

Abstract
Recently, a number of reports have shown that neurogenic inflammation may play a role in the secondary injury response following acute injury to the CNS, including traumatic brain injury (TBI) and stroke. In particular substance P (SP) release appears to be critically involved. Specifically, the expression of the neuropeptide SP is increased in acute CNS injury, with the magnitude of SP release being related to both the frequency and magnitude of the insult. SP release is associated with an increase in blood–brain barrier permeability and the development of vasogenic oedema as well as neuronal injury and worse functional outcome. Moreover, inhibiting the actions of SP through use of a NK 1 receptor antagonist is highly beneficial in both focal and diffuse models of TBI, as well as in ischaemic stroke, with a therapeutic window of up to 12 h. We propose that NK 1 receptor antagonists represent a novel therapeutic option for treatment of neurogenic inflammation following acute CNS injury.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- BBB
blood–brain barrier
- ICP
intracranial pressure
- MCA
middle cerebral artery
- NAT
n‐acetyl‐L‐tryptophan
- ROS
reactive oxygen species
- SP
substance P
- TBI
traumatic brain injury
- tPA
tissue plasminogen activator
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes d |
| NK1 receptor | ACE |
| NK2 receptor | MMP‐2 |
| NK3 receptor | MMP‐9 |
| Ligand‐gated ion channels b | NOS |
| NMDA receptors | PKC |
| Transporters c | Tissue plasminogen activator (tPA) |
| Calcium ATPase |
| LIGANDS | |
|---|---|
| 5‐HT | IL‐6 |
| ATP | IL‐12 |
| Bradykinin | Neurokinin A |
| Capsaicin | Neurokinin B |
| CGRP | Nitric oxide (NO) |
| Glutamate | Septide |
| Histamine | Substance P |
| IFN‐γ | TNF‐α |
These tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a, 2013b, 2013c, 2013d).
Experiments
All animal experiments were approved by the local institutional ethics committee and conducted according to the National Health and Medical Council (Australia) guidelines for the use of laboratory animals in experimental research. All animal care and experimental procedures were in accordance with the ARRIVE guidelines.
CNS injury
Acute insults to the CNS, such as traumatic brain injury (TBI) and ischaemic stroke, are a major cause of morbidity and mortality today representing a significant public health issue worldwide. An estimated 10 million people are affected annually by a TBI serious enough to result in death or hospitalization, with a mortality rate of 15–30 per 100 000 reported in industrialized countries (Finfer and Cohen, 2001; Tagliaferri et al., 2006). Stroke affects some 15 million individuals worldwide each year, of which 1/3 die and 1/3 are left permanently disabled and dependent for daily living (World Health Organization, 2004).
In both TBI and ischaemic stroke, cell death is caused by both primary (the initial insult) and secondary injury mechanisms. While the nature of primary injury differs between TBI and ischaemic stroke, the secondary injury cascade shares many similarities, with inflammation, and more recently neurogenic inflammation, shown to be a key determinant of neuronal injury and functional outcome in both conditions. The current article provides data supporting a critical role for neurogenic inflammation, and particularly substance P (SP) release, in the secondary injury response following TBI and ischaemic stroke, and discusses these data in the context of previously published reports.
Traumatic brain injury
TBI results from the head impacting with an object or from acceleration/deceleration forces that produce vigorous movement of the brain within the skull, with the resultant mechanical forces potentially damaging glia, neurones, axons and blood vessels. The type and severity of the injury depends upon the nature of the initiating force, as well as its site, direction and magnitude (Smith et al., 2003). Contact forces generated when the head strikes or is struck by an object generally produce focal injuries such as skull fractures, extradural haemorrhages, and contusions. In contrast, acceleration/deceleration forces that result from violent unrestrained head movement are associated with diffuse axonal injury (Blumbergs et al., 2008). Focal injuries such as contusions lead to the development of an ischaemic area of dead tissue (the necrotic core), which is surrounded by a penumbral area. This penumbral area may also experience ischaemia due to haemorrhage, disruptions in venous drainage and vasoconstriction, which contributes to the secondary injury process in this region (Leker and Shohami, 2002).
Ischaemic stroke
In ischaemic stroke, the primary injury is the sudden and profound reduction of blood flow to an area of the brain, most commonly because of atherothrombosis of cerebral vessels or emboli (Hademenos and Massoud, 1997). The amount of cell death is dependent upon the severity and duration of the resultant ischaemia, in addition to the availability of collateral blood supply. The ensuing infarct may be divided into two regions: the core and the penumbra. Within the core of the infarct, blood flow falls to less than 20% causing rapid cell death due to insufficient supply of substrate to maintain cellular energy production. The penumbra surrounding the core is an area of reduced perfusion in which cells are still viable. Their survival is only possible for a limited time due to the initiation of a number of secondary, pathological processes that are similar to that seen following TBI (Bouts et al., 2013).
Secondary injury
As outlined above, TBI results primarily from a mechanical impact that disrupts the brain parenchyma and causes shearing and tearing of blood vessels and axons (Bramlett and Dietrich, 2004). In contrast, ischaemic stroke results from a cessation of blood supply leading to cellular death due to a deprivation of oxygen and glucose (Leker and Shohami, 2002). Despite the differences in these primary injury mechanisms, they both lead to the activation of molecular and cellular responses that lead to secondary injury, with similarities noted in the harmful pathways that lead to secondary cell death in the penumbral ischaemic zone and in the area exposed to secondary post‐traumatic brain injury. These similarities include an increase in extracellular levels of glutamate leading to excitotoxicity, the onset of oxidative stress, disruption to the blood–brain barrier (BBB), cerebral oedema formation and inflammation (Figure 1), all of which exacerbate injury and tissue damage (Saatman et al., 1996). Of particular interest is the role that inflammation can play in this secondary injury cascade and whether modification of this inflammatory process has the potential to improve outcome.
Figure 1.
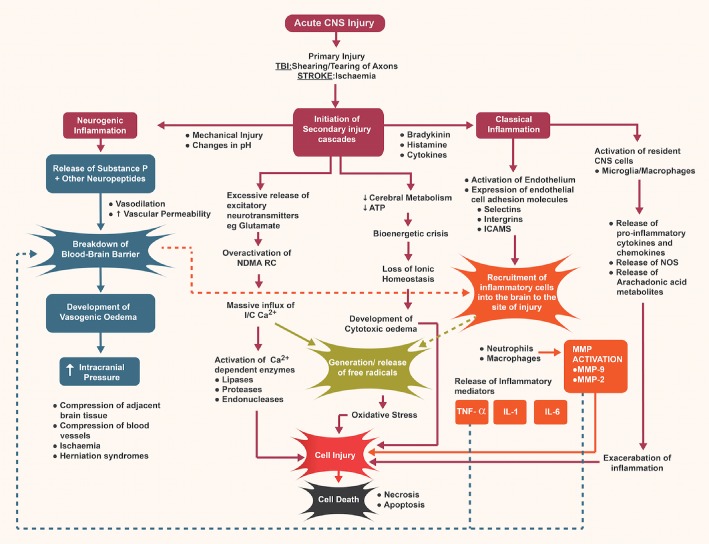
Overview of neurogenic and classical inflammation following acute CNS injury.
Inflammation
A marked inflammatory reaction accompanies any acute insult to the CNS, including ischaemic stroke and TBI (Danton and Dietrich, 2003). In the immediate phase following the initial insult this inflammatory response exacerbates cell injury and worsens outcome. Briefly, such an inflammatory response is characterized by glial activation, proliferation of microglia, leukocyte recruitment and up‐regulation and secretion of mediators such as cytokines and chemokines (Ziebell and Morganti‐Kossmann, 2010). Microglial cells become activated and release a range of neurotoxic factors including pro‐inflammatory cytokines, reactive oxygen species (ROS), NO and metalloproteinases (Brown and Neher, 2010). Cytokines like IL‐1 play a key role in orchestrating the release of other neurotoxic mediators such as ROS, proteases and PGs, while TNF promotes the release of proteolytic enzymes, breakdown of the BBB and induction of cell death (Shohami et al., 1999). While the role of the classical inflammatory response has been well characterized (Morganti‐Kossmann et al., 2002; Danton and Dietrich, 2003), the important role that neurogenic inflammation plays in exacerbating the secondary injury cascade has only recently begun to be recognized.
Neurogenic inflammation
Neurogenic inflammation is a neurally elicited inflammatory response characterized by the release of neuropeptides, including SP and CGRP, vasodilation, plasma extravasation, tissue swelling and mast cell degranulation (Severini et al., 2002). It results from the stimulation of capsaicin‐sensitive, neuronal C fibres by factors such as 5‐HT, histamine and leukotrienes, as well as by changes in pH, extremes of temperature and by mechanical injury (Saria and Lundberg, 1983; Harrison and Geppetti, 2001).
Substance P
SP is thought to be the most potent initiator of neurogenic inflammation due to its association with increased vascular permeability and subsequent plasma protein extravasation (Holzer, 1998). It also potentiates classical inflammation by stimulating the production of inflammatory mediators such as histamine, NO, cytokines (such as IL‐6) and kinins, in addition to interacting with adhesion molecules causing leukocyte migration (Averbeck and Reeh, 2001).
SP is a member of the tachykinin family, a group of structurally related peptides sharing the same carboxyl terminal sequence, which also includes neurokinin A and neurokinin B (Schaffer et al., 1998). It is derived from the preprotachykinin‐A (PPT‐A) gene, which is spliced to form four different mRNA forms, namely α‐PPT, β‐PPT, γ‐PPT and δ‐PPT (Harrison and Geppetti, 2001). The α‐PPT and β‐PPT forms encode for SP alone while the γ‐PPT and δ‐PPT encode for both SP and neurokinin A (Severini et al., 2002). The α‐PPT is more abundant within the brain, while the β‐PPT form is found primarily within the peripheral nervous system (PNS; Ribeiro‐da‐Silva and Hokfelt, 2000). Indeed, SP is widely distributed throughout the CNS, PNS and enteric nervous systems. In the CNS, it is present in dorsal root ganglion (primary sensory) neurons (Hokfelt et al., 2001) of many regions including the hippocampus, cortex, basal ganglia, hypothalamus, amygdala, caudate nucleus and spinal cord, and seems to be more abundant in the grey matter compared with white matter (Ebner and Singewald, 2006).
SP is synthesized in the ribosome as a large protein and transported to the terminal endings, where it is enzymatically converted into the active form and stored in vesicles ready for release (Lundy and Linden, 2004). Its actions are primarily mediated through the tachykinin NK1 receptor to which it preferentially binds, although it also binds to the other tachykinin receptors, NK2 and NK3, with varying affinity depending upon receptor and ligand availability (Regoli et al., 1994). The NK1 receptor is a typical GPCR with seven transmembrane domains (Garland et al., 1994) and is found on neurons and glia in the CNS, smooth muscle cells, endothelial cells, fibroblasts, keratinocytes and various circulating immune and inflammatory cells (Schaffer et al., 1998). On unstimulated neurons, NK1 receptors are localized to the plasma membrane of both the cell body and dendrites, but following SP binding, they are rapidly internalized into the cytoplasm via endosomes (Mantyh, 2002). This internalization is readily reversible with complete return of internalized receptors to the surface within 30 min.
The intracellular signalling pathways activated by the NK1 receptor appear to be cell‐dependent. In endothelial cells, SP binding to the NK1 receptor promotes phosphorylation of myosin by myosin light chain kinase, which in turn promotes the interaction of actin and myosin, leading to cell retraction and the formation of gaps between endothelial cells (Holzer, 1998; van Hinsbergh and van Nieuw Amerongen, 2002). In contrast, in vascular smooth muscle, the vasodilator actions of SP involve activation of adenylate cyclase producing cAMP, which activates calcium ATPases, reducing intracellular calcium levels and promoting relaxation and dilation of vascular smooth muscle (Maggi, 1995). In other cell types such as astrocytes and microglia, binding of SP to the NK1 receptor leads to activation of PLC, which catalyses the hydrolysis of phosphoinositides into inositol 1,4,5‐trisphosphate and diacylglycerol, which then allow mobilization of calcium from internal stores (O'Connor et al., 2004). This increase in calcium levels within primary afferents is thought to be involved in the decrease in the threshold for signalling of nociceptive information following injection of SP into the skin.
The role of SP following TBI
Previous research in our laboratory has found that SP plays an integral role in the secondary injury cascade following TBI (Nimmo et al., 2004; Donkin et al., 2009). Following diffuse TBI in rats, an increase in SP immunoreactivity is noted at both 5 and 24 h following injury, with a gradual return to sham levels by 7 days post‐injury (Donkin et al., 2009). Consistent with the animal studies, in human TBI, post‐mortem SP immunoreactivity was increased perivascularly as well as in cortical neurones and astrocytes in the majority of cases examined (Zacest et al., 2010). This release of SP appears to be both intensity‐ and frequency‐dependent, with a graded increase in SP immunoreactivity seen with increasing severity of injury. Consistent with this, experimental studies conducted in our laboratory (Figure 2) show that the highest level of SP immunoreactivity is observed following a 2 m (severe) impact TBI, while a 0.5 m (mild) impact TBI causes only a minimal increase in SP immunoreactivity. Increased axonal injury and worsening of motor outcome is also observed as the severity of injury increases (Marmarou et al., 1994), suggesting that SP may play a role in the heightened secondary injury response with increased injury severity. Notably, we have previously shown that axonal injury is reduced and functional outcome improved with administration of an NK1 receptor antagonist (Donkin et al., 2011). Previous studies examining SP release following thermal stimuli have also shown that stepwise increases at the extremes of temperature (<10°C and >43°C) results in a graduated release of SP and associated internalization of the NK1 receptors (Allen et al., 1997).
Figure 2.
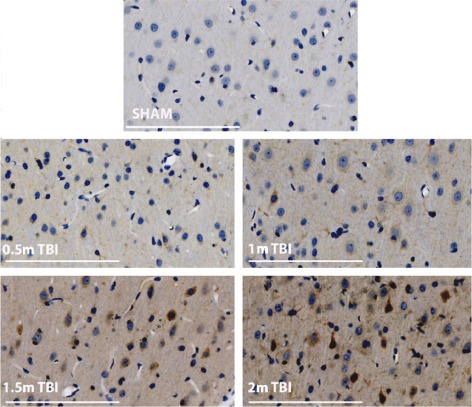
SP immunoreactivity following diffuse TBI of varying severity. Rats were injured using the Marmarou impact acceleration model as previously described (Donkin et al., 2009; Carthew et al., 2012) with a 450 g brass weight released from a height of either 0.5, 1, 1.5 or 2 m onto a metal helmet that was adhered centrally to the animal's skull. 24 h following injury, the brains were processed and then stained for SP (scale bar = 100 μm; images are representative of n = 3–4 per group). Note the increased intensity of SP immunoreactivity (brown staining) with increasing severity of injury.
Repeated exposure to a stimulus has also been shown to cause increased SP release and greater activation of NK1 receptor expressing cells (Mantyh, 2002), with diffusion of SP away from the site of release causing more widespread activation, estimated to be 3–5 times that of a single exposure. The brain is known to be more vulnerable to repeated injury, with rodents demonstrating prolonged cognitive deficits and enhanced axonal injury not seen with a single impact (Longhi et al., 2005). This suggests that enhanced release of SP may potentially play a role in the vulnerability of the brain to repeated concussions.
Antagonism of SP following TBI
The important role that SP plays in the secondary injury process following TBI has been confirmed in a number of studies that have shown that attenuating SP activity is beneficial to outcome (Vink and van den Heuvel, 2010). The first demonstration of neurogenic inflammation in TBI showed that depletion of sensory neuropeptides by pretreatment with capsaicin results in the attenuation of post‐traumatic BBB permeability, oedema formation and improved functional outcome (Nimmo et al., 2004). While not identifying the sensory neuropeptide responsible for these post‐traumatic effects, subsequent studies identified increased SP immunoreactivity as a possible therapeutic target. Accordingly, treatment with a NK1 receptor antagonist, targeting SP binding, was utilized after TBI in both male (Donkin et al., 2009) and female rats (Corrigan et al., 2012) which led to a significant attenuation of post‐traumatic BBB permeability with a significant reduction in oedema formation. This was accompanied by a reduction in axonal injury and associated improvement in both motor and cognitive outcome. Further evidence supporting a role for SP in TBI was provided in studies that administered an ACE inhibitor, which inhibits the breakdown of SP. Following ACE inhibitor administration, levels of SP were significantly increased after TBI, exacerbating neuronal injury and motor deficits (Harford‐Wright et al., 2010). In terms of the therapeutic window for neuroprotective action, treatment up to 12 h post‐injury with a centrally acting NK1 receptor antagonist was still able to provide significant benefit, with marked improvements noted in motor performance (Donkin et al., 2011).
All of the initial TBI studies characterizing neurogenic inflammation utilized the Marmarou impact acceleration model of TBI, which causes diffuse injury with widespread axonal damage, particularly in the long tracts of the brain stem, as well as within the corpus callosum and internal capsule (Marmarou et al., 1994). As human TBI covers the spectrum from diffuse through to focal injury, it is important to determine whether therapies targeting SP are equally efficacious in both types of injury. The fluid percussion model of TBI utilizes the application of a rapid pressure pulse onto the exposed and intact dura to cause a brief displacement and focal deformation of the brain tissue (Corrigan et al., 2011). This produces primarily a focal contusion, accompanied by axonal injury in the fimbria and internal capsule (Thompson et al., 2005). It is thus a highly suitable model to examine the effects of NK1 receptor antagonists on focal TBI. Figure 3 demonstrates that administration of an NK1 receptor antagonist, n‐acetyl‐L‐tryptophan (NAT; 2.5 mg·kg−1 i.v.) at 30 min post‐injury following fluid percussion‐induced TBI led to a similar significant improvement in motor outcome on the rotarod as that seen previously following diffuse injury. Specifically, NAT‐treated rats performed significantly better (P < 0.05) than saline vehicle‐treated rats on days 1, 2, 4 and 5 post‐injury, and had in fact returned to sham levels by day 4, while the vehicle‐treated rats still showed significant motor deficits on day 7, the final day of testing. Thus, administration of an NK1 receptor antagonist significantly improved outcome after TBI, irrespective of the focal or diffuse nature of the injury.
Figure 3.
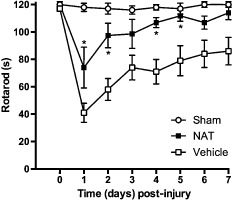
Effect of an NK 1 receptor antagonist on motor outcome following fluid percussion‐induced TBI in rats. Following moderate fluid percussion injury (Faden et al., 1989), rats (n = 6 per group) were treated with 2.5 mg·kg−1 i.v. NAT at 30 min post‐trauma and then assessed for motor outcome on the rotarod (Heath and Vink, 1999) daily for 7 days. Note that the NK 1 receptor antagonist significantly improved outcome (*P < 0.05; mean ± SEM; repeated anova followed by Student Neuman–Keuls tests) when compared with vehicle (saline) treated controls.
While the role of neurogenic inflammation had been confirmed in both focal and diffuse TBI, and the NK1 receptor antagonist had proven efficacious in both male and female animals, all pharmacological studies had been limited to rodents, which have historically led to few successful therapeutic translations to the clinic, particularly in the area of acute CNS injury. To increase the likelihood of successful clinical translation, it is important to test therapeutics developed in rodent models in large animal models given the anatomical differences between the small lissencephalic rodent brain and the larger gyrencephalic human brain. This has been a particular focus of our laboratory in recent years, which has developed models of both TBI and stroke in sheep (Van Den Heuvel et al., 2004; Finnie et al., 2012; Wells et al., 2012). As with the rodent studies, sheep demonstrated an increased SP immunoreactivity after TBI associated with increased BBB permeability and marked oedema formation, as well as significant increases in intracranial pressure (ICP) that were associated with brain water accumulation (Byard et al., 2009). Administration of an NK1 receptor antagonist in this sheep TBI model was able to significantly reduce ICP levels to normal within 4 h of drug administration as compared with the sustained elevation in ICP in the vehicle‐treated sheep (Gabrielian et al., 2013). This reduction in ICP is consistent with the ability of the NK1 receptor antagonists to reduce brain water concentration to normal levels in the previously published rodent studies (Vink and van den Heuvel, 2010).
The role of SP following ischaemic stroke
We have described above that neurogenic inflammation involving SP release is a ubiquitous feature of TBI, regardless of whether the insult is mild, moderate or severe or whether it is diffuse or focal in nature (see Figure 2). Many of the secondary injury pathways that feature in TBI also significantly contribute to the development of infarction following stroke. Indeed, we have reported a significant SP response in ischaemic tissue following experimental stroke in rodents (Turner et al., 2006). However, in the setting of cerebral ischaemia, it appears that SP release may not be as ubiquitous a feature of stroke as it is in TBI, and may be dependent upon the reperfusion status of the tissue.
SP immunoreactivity is particularly evident in the perivascular tissue of the ischaemic penumbra, as well as in neuronal and glial cells within this region (Turner et al., 2006; 2011). Levels of SP in the penumbra gradually increase with time following reperfusion, reaching a nadir at 24 h post‐reperfusion (Turner et al., 2011). In contrast, the core of the stroke lesion does not demonstrate appreciable SP immunoreactivity. Thus, there appears to be a difference in the SP response that is dependent upon the perfusion status of the tissue. To further demonstrate this effect, our laboratory has shown in rodents that 24 h of permanent middle cerebral artery (MCA) occlusion results in only modest elevations in SP immunoreactivity beyond sham levels. In contrast, 2 h of transient MCA occlusion followed by 22 h reperfusion is associated with profound increases in perivascular SP immunoreactivity within the penumbral tissue. These alterations in SP levels were confirmed using elisa assays of the ischaemic hemisphere (Turner et al., 2011). The results suggest that in regions where infarction has occurred, there is no SP response, whereas in the surrounding surviving but compromised penumbral tissue, SP is playing a role in the development of secondary injury. We posit that SP release may be a feature of reperfusion injury and that neurogenic inflammation may contribute to and exacerbate injury to the brain following stroke. It must be acknowledged that these findings may be model and species dependent. However, no data describing a difference in the SP response exists to date for other experimental models of stroke or in clinical samples.
Following transient ischaemia, SP immunoreactivity is maximal at 24 h following stroke, and declines thereafter over the next 7 days. Increased BBB permeability has been reported at various time points following stroke, and the 24 h time point when SP was markedly increased in our studies is consistent with such reports (Preston et al., 1993). Indeed, the 24 h time point in our stroke model was associated with profound disruption of the BBB (Turner et al., 2006), together with development of cerebral oedema and persistent functional deficits (Turner et al., 2011). SP therefore offers an attractive pharmacological target to reduce BBB permeability and the development of oedema following transient ischaemia.
Antagonism of SP following ischaemic stroke
A number of studies have now demonstrated that inhibition of neurogenic inflammation, and in particular SP activity, significantly improves outcome following transient ischaemia. In a rodent model of transient MCA occlusion, administration of the NK1 receptor antagonist NAT at 4 h post‐occlusion significantly attenuated BBB permeability and in turn completely ameliorated cerebral oedema and improved functional outcome (Turner et al., 2011). Moreover, NK1 receptor antagonists can be safely combined with thrombolysis (Turner and Vink, 2012a), which is of particular importance given the integral role that thrombolysis plays in clinical stroke management (Medcalf and Davis, 2012). Specifically, combination treatment of tissue plasminogen activator (tPA; alteplase) with NAT significantly improved BBB function as well as functional outcome, and importantly reduced the incidence of intracerebral haemorrhage associated with tPA administration. Presumably, the NK1 receptor antagonist prevented vascular disruption induced by tPA by maintaining the integrity of the BBB.
In terms of the therapeutic window, studies of transient (2 h) MCA occlusion in rats conducted in our laboratory (Figure 4) have demonstrated that an NK1 receptor antagonist (NAT; 25 μmoles·kg−1 i.v.) can be administered up to 8 h following the onset of stroke and still result in a significant improvement in rotarod motor function, and up to 12 h following stroke onset with a significant improvement in sensory function (bilateral asymmetry). Moreover, the treatment at 8 h was effective irrespective of the period of MCA occlusion (Figure 5), thus demonstrating efficacy over a range of mild to severe stroke. The 8–12 h therapeutic window is both highly relevant and achievable, especially when considered against the 4.5 h window of opportunity currently considered safe for clinical thrombolysis with tPA (Medcalf and Davis, 2012). Notably, a comparable therapeutic window of 12 h exists for the administration of NK1 receptor antagonists in TBI (Donkin et al., 2011).
Figure 4.
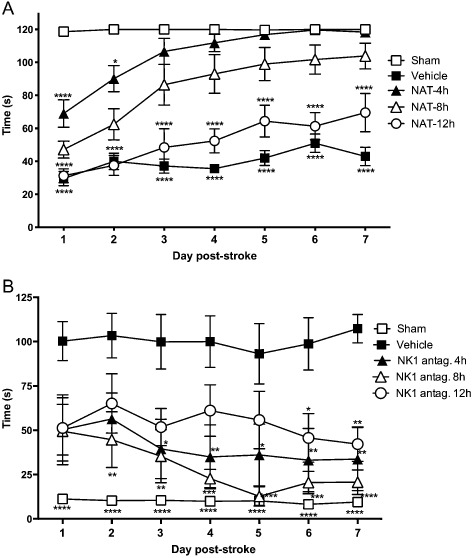
Therapeutic window for administration of a NK 1 receptor antagonist following ischaemic stroke. NK 1 receptor antagonist treatment (NAT; 25 μmol·kg−1 i.v.) improved (A) motor outcome as assessed using a rotarod device when administered up to 8 h following stroke and (B) sensory function as assessed using the bilateral asymmetry test (Turner and Vink, 2014) when administered up to 12 h following MCA occlusion in the rat (n = 6 per group; mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with saline vehicle; repeated anova followed by Student Neuman–Keuls tests).
Figure 5.
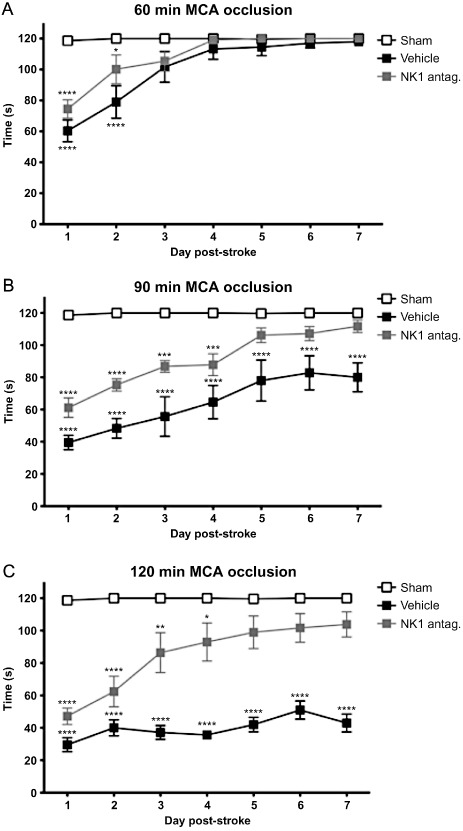
Effects of an NK1 receptor antagonist on motor outcome following mild to severe stroke. NK1 receptor antagonist treatment (NAT; 25 μmol·kg−1 in saline, i.v.) significantly improved motor function as assessed using the rotarod test when administred at 8 h following (A) 60 min, (B) 90 min or (C) 120 min of MCA occlusion in the rat (n = 6 per group; mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with shams; repeated anova followed by Student Neuman–Keuls tests).
The role of SP in the secondary injury response
The exact mechanisms by which SP influences outcome following TBI and stroke are yet to be fully characterized, although the neuropeptide is known to influence a number of secondary injury factors that have been well described following acute CNS injury, including classical inflammation, BBB breakdown, excitotoxicity and magnesium homeostasis (Vink and van den Heuvel, 2010).
Classical inflammation
It is widely accepted that treatments that limit the inflammatory response during the acute phase of experimental CNS injury have beneficial effects on outcome, and considerable effort has been directed at developing more effective anti‐inflammatory approaches (Nimmo and Vink, 2009). In this respect, the effect of SP on classical inflammation is highly relevant and is one mechanism by which NK1 receptor antagonists may ameliorate secondary injury and improve outcome. SP induces and augments many aspects of the classical inflammatory response including leukocyte activation, endothelial cell adhesion molecule expression, cytokine production and mast cell activation (Quinlan et al., 1999). SP also promotes secretion of cytokines including IL‐1, IL‐6, IL‐12 and TNF‐α by cells of the myeloid lineage, IFN‐γ by T‐cells and down‐regulates anti‐inflammatory cytokines such as TGF‐β (McCormack et al., 1996; Marriott and Bost, 2001). Following activation of NK1 receptors on endothelial cells, adhesion molecules are rapidly mobilized to the surface (Zimmerman et al., 1992), with SP exerting direct chemotactic actions on neutrophils (Carolan and Casale, 1993) and also priming them for oxidative metabolism, increasing the production of free radicals (Hafstrom et al., 1989). Astrocytes also express NK1 receptors following injury, and their activation is thought to contribute to the transformation to reactive astrocytes, with the resultant production of inflammatory mediators (Lin, 1995). Following TBI, NK1 receptor antagonists have been shown to significantly reduce production of the pro‐inflammatory cytokine IL‐6 (Reardon et al., 2004) as well as to decrease microglial proliferation (Carthew et al., 2012). Similar reductions in IL‐6 as well as TNF‐α have also been reported in a mouse model of bacterial meningitis in response to administration of an NK1 receptor antagonist (Chauhan et al., 2008).
Effects of SP on the BBB
The BBB is a highly selective barrier formed by a monolayer of endothelial cells that are joined together by tight junctions including proteins such as claudins, occludin, junctional adhesion molecules and zonula occludens proteins (ZOs; Huber et al., 2001). This endothelial layer is supported by the end‐feet of astrocytes that act to support and enhance the tight junctions. Any increase in the permeability of the BBB following acute CNS injury permits the extravasation of proteins and solutes from the cerebral vasculature into the extracellular space within the brain, leading to a net movement of water in the same direction and a subsequent increase in brain volume (Kimelberg, 1995). Due to the limited capacity of the skull to accommodate excess fluid, this increase in volume with the entry of water has deleterious consequences, including an increase in ICP. Such an increase in ICP inevitably decreases cerebral perfusion pressure and can ultimately lead to brain herniation and its associated adverse events including localized ischaemia and even cessation of respiration. The formation of cerebral oedema is thus a major cause of clinical deterioration, being associated with increased mortality and morbidity following TBI (Rhine et al., 2012) and the leading cause of death within the first week of an ischaemic stroke (Hacke et al., 1996).
SP is known to promote increased permeability of the BBB leading to increased extravasation of vascular protein into the brain extracellular space, both via transcellular and paracellular mechanisms. Indeed, previous studies have shown that intravenous injection of SP increases the permeability of dural blood vessels, with widening of the junctions between endothelial cells (Ghabriel et al., 1999). It also has effects on tight junction proteins, with decreased levels of ZO‐1 and claudin 5 reported in cerebral capillary endothelial cells upon application of SP (Lu et al., 2008). Not only does SP directly increase the permeability of the BBB, but it also leads to up‐regulation of adhesion molecules and MHC class II antigens leading to recruitment and migration of inflammatory cells across the BBB, thereby potentiating the inflammatory response and exacerbating tissue injury (Annunziata et al., 2002). The ability of SP to activate microglia and astrocytes also contributes to the increased permeability of the BBB. Increased permeability of the BBB has been repeatedly demonstrated following acute brain injury, and notably, administration of an NK1 antagonist significantly attenuates the BBB permeability back to normal levels, as measured by Evan's blue extravasation, in both TBI (Donkin et al., 2009) and stroke (Turner et al., 2011; Turner and Vink, 2013; 2014). This reduction in BBB permeability is associated with reduced vasogenic oedema formation and reduced ICP.
Excitotoxicity
Levels of extracellular glutamate profoundly increase immediately after both TBI and stroke (Faden et al., 1989; Bullock et al., 1998; Hazell, 2007) leading to excitotoxic cell death through increased calcium flux into neurons. The increase in extracellular glutamate is due to uncontrolled release of the excitatory amino acid with neuronal depolarization, as well as a decrease in its reuptake caused by impaired glutamate transporter activity (Yi and Hazell, 2006) and a reduction in the numbers of astrocytic glutamate transporters (van Landeghem et al., 2006). Accumulation of intracellular calcium activates a number of calcium‐dependent enzymes including proteases, lipases, translocases and endonucleases, which degrade cellular structures and eventually cause neuronal degeneration (Maas et al., 2008).
SP and glutamate have been shown to be co‐localized within the dorsal horn (Battaglia and Rustioni, 1988; Merighi et al., 1991), parts of the brainstem (Liu et al., 2004; Guiard et al., 2007) and striatum (Penny et al., 1986). Previous research has indicated a bidirectional relationship with glutamate able to stimulate release of SP via activation of presynaptically located NMDA receptors (Liu et al., 1997), while SP also facilitates glutamate release (Kangrga et al., 1990; Guiard et al., 2007), through a NK1 receptor‐dependent fashion, most likely through mobilization of intracellular calcium stores (Stacey et al., 2002). Indeed, in an organotypic brain slice culture model, application of SP, as well as another specific NK1 receptor agonist, septide, stimulated glutamate release leading to an increase in spontaneous excitatory synaptic currents (Stacey et al., 2002). SP can also alter the activity of NMDA receptors, which are primarily responsible for calcium influx following the binding of glutamate. Pre‐application of SP led to a sustained potentiation of the current mediated by NMDA receptors in cultured rat dorsal root ganglion neurons, an effect that could be reversed with application of an NK1 receptor antagonist (Wu et al., 2004; Castillo et al., 2011). A potential mechanism was suggested to involve NK1 receptor activation induced hydrolysis of phosphoinositide to diacylglycerol leading to the activation of PKC (Castillo et al., 2011). PKC is known to produce a positive phosphorylation modulation of the NMDA receptor, enhancing its activity (Lan et al., 2001). These studies support a SP‐NK1 receptor role in the release of glutamate and the activity of the NMDA receptor, but the direct effects of NK1 receptor antagonism on measures of excitotoxicity following TBI or stroke have yet to be examined.
Magnesium
Acute insults to the CNS decrease levels of free magnesium both within the brain and the blood, with the magnitude of the decline linked to the severity of the injury as assessed by functional outcome (Heath and Vink, 1999; Turner et al., 2012b). This is consistent with the critical role that optimal magnesium concentration plays in diverse biological processes including energy transduction, phosphorylation reactions, protein synthesis, DNA synthesis, preservation of membrane integrity, maintenance of ionic gradients, and in regulation of calcium transport and accumulation. The ion also has a gating function with respect to the NMDA receptor, with low concentrations of intracellular magnesium potentiating NMDA receptor activity and calcium entry. A decline in magnesium post‐injury has therefore been associated with exacerbation of other secondary injury pathways such as excitotoxicity, mitochondrial dysfunction, apoptosis, bioenergetic failure and oxidative stress (Turner et al., 2012b). Critically, administration of an NK1 receptor antagonist after TBI is able to reverse this decline in magnesium with treated rats demonstrating normal magnesium levels (Vink et al., 2004), with associated improvement in histological and functional outcome. The exact mechanism whereby SP promotes normal magnesium homeostasis is yet to be determined, with the possibility that restored magnesium levels simply reflect improved neuronal function. However, there may also be a direct relationship between SP and magnesium given that magnesium deficiency is associated with elevations in the plasma concentrations of SP (Weglicki and Phillips, 1992) and development of neurogenic inflammation (Weglicki et al., 1994).
Conclusion
The current overview and additional data have highlighted the critical role that neurogenic inflammation plays in the secondary injury process following acute brain injury, with particular focus on TBI and ischaemic stroke. An increase in SP release appears to be a ubiquitous feature of TBI and critical to cellular outcome within the penumbral region following ischaemic stroke, as well as within those areas experiencing reperfusion after stroke. Neurogenic inflammation appears to exacerbate neuronal damage through a number of mechanisms including promoting BBB breakdown, with the subsequent development of vasogenic oedema, augmenting the inflammatory response and enhancing excitotoxicity. Preventing the actions of SP through the use of an NK1 receptor antagonist has been shown to be a promising therapeutic agent in experimental models of TBI and stroke, and offers a novel approach to clinical management of acute CNS injury.
Author contributions
F. C. contributed to the design of the TBI experiments, participated in the acquisition, analysis and interpretation of the TBI experiments, and contributed to the drafting and final approval of the manuscript. R. J. T. contributed to the conception and design of the stroke experiments, participated in the acquisition, analysis and interpretation of the stroke experiments, and contributed to the drafting and final approval of the manuscript. R. V. contributed to the conception and design of all experiments, participated in the analysis and interpretation of all experimental data, and contributed to the drafting and final approval of the manuscript.
Conflict of interest
The authors have no conflicts of interest to declare.
Supporting information
File S1 Methods.
Acknowledgements
We thank Kelly McAteer for contributing the data shown in Figure 2. All studies were supported by grants from the National Health and Medical Research Council (Australia) and the Neurosurgical Research Foundation (Australia).
Corrigan, F. , Vink, R. , and Turner, R. J. (2016) Inflammation in acute CNS injury: a focus on the role of substance P . British Journal of Pharmacology, 173: 703–715. doi: 10.1111/bph.13155.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: ligand‐gated ion channels. Br J Pharmacol 170: 1582–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol 170: 1706–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013d). The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BJ, Rogers SD, Ghilardi JR, Menning PM, Kuskowski MA, Basbaum AI et al (1997). Noxious cutaneous thermal stimuli induce a graded release of endogenous substance P in the spinal cord: imaging peptide action in vivo . J Neurosci 17: 5921–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziata P, Cioni C, Santonini R, Paccagnini E (2002). Substance P antagonist blocks leakage and reduces activation of cytokine‐stimulated rat brain endothelium. J Neuroimmunol 131: 41–49. [DOI] [PubMed] [Google Scholar]
- Averbeck B, Reeh PW (2001). Interactions of inflammatory mediators stimulating release of calcitonin gene‐related peptide, substance P and prostaglandin E(2) from isolated rat skin. Neuropharmacology 40: 416–423. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Rustioni A (1988). Coexistence of glutamate and substance P in dorsal root ganglion neurons of the rat and monkey. J Comp Neurol 277: 302–312. [DOI] [PubMed] [Google Scholar]
- Blumbergs PC, Reilly PL, Vink R (2008). Trauma In: Love S, Louis DN, Ellison DW. (eds). Greenfield's Neuropathology, 8th edn Hodder Arnold Publishers: London, pp. 733–832. [Google Scholar]
- Bouts MJ, Tiebosch IA, van der Toorn A, Viergever MA, Wu O, Dijkhuizen RM (2013). Early identification of potentially salvageable tissue with MRI‐based predictive algorithms after experimental ischemic stroke. J Cereb Blood Flow Metab 33: 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD (2004). Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab 24: 133–150. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2010). Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41: 242–247. [DOI] [PubMed] [Google Scholar]
- Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD et al (1998). Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg 89: 507–518. [DOI] [PubMed] [Google Scholar]
- Byard RW, Bhatia KD, Reilly PL, Vink R (2009). How rapidly does cerebral swelling follow trauma? Observations using an animal model and possible implications in infancy. Leg Med (Tokyo) 11: S128–S131. [DOI] [PubMed] [Google Scholar]
- Carolan EJ, Casale TB (1993). Effects of neuropeptides on neutrophil migration through noncellular and endothelial barriers. J Allergy Clin Immunol 92: 589–598. [DOI] [PubMed] [Google Scholar]
- Carthew HL, Ziebell JM, Vink R (2012). Substance P‐induced changes in cell genesis following diffuse traumatic brain injury. Neuroscience 214: 78–83. [DOI] [PubMed] [Google Scholar]
- Castillo C, Norcini M, Baquero‐Buitrago J, Levacic D, Medina R, Montoya‐Gacharna JV et al (2011). The N‐methyl‐D‐aspartate‐evoked cytoplasmic calcium increase in adult rat dorsal root ganglion neuronal somata was potentiated by substance P pretreatment in a protein kinase C‐dependent manner. Neuroscience 177: 308–320. [DOI] [PubMed] [Google Scholar]
- Chauhan VS, Sterka DG Jr, Gray DL, Bost KL, Marriott I (2008). Neurogenic exacerbation of microglial and astrocyte responses to Neisseria meningitidis and Borrelia burgdorferi. J Immunol 180: 8241–8249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan F, Ziebell JM, Vink R (2011). Models of rodent, cortical traumatic brain injury In: Lane E, Dunnett SB. (eds). Neuromethods 62: Animal Models of Movement Disorders. Humana Press: New York, pp. 193–209. [Google Scholar]
- Corrigan F, Leonard A, Ghabriel M, Van Den Heuvel C, Vink R (2012). A substance P antagonist improves outcome in female Sprague Dawley rats following diffuse traumatic brain injury. CNS Neurosci Ther 18: 513–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD (2003). Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 62: 127–136. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC, Vink R (2009). Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab 29: 1388–1398. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Cernak I, Blumberg PC, Vink R (2011). A substance P antagonist reduces axonal injury and improves neurologic outcome when administered up to 12 hours after traumatic brain injury. J Neurotrauma 28: 218–224. [DOI] [PubMed] [Google Scholar]
- Ebner K, Singewald N (2006). The role of substance P in stress and anxiety responses. Amino Acids 31: 251–272. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R (1989). The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244: 798–800. [DOI] [PubMed] [Google Scholar]
- Finfer SR, Cohen J (2001). Severe traumatic brain injury. Resuscitation 48: 77–90. [DOI] [PubMed] [Google Scholar]
- Finnie JW, Blumbergs PC, Manavis J, Turner RJ, Helps S, Vink R et al (2012). Neuropathological changes in a lamb model of non‐accidental head injury (the shaken baby syndrome). J Clin Neurosci 19: 1159–1164. [DOI] [PubMed] [Google Scholar]
- Gabrielian L, Helps SC, Thornton E, Turner RJ, Leonard AV, Vink R (2013). Substance P antagonists as a novel intervention for brain edema and raised intracranial pressure. Acta Neurochir Suppl 118: 201–204. [DOI] [PubMed] [Google Scholar]
- Garland AM, Grady EF, Payan DG, Vigna SR, Bunnett NW (1994). Agonist‐induced internalization of the substance P (NK1) receptor expressed in epithelial cells. Biochem J 303: 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabriel MN, Lu MX, Leigh C, Cheung WC, Allt G (1999). Substance P‐induced enhanced permeability of dura mater microvessels is accompanied by pronounced ultrastructural changes, but is not dependent on the density of endothelial cell anionic sites. Acta Neuropathol (Berl) 97: 297–305. [DOI] [PubMed] [Google Scholar]
- Guiard BP, Guilloux JP, Reperant C, Hunt SP, Toth M, Gardier AM (2007). Substance P neurokinin 1 receptor activation within the dorsal raphe nucleus controls serotonin release in the mouse frontal cortex. Mol Pharmacol 72: 1411–1418. [DOI] [PubMed] [Google Scholar]
- Hacke W, Schwab S, Horn M, Spranger M, De Georgia M, von Kummer R (1996). ‘Malignant’ middle cerebral artery territory infarction: clinical course and prognostic signs. Arch Neurol 53: 309–315. [DOI] [PubMed] [Google Scholar]
- Hademenos GJ, Massoud TF (1997). Biophysical mechanisms of stroke. Stroke 28: 2067–2077. [DOI] [PubMed] [Google Scholar]
- Hafstrom I, Gyllenhammar H, Palmblad J, Ringertz B (1989). Substance P activates and modulates neutrophil oxidative metabolism and aggregation. J Rheumatol 16: 1033–1037. [PubMed] [Google Scholar]
- Harford‐Wright E, Thornton E, Vink R (2010). Angiotensin‐converting enzyme (ACE) inhibitors exacerbate histological damage and motor deficits after experimental traumatic brain injury. Neurosci Lett 481: 26–29. [DOI] [PubMed] [Google Scholar]
- Harrison S, Geppetti P (2001). Substance P. Int J Biochem Cell Biol 33: 555–576. [DOI] [PubMed] [Google Scholar]
- Hazell AS (2007). Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem Int 50: 941–953. [DOI] [PubMed] [Google Scholar]
- Heath DL, Vink R (1999). Concentration of brain free magnesium following severe brain injury correlates with neurologic motor outcome. J Clin Neurosci 6: 505–509. [DOI] [PubMed] [Google Scholar]
- van Hinsbergh VW, van Nieuw Amerongen GP (2002). Endothelial hyperpermeability in vascular leakage. Vascular Pharmacol 39: 171–172. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Pernow B, Wahren J (2001). Substance P: a pioneer amongst neuropeptides. J Intern Med 249: 27–40. [DOI] [PubMed] [Google Scholar]
- Holzer P (1998). Neurogenic vasodilatation and plasma leakage in the skin. Gen Pharmacol 30: 5–11. [DOI] [PubMed] [Google Scholar]
- Huber JD, Egleton RD, Davis TP (2001). Molecular physiology and pathophysiology of tight junctions in the blood‐brain barrier. Trends Neurosci 24: 719–725. [DOI] [PubMed] [Google Scholar]
- Kangrga I, Larew JS, Randic M (1990). The effects of substance P and calcitonin gene‐related peptide on the efflux of endogenous glutamate and aspartate from the rat spinal dorsal horn in vitro . Neurosci Lett 108: 155–160. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK (1995). Current concepts of brain edema. Review of laboratory investigations. J Neurosurg 83: 1051–1059. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC et al (2001). Protein kinase C modulates NMDA receptor trafficking and gating. Nature Neurosci 4: 382–390. [DOI] [PubMed] [Google Scholar]
- van Landeghem FK, Weiss T, Oehmichen M, von Deimling A (2006). Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma 23: 1518–1528. [DOI] [PubMed] [Google Scholar]
- Leker RR, Shohami E (2002). Cerebral ischemia and trauma‐different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Rev 39: 55–73. [DOI] [PubMed] [Google Scholar]
- Lin RC (1995). Reactive astrocytes express substance‐P immunoreactivity in the adult forebrain after injury. Neuroreport 7: 310–312. [PubMed] [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI (1997). NMDA‐receptor regulation of substance P release from primary afferent nociceptors. Nature 386: 721–724. [DOI] [PubMed] [Google Scholar]
- Liu YY, Wong‐Riley MT, Liu JP, Wei XY, Jia Y, Liu HL et al (2004). Substance P and enkephalinergic synapses onto neurokinin‐1 receptor‐immunoreactive neurons in the pre‐Botzinger complex of rats. Eur J Neurosci 19: 65–75. [DOI] [PubMed] [Google Scholar]
- Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF, Davis J et al (2005). Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery 56: 364–374, discussion 364–374. [DOI] [PubMed] [Google Scholar]
- Lu TS, Avraham HK, Seng S, Tachado SD, Koziel H, Makriyannis A et al (2008). Cannabinoids inhibit HIV‐1 Gp120‐mediated insults in brain microvascular endothelial cells. J Immunol 181: 6406–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundy FT, Linden GJ (2004). Neuropeptides and neurogenic mechanisms in oral and periodontal inflammation. Crit Reviews Oral Biol Med 15: 82–98. [DOI] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R (2008). Moderate and severe traumatic brain injury in adults. Lancet Neurol 7: 728–741. [DOI] [PubMed] [Google Scholar]
- Maggi CA (1995). The mammalian tachykinin receptors. Gen Pharmacol 26: 911–944. [DOI] [PubMed] [Google Scholar]
- Mantyh PW (2002). Neurobiology of substance P and the NK1 receptor. J Clin Psychiatry 63: 6–10. [PubMed] [Google Scholar]
- Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K (1994). A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg 80: 291–300. [DOI] [PubMed] [Google Scholar]
- Marriott I, Bost KL (2001). Substance P receptor mediated macrophage responses. Adv Exp Med Biol 493: 247–254. [DOI] [PubMed] [Google Scholar]
- McCormack RJ, Hart RP, Ganea D (1996). Expression of NK‐1 receptor mRNA in murine T lymphocytes. Neuroimmunomodulation 3: 35–46. [DOI] [PubMed] [Google Scholar]
- Medcalf RL, Davis SM (2012). Plasminogen activation and thrombolysis for ischemic stroke. Int J Stroke 7: 419–425. [DOI] [PubMed] [Google Scholar]
- Merighi A, Polak JM, Theodosis DT (1991). Ultrastructural visualization of glutamate and aspartate immunoreactivities in the rat dorsal horn, with special reference to the co‐localization of glutamate, substance P and calcitonin‐gene related peptide. Neuroscience 40: 67–80. [DOI] [PubMed] [Google Scholar]
- Morganti‐Kossmann MC, Rancan M, Stahel PF, Kossmann T (2002). Inflammatory response in acute traumatic brain injury: a double‐edged sword. Curr Opin Crit Care 8: 101–105. [DOI] [PubMed] [Google Scholar]
- Nimmo AJ, Vink R (2009). Recent patents in CNS drug discovery: the management of inflammation in the central nervous system. Recent Pat CNS Drug Discov 4: 86–95. [DOI] [PubMed] [Google Scholar]
- Nimmo AJ, Cernak I, Heath DL, Hu X, Bennett CJ, Vink R (2004). Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 38: 40–47. [DOI] [PubMed] [Google Scholar]
- O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP, Shanahan F (2004). The role of substance P in inflammatory disease. J Cell Physiol 201: 167–180. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny GR, Afsharpour S, Kitai ST (1986). The glutamate decarboxylase‐, leucine enkephalin‐, methionine enkephalin‐ and substance P‐immunoreactive neurons in the neostriatum of the rat and cat: evidence for partial population overlap. Neuroscience 17: 1011–1045. [DOI] [PubMed] [Google Scholar]
- Preston E, Sutherland G, Finsten A (1993). Three openings of the blood‐brain barrier produced by forebrain ischemia in the rat. Neurosci Lett 149: 75–78. [DOI] [PubMed] [Google Scholar]
- Quinlan KL, Naik SM, Cannon G, Armstrong CA, Bunnett NW, Ansel JC et al (1999). Substance P activates coincident NF‐AT‐ and NF‐kappa B‐dependent adhesion molecule gene expression in microvascular endothelial cells through intracellular calcium mobilization. J Immunol 163: 5656–5665. [PubMed] [Google Scholar]
- Reardon K, Heath DL, Nimmo AJ, Vink R, Whitfield K (2004). Inhibition of neurogenic inflammation attenuates the inflammatory response following traumatic brain injury in rats. In: 7th International Neurotrauma Symposium. Medimond S.r.l: Bologna, pp. 115–118.
- Regoli D, Boudon A, Fauchere JL (1994). Receptors and antagonists for substance P and related peptides. Pharmacol Rev 46: 551–599. [PubMed] [Google Scholar]
- Rhine T, Wade SL, Makoroff KL, Cassedy A, Michaud LJ (2012). Clinical predictors of outcome following inflicted traumatic brain injury in children. J Trauma Acute Care Surg 73: S248–S253. [DOI] [PubMed] [Google Scholar]
- Ribeiro‐da‐Silva A, Hokfelt T (2000). Neuroanatomical localisation of substance P in the CNS and sensory neurons. Neuropeptides 34: 256–271. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko‐Coyne D, Marcy V, Siman R, McIntosh TK (1996). Prolonged calpain‐mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropath Exp Neurol 55: 850–860. [DOI] [PubMed] [Google Scholar]
- Saria A, Lundberg JM (1983). Capsaicin pretreatment inhibits heat‐induced oedema in the rat skin. Naunyn Schmiedebergs Arch Pharmacol 323: 341–342. [DOI] [PubMed] [Google Scholar]
- Schaffer M, Beiter T, Becker HD, Hunt TK (1998). Neuropeptides: mediators of inflammation and tissue repair? Arch Surg 133: 1107–1116. [DOI] [PubMed] [Google Scholar]
- Severini C, Improta G, Falconieri‐Erspamer G, Salvadori S, Erspamer V (2002). The tachykinin peptide family. Pharmacol Rev 54: 285–322. [DOI] [PubMed] [Google Scholar]
- Shohami E, Ginis I, Hallenbeck JM (1999). Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev 10: 119–130. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF, Shull WH (2003). Diffuse axonal injury in head trauma. J Head Trauma Rehabil 18: 307–316. [DOI] [PubMed] [Google Scholar]
- Stacey AE, Woodhall GL, Jones RS (2002). Neurokinin‐receptor‐mediated depolarization of cortical neurons elicits an increase in glutamate release at excitatory synapses. Eur J Neurosci 16: 1896–1906. [DOI] [PubMed] [Google Scholar]
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J (2006). A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien) 148: 255–268. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA et al (2005). Lateral fluid percussion brain injury: a 15‐year review and evaluation. J Neurotrauma 22: 42–75. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Vink R (2012a). Combined tissue plasminogen activator and an NK1 tachykinin receptor antagonist: an effective treatment for reperfusion injury following acute ischemic stroke in rats. Neuroscience 220: 1–10. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Vink R (2013). The role of substance P in ischaemic brain injury. Brain Sci 3: 123–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Vink R (2014). NK1 tachykinin receptor treatment is superior to capsaicin pre‐treatment in improving functional outcome following acute ischemic stroke. Neuropeptides 48: 267–272. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Blumbergs PC, Sims NR, Helps SC, Rodgers KM, Vink R (2006). Increased substance P immunoreactivity and edema formation following reversible ischemic stroke. Acta Neurochir Suppl 96: 263–266. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Helps SC, Thornton E, Vink R (2011). A substance P antagonist improves outcome when administered 4 h after onset of ischemic stroke. Brain Res 1393: 84–90. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Corrigan F, Vink R (2012b). Magnesium in acute brain injury In: Li YV, Zhang JH. (eds). Metal Ions in Stroke. Springer.: New York, pp. 445–460. [Google Scholar]
- Van Den Heuvel C, Donkin JJ, Finnie JW, Blumbergs PC, Kuchel T, Koszyca B et al (2004). Downregulation of amyloid precursor protein (APP) expression following post‐traumatic cyclosporin‐A administration. J Neurotrauma 21: 1562–1572. [DOI] [PubMed] [Google Scholar]
- Vink R, van den Heuvel C (2010). Substance P antagonists as a therapeutic approach to improving outcome following traumatic brain injury. Neurother 7: 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, Donkin JJ, Cruz MI, Nimmo AJ, Cernak I (2004). A substance P antagonist increases brain intracellular free magnesium concentration after diffuse traumatic brain injury in rats. J Am Coll Nutr 23: 538S–540S. [DOI] [PubMed] [Google Scholar]
- Weglicki WB, Phillips TM (1992). Pathobiology of magnesium deficiency: a cytokine/neurogenic inflammation hypothesis. Am J Physiol 263: R734–R737. [DOI] [PubMed] [Google Scholar]
- Weglicki WB, Phillips TM, Mak IT, Cassidy MM, Dickens BF, Stafford R et al (1994). Cytokines, neuropeptides, and reperfusion injury during magnesium deficiency. nns NY Acad Sci 723: 246–257. [PubMed] [Google Scholar]
- Wells AJ, Vink R, Blumbergs PC, Brophy BP, Helps SC, Knox SJ et al (2012). A surgical model of permanent and transient middle cerebral artery stroke in the sheep. PLoS ONE 7: e42157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2004). Atlas of Heart Disease and Stroke. WHO: Geneva. [Google Scholar]
- Wu ZZ, Guan BC, Li ZW, Yang Q, Liu CJ, Chen JG (2004). Sustained potentiation by substance P of NMDA‐activated current in rat primary sensory neurons. Brain Res 1010: 117–126. [DOI] [PubMed] [Google Scholar]
- Yi JH, Hazell AS (2006). Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 48: 394–403. [DOI] [PubMed] [Google Scholar]
- Zacest AC, Vink R, Manavis J, Sarvestani GT, Blumbergs PC (2010). Substance P immunoreactivity increases following human traumatic brain injury. Acta Neurochir Suppl 106: 211–216. [DOI] [PubMed] [Google Scholar]
- Ziebell JM, Morganti‐Kossmann MC (2010). Involvement of pro‐ and anti‐inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurother 7: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman BJ, Anderson DC, Granger DN (1992). Neuropeptides promote neutrophil adherence to endothelial cell monolayers. Am J Physiol 263: G678–G682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1 Methods.