐type mice with established 1K/DOCA/salt‐induced hypertension. At day 10 of 1K/DOCA/salt treatment, mice had significantly elevated BP (Figure 8A). Initiation of treatment with MCC950 at this time‐point led to a gradual fall in BP such that by day 21, systolic BP had almost returned to baseline (Figure 7). By contrast, treatment with the vehicle for MCC950 did not affect systolic BP in hypertensive mice (Figure 8A). MCC950 treatment also decreased renal expression of the pro‐inflammatory cytokines IL‐17, TNF and osteopontin, and appeared to reduce IL‐6 expression, although this latter effect was not statistically significant (Figure 8B–E).
Figure 8.
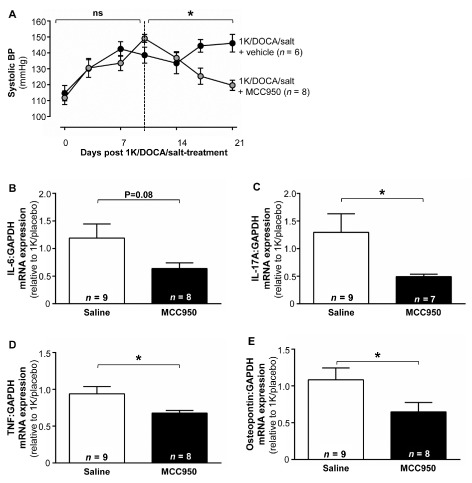
An NLRP3 inhibitor reverses BP and reduces renal expression of pro‐inflammatory cytokines in hypertensive mice. Hypertension was induced in mice by 1 kidney (1K)/DOCA/salt treatment and then after 10 days (dotted line) they were further treated with either MCC950 (10 mg·kg−1·day−1, s.c.) or vehicle (0.11 μL·h−1, s.c.) for the remainder of the experiment. Effect of intervention with MCC950 in hypertensive mice on systolic BP (A); and renal mRNA expression of pro‐inflammatory cytokines, IL‐6 (B), IL‐17A (C), TNF (D) and osteopontin (E). All values are expressed as mean ± SEM. *P ≤ 0.05, ns , not significant for two‐way repeated‐measures anova before and after the intervention, respectively (A), or Student's unpaired t‐test (B–E).
Discussion
This study highlights the crucial role of inflammasomes in the development of renal inflammation, fibrosis and elevated BP in 1K/DOCA/salt‐dependent hypertension in mice. Furthermore, it demonstrates that a small‐molecule inhibitor of inflammasome activity is effective at reversing BP and attenuating renal inflammation, even when administered to animals with established hypertension, thus providing proof‐of‐concept that this may be a viable strategy for the treatment of hypertension.
There is indirect evidence that inflammasome activity plays a role in the development of hypertension in humans. For example, hypertensive patients have elevated circulating levels of IL‐1β and IL‐18 (Dalekos et al., 1997; Rabkin, 2009; Barbaro et al., 2015) and serum levels of IL‐1β are positively correlated with BP (Dalekos et al., 1997). Omi et al. (2006) showed that a particular gain‐in‐function polymorphism in the NLRP3 gene occurs more frequently in hypertensive than in normotensive individuals. In addition, there appears to be a gene‐dose effect of the polymorphism on BP, whereby heterozygotes and homozygotes for the polymorphism display 2 and 5 mmHg increases in BP compared with wild‐type individuals respectively (Omi et al., 2006). These observations provide a rationale for further examining whether a cause–effect relationship exists between inflammasome activity and hypertension and thus whether inflammasomes might represent targets for novel anti‐hypertensive therapies.
Regarding preclinical studies, to our knowledge, there is no published data for increased circulating IL‐1β or IL‐18 in experimental models of hypertension. Indeed, we too found no evidence for elevated serum levels of these cytokines in mice with 1K/DOCA/salt hypertension. Nonetheless, a recent study showed that chronic treatment with an IL‐1β‐neutralizing antibody prevented renin‐dependent hypertension (two kidneys/one clip) in mice (Wang et al., 2014). These investigators also showed that hypertension was blunted in inflammasome‐deficient ASC−/− and NLRP3−/− mice (Wang et al., 2014). Thus, when considered alongside the current findings, these observations highlight the important role of inflammasome‐derived IL‐1β in the pathogenesis of hypertension.
In further support of a crucial role for inflammasome‐derived IL‐1β, several studies have shown that cytokines downstream of IL‐1β are up‐regulated and essential for the development of hypertension in experimental models. IL‐6 is released from macrophages and T‐cells in response to IL‐1β stimulation (Cahill and Rogers, 2008). IL‐6 levels are known to be elevated in hypertension (Humbert et al., 1995; Bautista et al., 2005; Lee et al., 2006) and genetic deletion of IL‐6 reduces BP, renal injury and fibrosis in angiotensin II‐treated mice (Lee et al., 2006; Zhang et al., 2012). IL‐1β and IL‐18, working in concert with IL‐23, can stimulate production of IL‐17A from T‐helper 17 and γδ T‐cells (Sutton et al., 2009; Lalor et al., 2011), and recent work demonstrated that IL‐17A−/− mice are resistant to angiotensin II‐dependent hypertension and vascular dysfunction (Madhur et al., 2010). In the present study, we showed that mRNA expression levels of pro‐IL‐1β, IL‐6 and IL‐17A, were all elevated in kidneys of wild‐type mice following 1K/DOCA/salt treatment. While expression of pro‐IL‐1β was similarly high in kidneys of IK/DOCA/salt‐treated ASC−/− mice, levels of IL‐6 and IL‐17A were markedly attenuated. Collectively, these findings are consistent with the trans‐activation events leading to increased IL‐1β versus IL‐6/IL‐17A occurring upstream and downstream, respectively, of inflammasome activation.
In addition to effects on IL‐6 and IL‐17A, ASC−/− deficiency prevented up‐regulation of several other inflammatory markers in the kidneys of 1K/DOCA/salt‐treated mice including CCL2, ICAM‐1 and VCAM‐1. These chemokines/adhesion molecules are crucial mediators of leukocyte extravasation and infiltration of tissues in numerous inflammatory conditions, including hypertension (Theuer et al., 2002; Chan et al., 2012). Consistent with such a role, immunohistochemical analysis revealed that ASC−/− mice were also protected from 1K/DOCA/salt‐induced macrophage accumulation in the kidneys.
Of note, we found no evidence of elevated IL‐18 levels, either in kidneys or blood of 1K/DOCA/salt‐treated mice. Moreover, expression of IFN‐γ, a cytokine activated directly downstream of IL‐18 (Dinarello, 1999), was also unchanged in kidneys from 1K/DOCA/salt versus control animals. Hence, the protective effects of inflammasome inhibition against hypertension are likely due to inhibition of IL‐1β rather than IL‐18 signalling pathways.
Inflammasome activity is regulated at two levels. First, the various components of the NLRP3 inflammasome complex and cytokine precursors are normally expressed at low levels, but undergo transcriptional upregulation or ‘priming’ following stimulation of the transcription factor NF‐κB (Bauernfeind et al., 2009). Here we observed that 1K/DOCA/salt‐induced hypertension in mice was associated with increased mRNA levels of NLRP3, ASC, pro‐caspase‐1 and pro‐IL‐1β, indicating priming of the system. Previous studies have shown that 1K/DOCA/salt‐dependent hypertension is associated with increased NF‐κB activity in the kidneys (Beswick et al., 2001; Elmarakby et al., 2008). Moreover, pro‐hypertensive stimuli such as aldosterone and angiotensin II can directly increase NF‐κB activity in myeloid cells (Chantong et al., 2012; Lee et al., 2014). Hence, we would suggest that these agonists represent strong candidates as the stimuli responsible for inflammasome priming in the present study.
In addition to priming, inflammasomes must undergo assembly to promote auto‐cleavage and activation of pro‐caspase‐1 and facilitate processing of IL‐1β and/or IL‐18 (Latz, 2010). Thus, protein expression levels of caspase‐1 p10/20 and IL‐1β p17 subunits provide measures of inflammasome activation (Mariathasan et al., 2006; Hornung et al., 2008). Here we used Western blotting and commercially available antibodies to measure caspase‐1 p10 and IL‐1β p17 subunits, and in both instances observed multiple immunoreactive bands. While the poor specificity of the antibodies is clearly a limitation of our study, based on the negative control sample (kidney proteins from caspase‐1−/− mice) and predicted molecular weights of the cleaved proteins, it would appear that 1K/DOCA/salt‐induced hypertension is associated with inflammasome activation in the kidneys. Assembly of the inflammasome complex occurs in response to DAMPs including microcrystals, protein aggregates and high extracellular ATP (Mariathasan et al., 2006; Martinon et al., 2006; Halle et al., 2008). At present, we have no evidence that 1K/DOCA/salt‐dependent hypertension is associated with increases in any of these DAMPs. However, it is noteworthy that even in control animals, we were able to detect low levels of both caspase‐1 p10 and IL‐1β p17 in the kidneys, suggesting that priming alone may have been sufficient to account for the increase in active caspase‐1 and IL‐1β in hypertensive versus normotensive animals. Indeed, this is supported by our observation that the ratio of cleaved : uncleaved caspase‐1 was not elevated in hypertensive versus normotensive animals.
Although our data indicate that priming may be more important for increased IL‐1β signalling in the 1K/DOCA/salt model, this does not mean that stimuli that promote inflammasome assembly could not contribute to hypertension in other settings. For example, gout and hyperuricaemia are associated with formation of urate microcrystals at multiple sites, including the kidneys, resulting in inflammasome activation (Wu et al., 1994; Martinon et al., 2006). The link between hyperuricaemia/gout and hypertension has been established for over a century (Haig, 1889; Bos et al., 2006), and treatment with urate‐lowering compounds such as allopurinol lowers BP in a large proportion of individuals (Feig et al., 2008). More recently, Mazzali et al. (2001) showed that induction of mild hyperuricemia in rats causes them to become hypertensive, further suggesting a causal link between the two conditions. Together with our new data, these observations raise the possibility that increased inflammasome activity in the kidneys, either resulting from priming and/or activation, may be a fundamental mechanism in the development of hypertension.
Our finding that ASC deficiency afforded protection against the chronic pressor, renal inflammatory and pro‐fibrotic actions of 1K/DOCA/salt, and the hypertensive effects of angiotensin II, implies that targeting the inflammasome/IL‐1β nexus may be a new approach for reducing BP and renal damage in hypertension. There are currently three drugs in clinical use for the treatment of autoimmune diseases such as rheumatoid arthritis and gout that target IL‐1β signalling including: anakinra, a recombinant human IL‐1 receptor antagonist; rilonacept, a fusion protein that binds and neutralizes IL‐1β; and canakinumab, a human monoclonal IL‐1β neutralizing antibody (Mertens and Singh, 2009; Schlesinger, 2014). A clinical trial is currently underway to evaluate canakinumab as a treatment for cardiovascular disease (Ridker et al., 2011); however, there is currently no clinical data on potential anti‐hypertensive effects of these aforementioned IL‐1β inhibitors. Recently, a novel small‐molecule inhibitor of NLRP3 activity, MCC950, was described (Coll et al., 2015) with an IC50 of 7.5–8.0 nM in vitro and efficacy in vivo in an experimental model of multiple sclerosis (Coll et al., 2015). We now show that MCC950 is similarly effective at reducing BP and expression of several markers of renal inflammation and damage, even when administered after the establishment of hypertension in mice. Together with the findings of Wang et al. (2014), these data provide proof‐of‐concept that pharmacological inhibition of the inflammasome/IL‐1β pathway is likely to be an effective strategy for the treatment of hypertension.
In conclusion, we have provided evidence that increased inflammasome activity in the kidney, leading to IL‐1β maturation and activation of downstream pro‐inflammatory pathways, plays a key role in the pathogenesis of hypertension. These findings add to the growing body of evidence suggesting that hypertension is an inflammatory disease, and also highlight new possibilities for therapies to treat hypertension and its sequelae of renal disease by specifically targeting inflammasome/IL‐1β signalling.
Author contributions
S. M. K., J. K. D., H. D., D. F., A. P. and T. D. H. performed the research. S. M. K., E. L., A. M., C. G. S. and G. R. D. designed the research study. A. A. B. R., M. A. C., A. M. contributed essential reagents or tools. S. M. K., T. D. H. and G. R. D. analysed the data. S. M. K., Y. H. L., C. T. C., M. M. K., C. S. S., A. V., T. V. A., B. K. K‐H., E. L., A. M., C. G. S. and G. R. D. wrote the paper.
Conflict of interest
The authors have no competing interests.
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC; APP1062721) and the Group‐of‐Eight Australia (Go8)/German Academic Exchange Service (DAAD) Joint Research‐Cooperation Scheme. G. R. D., C. G. S. and C. S. S. are supported by Senior Research Fellowships from the National Health and Medical Research Council of Australia (NHMRC; ID Nos. APP1006017, 350327 and APP1041766, respectively). S. M. K. is supported by a Monash Graduate Scholarship (ID No. 5129593). None of these funding sources had any role in the writing of the report or in the decision to submit the article for publication. We would like to thank Prof. Benjamin Kile (Walter and Eliza Hall Institute of Medical Research, Australia) for providing us with kidneys from caspase‐1−/− and caspase‐1+/+ mice.
Krishnan, S. M. , Dowling, J. K. , Ling, Y. H. , Diep, H. , Chan, C. T. , Ferens, D. , Kett, M. M. , Pinar, A. , Samuel, C. S. , Vinh, A. , Arumugam, T. V. , Hewitson, T. D. , Kemp‐Harper, B. K. , Robertson, A. A. B. , Cooper, M. A. , Latz, E. , Mansell, A. , Sobey, C. G. , and Drummond, G. R. (2016) Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt‐induced hypertension in mice. British Journal of Pharmacology, 173: 752–765. doi: 10.1111/bph.13230.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi W, Niisato N, Sawabe Y, Miyazaki H, Tokuda S, Nishio K et al (2007). Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt‐sensitively hypertensive rats. Cell Biol Int 31: 1288–1291. [DOI] [PubMed] [Google Scholar]
- Barbaro NR, Fontana V, Modolo R, De Faria AP, Sabbatini AR, Fonseca FH et al (2015). Increased arterial stiffness in resistant hypertension is associated with inflammatory biomarkers. Blood Press 24: 7–13. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V (2011). Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187: 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al (2009). Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista L, Vera L, Arenas I, Gamarra G (2005). Independent association between inflammatory markers (C‐reactive protein, interleukin‐6, and TNF‐alpha) and essential hypertension. J Hum Hypertens 19: 149–154. [DOI] [PubMed] [Google Scholar]
- Beswick RA, Zhang H, Marable D, Catravas JD, Hill WD, Webb RC (2001). Long‐term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension 37 (2 Pt 2): 781–786. [DOI] [PubMed] [Google Scholar]
- Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG (2003). Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int 63: 1791–1800. [DOI] [PubMed] [Google Scholar]
- Bos MJ, Koudstaal PJ, Hofman A, Witteman JC, Breteler MM (2006). Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam study. Stroke 37: 1503–1507. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Rogers JT (2008). Interleukin (IL) 1beta induction of IL‐6 is mediated by a novel phosphatidylinositol 3‐kinase‐dependent AKT/IkappaB kinase alpha pathway targeting activator protein‐1. J Biol Chem 283: 25900–25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Moore J, Budzyn K, Guida E, Diep H, Vinh A et al (2012). Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt‐treated mice. Hypertension 60: 1207–1212. [DOI] [PubMed] [Google Scholar]
- Chantong B, Kratschmar DV, Nashev LG, Balazs Z, Odermatt A (2012). Mineralocorticoid and glucocorticoid receptors differentially regulate NF‐kappaB activity and pro‐inflammatory cytokine production in murine BV‐2 microglial cells. J Neuroinflammation 9: 260: e1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz‐Planillo R, Inserra MC et al (2015). A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC (1997). Increased serum levels of interleukin‐1beta in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med 129: 300–308. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (1996). Biologic basis for interleukin‐1 in disease. Blood 87: 2095–2147. [PubMed] [Google Scholar]
- Dinarello CA (1999). Interleukin‐18. Methods 19: 121–132. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464: 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM (2008). TNF‐alpha inhibition reduces renal injury in DOCA‐salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol 294: R76–R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig DI, Soletsky B, Johnson RJ (2008). Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 300: 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig A (1889). On uric acid and arterial tension. Br Med J 1: 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol 9: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR et al (2011). Inflammation, immunity, and hypertension. Hypertension 57: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL et al (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot‐Keros L et al (1995). Increased interleukin‐1 and interleukin‐6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151: 1628–1631. [DOI] [PubMed] [Google Scholar]
- Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR (2005). Potentiation of caspase‐1 activation by the P2X7 receptor is dependent on TLR signals and requires NF‐kappaB‐driven protein synthesis. J Immunol 175: 7611–7622. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Hagaman JR, Smithies O (1995). A noninvasive computerized tail‐cuff system for measuring blood pressure in mice. Hypertension 25: 1111–1115. [DOI] [PubMed] [Google Scholar]
- Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR (2014). IL‐1beta and IL‐18: inflammatory markers or mediators of hypertension? Br J Pharmacol 171: 5589–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH (2011). Caspase‐1‐processed cytokines IL‐1beta and IL‐18 promote IL‐17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol 186: 5738–5748. [DOI] [PubMed] [Google Scholar]
- Latz E (2010). The inflammasomes: mechanisms of activation and function. Curr Opin Immunol 22: 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Chun J, Hwang SW, Kang SJ, Im JP, Kim JS (2014). Enalapril inhibits nuclear factor‐kappaB signaling in intestinal epithelial cells and peritoneal macrophages and attenuates experimental colitis in mice. Life Sci 95: 29–39. [DOI] [PubMed] [Google Scholar]
- Lee D, Sturgis L, Labazi H, Osborne J, Fleming C, Pollock J et al (2006). Angiotensin II hypertension is attenuated in interleukin‐6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940. [DOI] [PubMed] [Google Scholar]
- Madhur M, Lob H, McCann L, Iwakura Y, Blinder Y, Guzik T et al (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW et al (2009). Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA‐salt hypertension. Am J Physiol Renal Physiol 297: F740–F748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al (2004). Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 430: 213–218. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M et al (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006). Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241. [DOI] [PubMed] [Google Scholar]
- Mattson DL (2014). Infiltrating immune cells in the kidney in salt‐sensitive hypertension and renal injury. Am J Physiol Renal Physiol 307: F499–F508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL et al (2001). Elevated uric acid increases blood pressure in the rat by a novel crystal‐independent mechanism. Hypertension 38: 1101–1106. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Yamagiwa Y, Ueno Y, Patel T (2006). Over‐expression of interleukin‐6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol 44: 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens M, Singh JA (2009). Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol 36: 1118–1125. [DOI] [PubMed] [Google Scholar]
- Mezzano SA, Ruiz‐Ortega M, Egido J (2001). Angiotensin II and renal fibrosis. Hypertension 38 (3 Pt 2): 635–638. [DOI] [PubMed] [Google Scholar]
- Mills K, Dungan L, Jones S, Harris J (2013). The role of inflammasome‐derived IL‐1 in driving IL‐17 responses. J Leukoc Biol 93: 489–497. [DOI] [PubMed] [Google Scholar]
- Moore JP, Sakkal S, Bullen ML, Kemp‐Harper BK, Ricardo SD, Sobey CG et al (2013). A flow cytometric method for the analysis of macrophages in the vascular wall. J Immunol Methods 396: 33–43. [DOI] [PubMed] [Google Scholar]
- Niemi K, Teirila L, Lappalainen J, Rajamaki K, Baumann MH, Oorni K et al (2011). Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B‐sensitive pathway. J Immunol 186: 6119–6128. [DOI] [PubMed] [Google Scholar]
- Omi T, Kumada M, Kamesaki T, Okuda H, Munkhtulga L, Yanagisawa Y et al (2006). An intronic variable number of tandem repeat polymorphisms of the cold‐induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet 14: 1295–1305. [DOI] [PubMed] [Google Scholar]
- Ozaki E, Campbell M, Doyle SL (2015). Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 8: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR . (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabkin SW (2009). The role of interleukin 18 in the pathogenesis of hypertension‐induced vascular disease. Nat Clin Pract Cardiovasc Med 6: 192–199. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Thuren T, Zalewski A, Libby P (2011). Interleukin‐1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti‐inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J 162: 597–605. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Johnson RJ (2010). The role of renal microvascular disease and interstitial inflammation in salt‐sensitive hypertension. Hypertens Res 33: 975–980. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ (2012). Renal inflammation, autoimmunity and salt‐sensitive hypertension. Clin Exp Pharmacol Physiol 39: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger N (2014). Anti‐interleukin‐1 therapy in the management of gout. Curr Rheumatol Rep 16: 398. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008). Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010a). The inflammasomes. Cell 140: 821–832. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J (2010b). The NLRP3 inflammasome: a sensor for metabolic danger? Science 327: 296–300. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009). Interleukin‐1 and IL‐23 induce innate IL‐17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31: 331–341. [DOI] [PubMed] [Google Scholar]
- Theuer J, Dechend R, Muller DN, Park JK, Fiebeler A, Barta P et al (2002). Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc Disord 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AQ, So A, Nussberger J, Ives A, Bagnoudi N, Shaefer S et al (2014). Renin‐dependent hypertension in mice requires the NLRP3‐inflammasome. J Hypertens 3: e1–6. [Google Scholar]
- Wei Z, Spizzo I, Diep H, Drummond GR, Widdop RE, Vinh A (2014). Differential phenotypes of tissue‐infiltrating T cells during angiotensin II‐induced hypertension in mice. PLoS ONE 9: e114895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Ting J, O'Neill L (2012). A role for the NLRP3 inflammasome in metabolic diseases – did Warburg miss inflammation? Nat Immunol 13: 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wakamiya M, Vaishnav S, Geske R, Montgomery C Jr, Jones P et al (1994). Hyperuricemia and urate nephropathy in urate oxidase‐deficient mice. Proc Natl Acad Sci U S A 91: 742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C et al (2012). Interleukin 6 underlies angiotensin II‐induced hypertension and chronic renal damage. Hypertension 59: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]