

Abstract
Background and Purpose
Neutrophil elastase plays a crucial role in arthritis. Here, its potential in triggering joint inflammation and pain was assessed, and whether these effects were mediated by proteinase‐activated receptor‐2 (PAR2).
Experimental Approach
Neutrophil elastase (5 μg) was injected into the knee joints of mice and changes in blood perfusion, leukocyte kinetics and paw withdrawal threshold were assessed. Similar experiments were performed in animals pretreated with the neutrophil elastase inhibitor sivelestat, the PAR2 antagonist GB83, the p44/42 MAPK inhibitor U0126 and in PAR2 receptor knockout (KO) mice. Neutrophil elastase activity was also evaluated in arthritic joints by fluorescent imaging and sivelestat was assessed for anti‐inflammatory and analgesic properties.
Key Results
Intra‐articular injection of neutrophil elastase caused an increase in blood perfusion, leukocyte kinetics and a decrease in paw withdrawal threshold. Sivelestat treatment suppressed this effect. The PAR2 antagonist GB83 reversed neutrophil elastase‐induced synovitis and pain and these responses were also attenuated in PAR2 KO mice. The MAPK inhibitor U0126 also blocked neutrophil elastase‐induced inflammation and pain. Active neutrophil elastase was increased in acutely inflamed knees as shown by an activatable fluorescent probe. Sivelestat appeared to reduce neutrophil elastase activity, but had only a moderate anti‐inflammatory effect in this model.
Conclusions and Implications
Neutrophil elastase induced acute inflammation and pain in knee joints of mice. These changes are PAR2‐dependent and appear to involve activation of a p44/42 MAPK pathway. Blocking neutrophil elastase, PAR2 and p44/42 MAPK activity can reduce inflammation and pain, suggesting their utility as therapeutic targets.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- IVM
intravital microscopy
- LASCA
laser speckle contrast analysis
- PAR
proteinase‐activated receptor
- TRPV
transient receptor potential vanilloid
- VCAM
vascular cell adhesion molecule
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| PAR2 | Neutrophil elastase |
| Ion channels b | p44/42 MAPK |
| TRPV1 |
| LIGANDS |
|---|
| Sivelestat |
| U0126 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c).
Introduction
Acute joint inflammation is a physiological response to injury or pathogenic infection which, when chronic, leads to the development of arthritis. Joints typically initiate a natural inflammatory reaction by releasing a complex mixture of inflammatory mediators from peripheral nerves, synoviocytes and vascular endothelial cells within the joint. Persistent inflammation, however, leads to severe joint swelling, angiogenesis, reduced mobility, loss of articular cartilage and the eventual eburnation of subchondral bone. In addition to these morphological changes in the joint, articular afferent nerves become sensitized through the actions of various inflammatory mediators which culminates in the generation of joint pain (McDougall, 2006).
During inflammation, a variety of serine proteinases are released into the joint by resident mast cells and accumulated immune cells. For example, mast cells release trypsin and tryptase, while neutrophils release neutrophil elastase, cathepsin G and proteinase 3 (Bohm et al., 1996; Corvera et al., 1997; Knecht et al., 2007; Korkmaz et al., 2010). Many different effects are triggered by serine proteinases, such as cytokine, kinin, and growth factor generation and clustering of integrins (McPhail et al., 1992; Ramachandran et al., 2011). An increase in the levels of these degrading enzymes can destroy joint collagen and proteoglycans (Racine and Aaron, 2013) and this process is one of the main causes of joint destruction. Neutrophil elastase, for example, has a broad specificity for a number of substrates including connective tissue elastin, collagen, proteoglycan, fibronectin and other extracellular matrix proteins (Watanabe et al., 1990). This would mean that, in joints, neutrophil elastase could be responsible for the destruction of articular cartilage, menisci, ligaments and capsule. Neutrophil elastase levels are increased in rheumatoid arthritic joints (Momohara et al., 1997), suggesting the involvement of this enzyme in inflammatory joint disease.
In addition to their enzymic effects, serine proteinases can also cleave a family of GPCRs called proteinase‐activated receptors (PARs) and, to date, four PARs have been identified. A range of immune cells such as T‐cells, macrophages, neutrophils and mast cells express PAR2, and stimulation of these cells with pro‐inflammatory cytokines has been shown to up‐regulate PAR2 expression, suggesting their involvement in the inflammatory response (Vergnolle et al., 2003; Russell and McDougall, 2009). PAR2 also plays an important role in immune‐mediated joint inflammation (Busso et al., 2007), and antagonism of PAR2 has been shown to possess therapeutic potential in treating joint inflammation (Kelso et al., 2006). PAR2 is expressed on afferent neurons and its activation triggers neurogenic inflammation through the local release of CGRP and substance P (Steinhoff et al., 2000). The identification of PAR2 on joint sensory nerves (Russell et al., 2012) implies that activation of these receptors is involved in regulating pain transmission. Indeed, selective activation of articular PAR2 by synthetic peptides causes peripheral sensitization and the generation of joint pain in rodents (Helyes et al., 2010; Russell et al., 2010; 2011).
PAR2 activation by serine proteinases is widely considered to cause a pro‐inflammatory and pro‐nociceptive response in various pathological conditions (Fiorucci et al., 2001; Vergnolle et al., 2003; Singh et al., 2007; Laukkarinen et al., 2008) and can trigger the destruction of cartilage by increasing the levels of degradative enzymes. Activation of each of the PARs involves cleavage of an established site on the extracellular N terminal domain by thrombin (PARs 1, 3 and 4), trypsin (PARs 2 and 4) or other proteases to unmask the tethered ligand that activates signalling via Gq, Gi or G12/13 (Hollenberg et al., 2014). However, some proteinases cleave PARs at a different site on the extracellular domain, leading to activation of alternate second messenger pathways in a process termed biased signalling. Recently, it was found that PAR2 is capable of exhibiting biased signalling either via a Gq‐coupled calcium signal or via Gi or G12/13‐coupled p44/42 MAPK signal (Ramachandran et al., 2009). Neutrophil elastase has been shown to specifically cleave PAR2 (Zhou et al., 2013) and selectively activate the intracellular MAPK pathway (Ramachandran et al., 2011).
In this study, we hypothesized that neutrophil elastase would induce inflammation and pain by directly activating PAR2, and that blockade of this receptor would decrease joint inflammation and pain. We also attempted to characterize the signalling pathway downstream of the receptor and to investigate a possible role for neutrophil elastase in a model of inflammatory arthritis. Our results demonstrated that neutrophil elastase induced inflammation and pain in the knee joints of mice via a PAR2‐dependent mechanism.
Methods
Animals
All animal care and experimental procedures complied with the Canadian Council for Animal Care guidelines (http://www.ccac.ca/) and were approved by the Dalhousie University Committee on Laboratory Animals and the University of Pécs Ethics Committee on Animal Research (License No: BA 02/2000‐2/2012). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 232 animals were used in the experiments described here.
Experiments were performed on male C57Bl/6 mice (Charles River Laboratories Inc, QC, Canada) or wild‐type (PAR2+/+) and PAR2‐deficient (PAR2−/−) mice of either sex raised in‐house (original breeders developed on a C57Bl/6 background from Jackson Laboratories, Bar Harbor, ME, USA). Animal weights ranged from 20 to 30 g (8–14 weeks old). Mice were housed at 22 ± 2°C on a 12:12 h light : dark cycle. Animals were fed standard lab chow with water available ad libitum.
Surgical preparation for vascular assessment
Deep anaesthesia of mice was achieved using urethane (25%; 0.3–0.4 mL i.p.) and was confirmed by the absence of a hindpaw withdrawal reflex before any surgical procedures were performed. Mice were placed in dorsal recumbency on a heated blanket (SoftHeat HP710‐24‐3P‐S Electric Heating Pad, Kaz Inc., Southborough, MA, USA) to maintain body temperature. The ventral aspect of the neck was coated with mineral oil and a longitudinal incision was made in the skin to expose the trachea, which was cannulated using polyethylene tubing (0.76 mm internal diameter, 1.22 mm outer diameter; Clay Adams, Parsippany, NJ, USA) to allow unrestricted breathing. The right carotid artery and jugular vein were then isolated and cannulated with polyethylene tubing (0.28 mm internal diameter, 0.61 mm outer diameter; Clay Adams) filled with heparinized saline (1 U·mL−1). The carotid artery cannula was connected in series to a pressure transducer (Kent Scientific Corporation, Torrington, CT, USA) and mean arterial pressure was recorded on a differentially amplified BP monitor (BP‐1; World Precision Instruments, Sarasota, FL, USA). The skin covering both knee joints was excised (∼1 cm long × ∼0.5 cm wide) and all superficial fasciae removed to allow an unrestricted view of the joint microvasculature. Warm (37°C) physiological buffer was intermittently perfused over the surface of the knee to prevent tissue desiccation.
Measurement of leukocyte trafficking
Leukocyte trafficking in the mouse knee joint microvasculature was assessed using intravital microscopy (IVM), as previously described (Andruski et al., 2008). After surgical preparation, the synovial microcirculation was visualized under incident fluorescent light using a Leica DM2500 microscope with a HCX APO L 20X objective and a HC Plan 10X eyepiece (Leica Microsystems Inc., Richmond Hill, ON, Canada; final magnification 200×). Leukocytes were stained in vivo using 0.05% rhodamine 6G (0.06 mL) injected through the jugular vein cannula immediately before measurement. Straight, unbranched, postcapillary venules (diameter 20–50 μm), located directly on the knee joint capsule, were selected for analysis. Recordings of 1 min duration were made using a BC‐71 AVT camera (Horn Imaging, Aalen, Germany). Rolling leukocytes, which travel along the venular endothelium at a velocity less than the free flowing cells in the same vessel and the same radial position, were quantified over a 60 s period. Adherent leukocytes, which remain stationary for the duration of the 30 s measurement period, were quantified within a 100 μm length of venule. The videos from three different venules per knee joint were recorded and the values obtained were averaged.
Microvascular perfusion
Microvascular perfusion in the mouse knee joint was assessed using laser speckle contrast analysis (LASCA – PeriCam PSI System, Perimed Inc., Ardmore, PA, USA), as previously described (Krustev et al., 2014). This system monitors tissue blood perfusion in real time by measuring the backscatter of laser light which forms an interference pattern consisting of dark and bright areas in response to movement of erythrocytes. The speckle pattern is captured and converted to a measure of blood perfusion and assigned an arbitrary perfusion unit value. One minute recordings were taken at a working distance of 10 cm with a frame rate of 25 images per second. After conclusion of the final reading, the mouse was killed by injection of sodium pentobarbital (1000 mg·kg−1 i.p.). A ‘dead scan’ was recorded to obtain a ‘biological zero’ measurement, which was subtracted from all previous measurements for that animal.
Joint pain assessment
Ipsilateral hindpaw mechanosensitivity was assessed by plantar application of von Frey hair filaments using a modification of the Dixon's up‐down method (Chaplan et al., 1994). Animals were placed in elevated Plexiglas chambers on metal mesh flooring allowing access to the paws. After allowing the animal to acclimate until exploratory behaviour ceased, a von Frey hair was applied perpendicular to the plantar surface of the hindpaw (avoiding the toe pads) until the hair started to bend, and the hair was held in place for 3 s. If there was a positive response (withdrawal, shake or lick of the hindpaw), the next lower strength hair was applied; if there was no response, the next higher strength hair was applied up to a maximum cut‐off level, which corresponded to a 4 g bending force. After the first difference in response was observed, four more measurements were made and the pattern of responses was converted to a 50% withdrawal threshold calculated using the following formula: 10[Xf + kδ]/10 000; where Xf = value (in log units) of the final von Frey hair used, k = tabular value for the pattern of the last six positive/negative responses, and δ = mean difference (in log units) between stimuli. Animals were returned to their home cages for the interval between measurements.
Neutrophil elastase‐induced inflammation and pain
For induction of neutrophil elastase‐induced inflammation and pain, mice were anaesthetized (2–4% isoflurane; 100% oxygen at 1 L·min−1) and an acceptable plane of anaesthesia was confirmed by failure to produce a hindpaw withdrawal reflex. The right knee joint was shaved and baseline knee joint diameter was measured using a digital micrometre (Control Company, Friendswood, TX, USA). A single intra‐articular injection of 5 μg (4.4 U) neutrophil elastase (10 μL) was administered through the patellar ligament of the right knee using a 30 G needle. The knee was then manually extended and flexed for 30 s to disperse the neutrophil elastase throughout the joint. For IVM and LASCA experiments, the left (contralateral) knee was injected with 10 μL of physiological saline and measurements were subtracted from readings taken from the neutrophil elastase‐injected knee.
To confirm that the inflammatory changes were induced by neutrophil elastase, further experiments were conducted in which animals were pretreated with the neutrophil elastase inhibitor sivelestat (50 mg·kg−1 i.p.) 10 min before injection of neutrophil elastase. As neutrophil elastase produced a maximal effect at 4 h post‐administration across all parameters measured, including knee diameter, further testing focused on this time point.
The role of PAR2 receptors was investigated by treatment with the PAR2 antagonist GB83 (Barry et al., 2010) (5 μg X3 i.p.), administered 10 min before and 110 and 230 min after neutrophil elastase. Neutrophil elastase was also evaluated in PAR2 knockout mice. To elucidate the possible intracellular pathway downstream of PAR2, mice were treated with the p44/42 MAPK inhibitor U0126 (30 mg·kg−1 i.p.) 2 h after injection of neutrophil elastase.
Kaolin‐carrageenan induction of knee joint acute inflammation and pain
Mice were deeply anaesthetized with isoflurane and joint inflammation was induced as follows. Kaolin (2%, 10 μL ) was injected into the intra‐articular space of the right knee joint and the limb was flexed and extended for 10 min to disperse the substance throughout the joint and cause mechanical damage and irritation of the synovial space. Carrageenan (2%, 10 μL) was injected in the same manner and was followed by 30 s of hindlimb flexion and extension. Changes in the leukocyte kinetics, microvascular perfusion and behavioural pain were recorded at 24 h post injection.
To assess the role of neutrophil elastase in kaolin/carrageenan‐induced inflammation, further experiments were conducted in which animals were treated with the neutrophil elastase inhibitor sivelestat (50 mg·kg−1 i.p.) 20 h after injection of kaolin/carrageenan (i.e. 4 h before measurements).
In vivo optical imaging of neutrophil elastase enzyme activity
Acute knee joint inflammation was induced by kaolin‐carrageenan as described, and sivelestat (50 mg·kg−1 i.p.) or saline treatment was performed 18 h later. The contrast agent Neutrophil Elastase 680 FAST (NE680) in the dosage recommended by the manufacturer (4 nmol/100 μL/mouse in PBS) was retroorbitally injected under anaesthesia 30 min following sivelestat. NE680 is a commercially available fluorescence agent that is optically silent, unless enzymically cleaved. It was demonstrated previously that NE680 enables sensitive and selective detection of elastase activity during inflammation, and also that the cleavage of this contrast agent can be inhibited by sivelestat both in vitro and in vivo (Kossodo et al., 2011). The hair around the knee was removed, and the mice were imaged in reflectance mode 6 h post injection by the FMT 2000 optical imaging system (PerkinElmer, Waltham, MA, USA). Identical regions of interest were drawn around each knee, and the fluorescence intensity within the regions of interest was calculated as counts/energy.
Data analysis
All data are presented as means ± SEM and were analysed with the statistical software package GraphPad Prism v.5 (GraphPad Software Inc., San Diego, CA, USA). The data were first tested for normal distribution using the Kolmogorov–Smirnov test. Time courses of neutrophil elastase with and without sivelestat were compared by a two‐way anova. The time course of effect of neutrophil elastase was analysed by a one‐way anova, and the time point of maximal effect was determined by a Dunnett's post hoc test; further analysis focused on this time point (4 h). The remaining data were analysed by one‐way anova with Dunnett's post hoc test, comparing all experimental groups to the neutrophil elastase‐treated group. Kaolin‐carrageenan data were analysed by a one‐way anova with Dunnett's post hoc test, comparing all experimental groups to the inflamed control group. Mean arterial pressure data were analysed by a one‐way anova with Tukey's multiple comparison post hoc test. The fluorescence imaging results were analysed by Student's t‐test for paired comparison.
Materials
Neutrophil elastase purified from human sputum was obtained from Elastin Products (Owensville, MO, USA). Sivelestat (neutrophil elastase inhibitor; sodium N‐[2‐[4‐(2,2‐dimethylepropionyloxy)phenyl‐sulfonylamino]benzoil] aminoacetate) was obtained from Enzo Life Sciences (New York, NY, USA). GB83 (PAR2 antagonist; N‐((S)‐3‐cyclohexyl‐1‐((2S,3S)‐1‐(2,3‐dihydrospiro[indene‐1,4′‐piperidine]‐1′‐yl)‐3‐methyl‐1‐oxopentan‐2‐ylamino)‐1‐oxopropan‐2‐yl)isoxazole‐5‐carboxamide) was obtained from Axon Medchem (Groningen, The Netherlands). U0126 (MAPK inhibitor; 1,4‐diamino‐2,3‐dicyano‐1,4‐bis[2‐aminophenylthio]butadiene) was obtained from Cayman Chemicals (Ann Arbor, MI, USA). Rhodamine 6G, cremophor, DMSO, urethane, kaolin and carrageenan were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Neutrophil Elastase 680 FAST was purchased from PerkinElmer. Neutrophil elastase, sivelestat, kaolin, carrageenan and rhodamine 6G were dissolved in saline. GB83 and U0126 were dissolved in vehicle (1:1:8 DMSO : cremophor : saline). Physiological buffer (135 mM NaCl, 20 mM NaHCO3, 5 mM KCl, 1 mM MgSO4*7H2O, pH =7.4) was prepared in‐house.
Results
Knee diameter
At 4 h after neutrophil elastase injection, joint diameter was increased significantly in wild‐type animals compared with saline control (P < 0.001) or PAR2−/− animals (P < 0.05) (Figure 1; one‐way anova with Dunnett's post hoc test).
Figure 1.
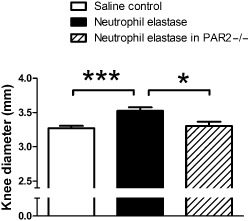
Knee diameter in mice. Knee joint diameters, measured 4 h following intra‐articular injection of neutrophil elastase, showing a significant increase in wild‐type mice, but not in PAR2 KO mice (PAR 2−/−), when compared with intra‐articular saline. Values represent mean ± SEM (n = 9–15/group). *P < 0.05, ***P < 0.001 compared with neutrophil elastase.
Mean arterial pressure
Mean arterial pressure was recorded during IVM and LASCA measurements and no significant differences were observed between treatment groups (Table 1).
Table 1.
Mean arterial pressure
| Treatment group | Mean arterial pressure, mmHg (mean ± SEM) |
|---|---|
| Neutrophil elastase | 40.23 ± 1.56 |
| Neutrophil elastase + sivelestat | 45.57 ± 6.98 |
| Neutrophil elastase + GB83 | 43.90 ± 5.11 |
| Neutrophil elastase in PAR2−/− | 39.00 ± 5.60 |
| Neutrophil elastase + U0126 | 53.80 ± 6.00 |
| Kaolin‐carrageenan | 42.10 ± 4.43 |
| Kaolin‐carrageenan + sivelestat | 53.40 ± 3.83 |
Mean arterial pressure values did not differ significantly between treatment groups. Values shown are means ± SEM for n = 9–15 per group.
Neutrophil elastase‐induced joint inflammation and pain
Intra‐articular injection of neutrophil elastase (5 μg) caused a progressive increase in microvascular perfusion which peaked at 4 h (Figure 2A, P = 0.01, one‐way anova). This hyperaemic effect of the protease was blocked by treatment with the neutrophil elastase inhibitor sivelestat (Figure 2A, P < 0.0001, two‐way anova). The number of rolling leukocytes (Figure 3A, P < 0.005, one‐way anova) and adherent leukocytes (Figure 3C, P < 0.0005, one‐way anova) also gradually increased, with maximal effect occurring 4 h after injection of neutrophil elastase. These inflammatory changes ultimately resolved by 24 h. Pretreatment of mice with sivelestat completely blocked the increase in leukocyte trafficking across the entire time course (Figure 3A and C, P = 0.002 and P = 0.012, respectively, two‐way anova).
Figure 2.
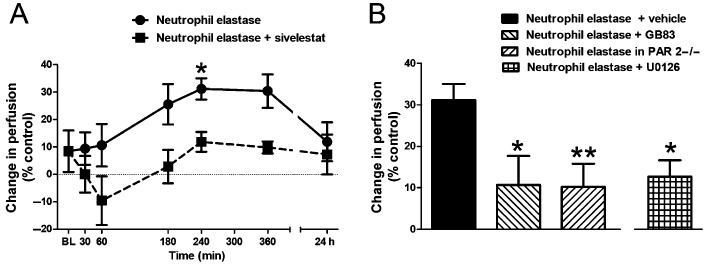
Changes in vascular perfusion. (A) Time course of the increase in knee joint blood perfusion following intra‐articular injection of neutrophil elastase and its reversal by systemic pretreatment with the neutrophil elastase inhibitor sivelestat (n = 6–7 per time point). *P < 0.05 compared with baseline (BL). (B) Mean perfusion in the knee joint microvasculature at 4 h following intra‐articular injection of neutrophil elastase showing the increased perfusion is blocked by the PAR2 antagonist GB83 and is absent in PAR2 KO mice (PAR 2−/−). The neutrophil elastase effect is also blocked by the MAPK inhibitor U0126. Values shown are the per cent difference in perfusion units compared with the saline‐injected contralateral knee and are means ± SEM (n = 9–15/group). *P < 0.05, **P < 0.01 compared with neutrophil elastase alone.
Figure 3.
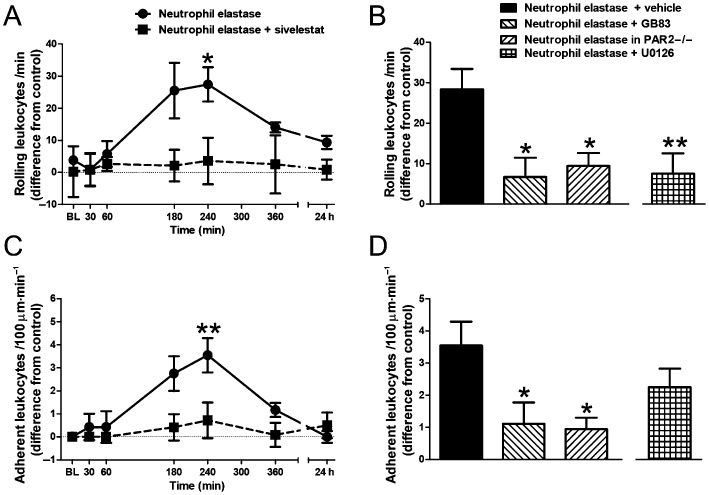
Changes in leukocyte trafficking. Time course of the increase in the number of (A) rolling and (C) adherent leukocytes following intra‐articular injection of neutrophil elastase and its reversal by systemic pretreatment with sivelestat (n = 6–7 per time point). *P < 0.05, **P < 0.01 compared with baseline (BL). Increases in the number of (B) rolling and (D) adherent leukocytes in the knee joint microvasculature at 4 h following intra‐articular injection of neutrophil elastase are blocked by the PAR2 antagonist GB83 and are absent in PAR2 KO mice (PAR 2−/−). The effect of neutrophil elastase on rolling leukocytes is also blocked by the MAPK inhibitor U0126. Values shown are the difference in number of cells compared with the saline‐injected contralateral knee and are means ± SEM (n = 9–15/group). *P < 0.05, **P < 0.01 compared with neutrophil elastase alone.
Intra‐articular injection of neutrophil elastase caused a significant decrease in hindpaw mechanical sensitivity, indicative of secondary allodynia (Figure 4A, P < 0.005, one‐way anova). The pain response peaked at 4 h after intra‐articular injection before returning to baseline levels by 24 h. Sivelestat significantly inhibited the increased pain sensitivity over the entire time course tested (Figure 4A, P < 0.0001, two‐way anova).
Figure 4.
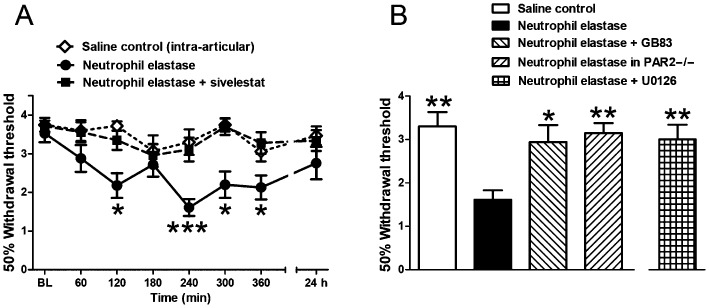
Changes in behavioural pain. (A) Time course of the decrease in withdrawal threshold following intra‐articular neutrophil elastase and its reversal by systemic pretreatment with the neutrophil elastase inhibitor sivelestat. The intra‐articular saline control did not show a change in withdrawal threshold. *P < 0.05, ***P < 0.001 compared with baseline (BL). (B) Withdrawal thresholds at 4 h following intra‐articular neutrophil elastase showing the induced change is blocked by pretreatment with the PAR2 antagonist GB83 and is absent in PAR2 KO mice (PAR 2−/−). The neutrophil elastase effect is also blocked by the MAPK inhibitor U0126. Values shown are von Frey 50% withdrawal thresholds and are mean ± SEM (n = 9–11/group). *P < 0.05, **P < 0.01 compared with neutrophil elastase alone.
Involvement of PAR2 in mediating neutrophil elastase‐induced joint inflammation and pain
To examine the involvement of PAR2 in mediating neutrophil elastase‐induced joint inflammation and pain, we repeated the experiments with neutrophil elastase in the presence of the PAR2 antagonist GB83 and in PAR2 knockout mice. GB83 significantly blocked the neutrophil elastase‐induced increase in vascular perfusion (Figure 2B, P < 0.05, one‐way anova with Dunnett's post hoc test), as well as the number of rolling leukocytes (Figure 3B, P < 0.05, one‐way anova with Dunnett's post hoc test) and the number of adherent leukocytes (Figure 3D, P < 0.05, one‐way anova with Dunnett's post hoc test) at 4 h. In pain assessment experiments, GB83 also significantly attenuated hindpaw allodynia (Figure 4B, P < 0.05, one‐way anova with Dunnett's post hoc test).
In PAR2 knockout mice, neutrophil elastase failed to induce a change in vascular perfusion (Figure 2B), leukocyte trafficking (Figure 3B and D) or pain (Figure 4B).
Intracellular mechanism of PAR2 activation
To elucidate the downstream signalling pathway following PAR2 activation by neutrophil elastase, the p44/42 MAPK inhibitor U0126 was tested. Systemic treatment of mice with U0126 blocked the hyperaemic effect of neutrophil elastase (Figure 2B, P < 0.05, one‐way anova with Dunnett's post hoc test) as well as the increase in rolling leukocytes (Figure 3B, P < 0.01, one‐way anova with Dunnett's post hoc test). Leukocyte adhesion was slightly blocked by U0126 treatment (Figure 3D); however, this was found to be not statistically significant. U0126 also significantly inhibited neutrophil elastase‐induced secondary allodynia (Figure 4B, P < 0.01, one‐way anova with Dunnett's post hoc test).
Neutrophil elastase activity in acutely inflamed joints
One day after the induction of knee joint inflammation with kaolin‐carrageenan, elastase activity was measured in vivo by imaging of the fluorescence contrast agent NE680. Inflamed mice treated with systemic saline demonstrated a moderate, but significant increase of signal in the inflamed joint when compared with the contralateral control joint (Figure 5, P < 0.05, paired Student's t‐test). Treatment with systemic sivelestat tended to reduce neutrophil elastase activity in the inflamed knee but the observed difference was not statistically significant (Figure 5).
Figure 5.
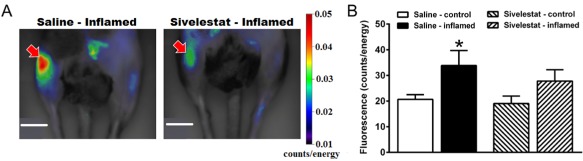
In vivo imaging of neutrophil elastase enzyme activity. (A) Representative fluorescence images taken 24 h after unilateral induction of knee joint inflammation with kaolin‐carrageenan (right knee, indicated by arrow) compared with the untreated contralateral joint (left knee). Arthritic animals were treated with either systemic saline (left image) or systemic sivelestat (right image). Sivelestat significantly reduced neutrophil elastase activity in inflamed joints (arrow in right image). Scale bar = 1 cm. (B) Fluorescence intensity in the inflamed and control knee joints. Values shown are means ± SEM (n = 5–6 per group). *P < 0.05 compared with saline control.
Effect of neutrophil elastase inhibition on joint inflammation and pain
In order to examine a possible role for neutrophil elastase in inflammatory arthritis, acutely inflamed mice were treated with sivelestat 4 h before vascular and pain testing. Drug treatment reduced the increase in vascular perfusion (Figure 6A, P < 0.05, one‐way anova with Dunnett's post hoc test) and adherent leukocytes (Figure 6C, P < 0.01, one‐way anova with Dunnett's post hoc test) in acutely inflamed animals. However, sivelestat had no discernible effect on leukocyte rolling (Figure 6B) or pain (Figure 6D) in inflamed mice.
Figure 6.
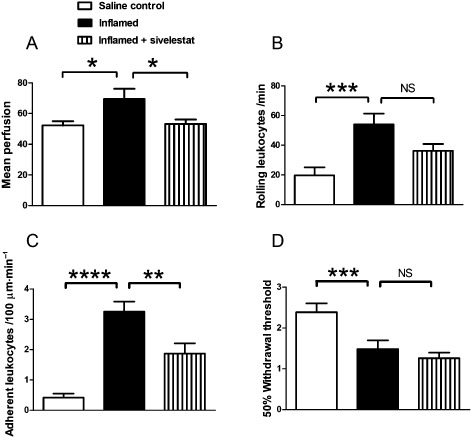
Kaolin/carrageenan‐induced inflammation and pain. Changes in (A) mean perfusion, (B) rolling and (C) adherent leukocytes, and (D) secondary allodynia in the knee joint 24 h following intra‐articular administration of saline (control), or kaolin‐carrageenan (inflamed) with or without systemic administration of sivelestat. Sivelestat treatment significantly inhibited joint inflammation but not joint pain. Values shown are means ± SEM (n = 10–12 per group). NS = not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with inflamed control.
Discussion
The present study shows for the first time that local administration of neutrophil elastase induces pro‐inflammatory and pro‐nociceptive changes in the knee joints of mice. These effects are mediated by activation of PAR2 with subsequent downstream signalling via the p44/42 MAPK pathway.
Pro‐inflammatory effects of neutrophil elastase
Neutrophil elastase has been shown to be involved in the pathogenesis of a variety of inflammatory diseases, including idiopathic pulmonary fibrosis (Song et al., 2009), rheumatoid arthritis (Adeyemi et al., 1990; Kakimoto et al., 1995; Momohara et al., 1997), respiratory distress syndrome (Doring, 1994), lung emphysema, cystic fibrosis (Hentschel et al., 2015) and sepsis (Tsujimoto et al., 2005). The objective of this study was to assess the role of neutrophil elastase in inflammation of the mouse knee joint. Leukocyte extravasation and increased microvascular perfusion at the affected site are key characteristics of inflammation. In response to intra‐articular injection of neutrophil elastase, the number of rolling and adherent leukocytes increased within synovial venules and the joint gradually became hyperaemic. Extravasation is regulated by release of different cytokines and chemokines which cause an increase in expression of adhesion molecules on the surface of endothelial cells that promote the recruitment of leukocytes. Subsequently, endothelial surface enzymes like vascular adhesion protein‐1 and CD73 cause leukocyte extravasation (Jalkanen and Salmi, 2008); whether this process occurs following neutrophil elastase exposure requires further investigation. Previous studies indicate that neutrophil elastase triggers the release of a variety of chemokines and cytokines (e.g. TNF‐α, GM‐CSF, IL‐8 and IFN‐γ) in various tissues that could drive the inflammation observed here (Hallett and Lloyds, 1995; Wright et al., 2010). Conversely, leukocyte infiltration can be limited by decreasing levels of pro‐inflammatory cytokines like TNF‐α and IL‐1β which prevents the up‐regulation of adhesion molecules (vascular cell adhesion molecule, VCAM‐1; P‐selectin) and activation of macrophages. When mice were pretreated with the neutrophil elastase inhibitor sivelestat, the altered leukocyte kinetics and hyperaemia induced by neutrophil elastase were no longer observed. Sivelestat inhibits the enzymic action of neutrophil elastase directly by a reversible ‘acylation‐deacylation’ mechanism (Nakayama et al., 2002) and has been shown to reverse inflammatory changes induced by neutrophil elastase (Kakimoto et al., 1995; Fukatsu et al., 2010; Nomura et al., 2013).
Activation of PAR2 by different serine proteinases results in conversion of arachidonic acid into prostaglandins, which are potent inflammatory mediators (Kong et al., 1997; Frungieri et al., 2005). Whether neutrophil elastase similarly causes the release of inflammatory prostaglandins in joints is currently unknown. PAR2 has also been shown to mediate inflammatory changes in the gut, lungs and joints by neurogenic mechanisms (Dulon et al., 2003; Russell and McDougall, 2009). In knee joints, activation of PAR2 with a synthetic peptide agonist promotes leukocyte trafficking, joint oedema and synovial hyperaemia (Ferrell et al., 2003; Helyes et al., 2010; Russell et al., 2012). In PAR2 knockout mice, the onset of inflammation after surgical trauma is delayed and correlates with decreased leukocyte rolling (Lindner et al., 2000). In the present study, neutrophil elastase‐induced inflammation was blocked by treatment with the PAR2 antagonist GB83 and was absent in PAR2 knockout mice. Thus, neutrophil elastase has the capacity to cleave PAR2, leading to joint inflammation.
Molecular studies have determined that neutrophil elastase activates PAR2 by an atypical mechanism that differs from other serine proteinases. Recent evidence shows that neutrophil elastase acts as a biased agonist for PAR2 by cleaving the receptor at a non‐canonical site in the extracellular N terminal. This biased signalling leads to activation of the intracellular p44/42 MAPK pathway without triggering calcium release (Ramachandran et al., 2011). Biased activation of this pathway leads to sensitization of transient receptor potential vanilloid (TRPV) 4 channels (Sostegni et al., 2014), which contributes to tissue inflammation (Denadai‐Souza et al., 2012) and pain (Grant et al., 2007). Luo et al. (2010) showed that TNF‐α‐induced VCAM‐1 expression is mediated through activation of the p44/42 MAPK and NF‐κB pathways in rheumatoid arthritis synovial fibroblasts, confirming the role of these pathways in joint inflammation. Here, neutrophil elastase‐induced inflammation was attenuated following treatment with the p44/42 MAPK inhibitor U0126. This finding indicates that cleavage of PAR2 by neutrophil elastase leads to signalling via the p44/42 MAPK cascade.
The present study examined the role of endogenous neutrophil elastase in driving inflammation by administering sivelestat in the kaolin‐carrageenan in vivo model of acute inflammation. Our results showed that sivelestat had moderate anti‐inflammatory actions. This finding is in accordance with a previous report where inactivation of various neutrophil proteinases resulted in anti‐inflammatory activity in a series of in vivo experimental models including carrageenan‐induced paw oedema and carrageenan‐induced pleurisy (Oliveira et al., 2010).
Pro‐nociceptive effects of neutrophil elastase
Pain is a key feature of joint inflammation and the greatest concern for arthritic patients. Sensory nerves are sensitized during inflammation which results in pain responses to normally innocuous stimuli (McDougall, 2011). The results presented here show that intra‐articular neutrophil elastase can induce tactile sensitivity of the plantar surface of the hindpaw (secondary allodynia). A clinical study found a significantly increased concentration of neutrophil elastase in the urine of interstitial cystitis patients with pain compared with those without bladder pain as the predominant symptom (Kuromitsu et al., 2008). These findings suggest an important role for neutrophil elastase in the development of pain in various disease states.
Serine proteases, such as mast cell tryptase, can cause neurogenic inflammation and pain by activating PAR2 on nociceptive neurons (Steinhoff et al., 2000; Vergnolle et al., 2001; Veldhuis and Bunnett, 2013). In joints, PAR2 is functionally coupled to TRPV1 ion channels which, when opened, leads to a pain response possibly orchestrated by local release of CGRP and substance P (Helyes et al., 2010; Russell et al., 2012). The current study confirms a role for PAR2 in neutrophil elastase‐induced secondary allodynia, as treatment with a PAR2 antagonist or absence of PAR2 in knockout animals showed a reduction in hindlimb pain. Whether cleavage of PAR2 by neutrophil elastase also leads to TRPV1‐dependent release of algesic neuropeptides is likely, but not assessed here.
Neuronal and glial cells contribute to inflammatory pain via ERK activation (Ji et al., 1999; Hu and Gereau, 2003; Lever et al., 2003), and it has been found that the neuronal MAPK‐ERK pathway is indeed an important intracellular cascade associated with formalin‐induced inflammatory pain and thermal hyperalgesia (Karim et al., 2006). In the present set of experiments, p44/42 MAPK inhibition was able to reverse neutrophil elastase‐induced secondary allodynia suggesting involvement of the p44/42 MAPK pathway. Thus, as with the inflammatory findings, proteolytic cleavage of PAR2 by neutrophil elastase causes biased activation of the p44/42 MAPK pathway leading to the generation of pain.
The data presented here show for the first time that neutrophil elastase is proteolytically active in the kaolin‐carrageenan model of acute synovitis. Treatment of acutely inflamed animals with sivelestat ameliorated neutrophil elastase bioactivity, but did not reverse it. Correspondingly, the secondary allodynia observed with kaolin‐carrageenan treatment was not alleviated by sivelestat suggesting that neutrophil elastase may not be a primary contributor to joint pain in this acute model.
In conclusion, the present findings demonstrate that neutrophil elastase has pro‐inflammatory and pro‐nociceptive activity in the knee joints of mice which is mediated by PAR2. Results also implicate the p44/42 MAPK pathway, which could be selectively activated by biased agonism of PAR2 by neutrophil elastase. Futher work is required to identify the exact mechanisms for generation of pain by neutrophil elastase in animals; the results shown here may be due solely to inflammation, but other mechanisms could be involved. Nevertheless, this study indicates that neutrophil elastase, PAR2 and p44/42 MAPK should be considered as potential targets for the development of novel drugs to treat joint inflammation and pain.
Author contributions
M. M. M. carried out the vascular experiments, analysed and interpreted the resulting data, and contributed to writing the manuscript. A. R. R. carried out the pain behaviour experiments, analysed and interpreted the resulting data, and contributed to writing the manuscript. B. B. and K. B. carried out the fluorescent imaging experiments, and analysed and interpreted the resulting data. Z. H. helped design the fluorescent imaging experiments, interpreted the resulting data and contributed to writing the manuscript. J. J. M. designed all experiments, helped analyse and interpret the resulting data, and contributed to writing the manuscript.
Conflict of interest
The authors state no conflicts of interest.
Acknowledgements
This work was supported by an operating grant from the Canadian Institutes for Health Research and a CIHR team grant (REACH) (J. J. McD.). M. M. M. holds Graduate Research Scholar awards from the Arthritis Society of Canada, the Nova Scotia Health Research Foundation, and the Nova Scotia Innovation Fund. This work was also supported by KTIA_NAP_13‐2014‐0022 identification number: 888819 to Z. H. The authors also acknowledge Eugene Krustev for his technical support.
Muley, M. M. , Reid, A. R. , Botz, B. , Bölcskei, K. , Helyes, Z. , and McDougall, J. J. (2016) Neutrophil elastase induces inflammation and pain in mouse knee joints via activation of proteinase‐activated receptor‐2. British Journal of Pharmacology, 173: 766–777. doi: 10.1111/bph.13237.
References
- Adeyemi EO, Campos LB, Loizou S, Walport MJ, Hodgson HJF (1990). Plasma lactoferrin and neutrophil elastase in rheumatoid arthritis and systemic lupus erythematosus. Br J Rheumatol 29: 15–20. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The concise guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruski B, McCafferty DM, Ignacy T, Millen B, McDougall JJ (2008). Leukocyte trafficking and pain behavioral responses to a hydrogen sulfide donor in acute monoarthritis. Am J Physiol Regul Integr Comp Physiol 295: R814–R820. [DOI] [PubMed] [Google Scholar]
- Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP (2010). Novel agonists and antagonists for human protease activated receptor 2. J Med Chem 53: 7428–7440. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A et al (1996). Molecular cloning, expression and potential functions of the human proteinase‐activated receptor‐2. Biochem J 314: 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso N, Frasnelli M, Feifel R, Cenni B, Steinhoff M, Hamilton J et al (2007). Evaluation of protease‐activated receptor 2 in murine models of arthritis. Arthritis Rheum 56: 101–107. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Corvera CU, Dery O, McConalogue K, Bohm SK, Khitin LM, Caughey GH et al (1997). Mast cell tryptase regulates rat colonic myocytes through proteinase‐activated receptor 2. J Clin Invest 100: 1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denadai‐Souza A, Martin L, de Paula MA, de Avellar MC, Muscará MN, Vergnolle N et al (2012). Role of transient receptor potential vanilloid 4 in rat joint inflammation. Arthritis Rheum 64: 1848–1858. [DOI] [PubMed] [Google Scholar]
- Doring G (1994). The role of neutrophil elastase in chronic inflammation. Am J Respir Crit Care Med 150: S114–S117. [DOI] [PubMed] [Google Scholar]
- Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D (2003). Proteinase‐activated receptor‐2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol 28: 339–346. [DOI] [PubMed] [Google Scholar]
- Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE et al (2003). Essential role for proteinase‐activated receptor‐2 in arthritis. J Clin Invest 111: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Mencarelli A, Palazzetti B, Distrutti E, Vergnolle N, Hollenberg MD et al (2001). Proteinase‐activated receptor 2 is an anti‐inflammatory signal for colonic lamina propria lymphocytes in a mouse model of colitis. Proc Natl Acad Sci USA 98: 13936–13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frungieri MB, Albrecht M, Raemsch R, Mayerhofer A (2005). The action of the mast cell product tryptase on cyclooxygenase‐2 (COX2) and subsequent fibroblast proliferation involves activation of the extracellular signal‐regulated kinase isoforms 1 and 2 (erk1/2). Cell Signal 17: 525–533. [DOI] [PubMed] [Google Scholar]
- Fukatsu K, Tanabe K, Maeshima Y, Omata J, Yasuhara H, Saitoh D (2010). Neutrophil elastase inhibitor restores gut ischemia reperfusion‐induced impairment of gut immunity with reduced plasma interleukin‐6 concentrations in mice. Surg Infect (Larchmt) 11: 517–522. [DOI] [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S et al (2007). Protease‐activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 78: 715–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett MB, Lloyds D (1995). Neutrophil priming: the cellular signals that say ‘amber’ but not ‘green’. Immunol Today 16: 264–268. [DOI] [PubMed] [Google Scholar]
- Helyes Z, Sandor K, Borbely E, Tekus V, Pinter E, Elekes K et al (2010). Involvement of transient receptor potential vanilloid 1 receptors in protease‐activated receptor‐2‐induced joint inflammation and nociception. Eur J Pain 14: 351–358. [DOI] [PubMed] [Google Scholar]
- Hentschel J, Fischer N, Janhsen WK, Markert UR, Lehmann T, Sonnemann J et al (2015). Protease‐antiprotease imbalances differ between Cystic Fibrosis patients' upper and lower airway secretions. J Cyst Fibros 14: 324–333. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP et al (2014). Biased signalling and proteinase‐activated receptors (PARs): targeting inflammatory disease. Br J Pharmacol 171: 1180–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Gereau RW 4th (2003). ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol 90: 1680–1688. [DOI] [PubMed] [Google Scholar]
- Jalkanen S, Salmi M (2008). VAP‐1 and CD73, endothelial cell surface enzymes in leukocyte extravasation. Arterioscler Thromb Vasc Biol 28: 18–26. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ (1999). Nociceptive‐specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci 2: 1114–1119. [DOI] [PubMed] [Google Scholar]
- Kakimoto K, Matsukawa A, Yoshinaga M, Nakamura H (1995). Suppressive effect of a neutrophil elastase inhibitor on the development of collagen‐induced arthritis. Cell Immunol 165: 26–32. [DOI] [PubMed] [Google Scholar]
- Karim F, Hu HJ, Adwanikar H, Kaplan D, Gereau RW 4th (2006). Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron‐specific dominant negative mitogen activated protein kinase (MEK). Mol Pain 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD et al (2006). Therapeutic promise of proteinase‐activated receptor‐2 antagonism in joint inflammation. J Pharmacol Exp Ther 316: 1017–1024. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K et al (2007). Trypsin IV or mesotrypsin and p23 cleave protease‐activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem 282: 26089–26100. [DOI] [PubMed] [Google Scholar]
- Kong W, McConalogue K, Khitin LM, Hollenberg MD, Payan DG, Böhm SK et al (1997). Luminal trypsin may regulate enterocytes through proteinase‐activated receptor 2. Proc Natl Acad Sci U S A 94: 8884–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz B, Horwitz MS, Jenne DE, Gauthier F (2010). Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62: 726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossodo S, Zhang J, Groves K, Cuneo GJ, Handy E, Morin J et al (2011). Noninvasive in vivo quantification of neutrophil elastase activity in acute experimental mouse lung injury. Int J Mol Imaging 2011: 581406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krustev E, Reid A, McDougall JJ (2014). Tapping into the endocannabinoid system to ameliorate acute inflammatory flares and associated pain in mouse knee joints. Arthritis Res Ther 16: 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromitsu S, Yokota H, Hiramoto M, Morita S, Mita H, Yamada T (2008). Increased concentration of neutrophil elastase in urine from patients with interstitial cystitis. Scand J Urol Nephrol 42: 455–461. [DOI] [PubMed] [Google Scholar]
- Laukkarinen JM, Weiss ER, van Acker GJ, Steer ML, Perides G (2008). Protease‐activated receptor‐2 exerts contrasting model‐specific effects on acute experimental pancreatitis. J Biol Chem 283: 20703–20712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever IJ, Pezet S, McMahon SB, Malcangio M (2003). The signaling components of sensory fiber transmission involved in the activation of ERK MAP kinase in the mouse dorsal horn. Mol Cell Neurosci 24: 259–270. [DOI] [PubMed] [Google Scholar]
- Lindner JR, Kahn ML, Coughlin SR, Sambrano GR, Schauble E, Bernstein D et al (2000). Delayed onset of inflammation in protease‐activated receptor‐2‐deficient mice. J Immunol 165: 6504–6510. [DOI] [PubMed] [Google Scholar]
- Luo SF, Fang RY, Hsieh HL, Chi PL, Lin CC, Hsiao LD et al (2010). Involvement of MAPKs and NF‐kappaB in tumor necrosis factor alpha‐induced vascular cell adhesion molecule 1 expression in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 62: 105–116. [DOI] [PubMed] [Google Scholar]
- McDougall JJ (2006). Arthritis and pain. Neurogenic origin of joint pain. Arthritis Res Ther 8: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall JJ (2011). Peripheral analgesia: hitting pain where it hurts. Biochim Biophys Acta 1812: 459–467. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail LC, Strum SL, Leone PA, Sozzani S (1992). The neutrophil respiratory burst mechanism. Immunol Ser 57: 47–76. [PubMed] [Google Scholar]
- Momohara S, Kashiwazaki S, Inoue K, Saito S, Nakagawa T (1997). Elastase from polymorphonuclear leukocyte in articular cartilage and synovial fluids of patients with rheumatoid arthritis. Clin Rheumatol 16: 133–140. [DOI] [PubMed] [Google Scholar]
- Nakayama Y, Odagaki Y, Fujita S, Matsuoka S, Hamanaka N, Nakai H et al (2002). Clarification of mechanism of human sputum elastase inhibition by a new inhibitor, ONO‐5046, using electrospray ionization mass spectrometry. Bioorg Med Chem Lett 12: 2349–2353. [DOI] [PubMed] [Google Scholar]
- Nomura N, Asano M, Saito T, Nakayama T, Mishima A (2013). Sivelestat attenuates lung injury in surgery for congenital heart disease with pulmonary hypertension. Ann Thorac Surg 96: 2184–2191. [DOI] [PubMed] [Google Scholar]
- Oliveira C, Navarro‐Xavier RA, Anjos‐Vallota EA, Martins JO, Silveira VL, Goncalves LR et al (2010). Effect of plant neutrophil elastase inhibitor on leucocyte migration, adhesion and cytokine release in inflammatory conditions. Br J Pharmacol 161: 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine J, Aaron RK (2013). Pathogenesis and epidemiology of osteoarthritis. R I Med J (2013) 96: 19–22. [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K et al (2009). Agonist‐biased signaling via proteinase activated receptor‐2: differential activation of calcium and mitogen‐activated protein kinase pathways. Mol Pharmacol 76: 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA (2011). Neutrophil elastase acts as a biased agonist for proteinase‐activated receptor‐2 (PAR2). J Biol Chem 286: 24638–24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell FA, McDougall JJ (2009). Proteinase activated receptor (PAR) involvement in mediating arthritis pain and inflammation. Inflamm Res 58: 119–126. [DOI] [PubMed] [Google Scholar]
- Russell FA, Veldhoen VE, Tchitchkan D, McDougall JJ (2010). Proteinase‐activated receptor‐4 (PAR4) activation leads to sensitization of rat joint primary afferents via a bradykinin B2 receptor‐dependent mechanism. J Neurophysiol 103: 155–163. [DOI] [PubMed] [Google Scholar]
- Russell FA, Schuelert N, Veldhoen VE, McDougall JJ (2011). Cathepsin G has an anti‐nociceptive effect in normal rat knee joints. Inflamm Res 60 (Suppl. 1): S293–S295. [Google Scholar]
- Russell FA, Schuelert N, Veldhoen VE, Hollenberg MD, McDougall JJ (2012). Activation of PAR(2) receptors sensitizes primary afferents and causes leukocyte rolling and adherence in the rat knee joint. Br J Pharmacol 167: 1665–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh VP, Bhagat L, Navina S, Sharif R, Dawra RK, Saluja AK (2007). Protease‐activated receptor‐2 protects against pancreatitis by stimulating exocrine secretion. Gut 56: 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JS, Kang CM, Rhee CK, Yoon HK, Kim YK, Moon HS et al (2009). Effects of elastase inhibitor on the epithelial cell apoptosis in bleomycin‐induced pulmonary fibrosis. Exp Lung Res 35: 817–829. [DOI] [PubMed] [Google Scholar]
- Sostegni S, Diakov A, McIntyre P, Bunnett N, Korbmacher C, Haerteis S (2014). Sensitisation of TRPV4 by PAR2 is independent of intracellular calcium signalling and can be mediated by the biased agonist neutrophil elastase. Pflugers Arch 467: 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS et al (2000). Agonists of proteinase activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 6: 151–158. [DOI] [PubMed] [Google Scholar]
- Tsujimoto H, Ono S, Majima T, Kawarabayashi N, Takayama E, Kinoshita M et al (2005). Neutrophil elastase, MIP‐2, and TLR‐4 expression during human and experimental sepsis. Shock 23: 39–44. [DOI] [PubMed] [Google Scholar]
- Veldhuis NA, Bunnett NW (2013). Proteolytic regulation of TRP channels: implications for pain and neurogenic inflammation. Proceed Aust Physiol Soc 44: 101–108. [Google Scholar]
- Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD (2001). Protease‐activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol Sci 22: 146–152. [DOI] [PubMed] [Google Scholar]
- Vergnolle N, Ferazzini M, D'Andrea MR, Buddenkotte J, Steinhoff M (2003). Proteinase‐activated receptors: novel signals for peripheral nerves. Trends Neurosci 26: 496–500. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Hattori S, Katsuda S, Nakanishi I, Nagai Y (1990). Human neutrophil elastase: degradation of basement membrane components and immunolocalization in the tissue. J Biochem 108: 753–759. [DOI] [PubMed] [Google Scholar]
- Wright HL, Moots RJ, Bucknall RC, Edwards SW (2010). Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 49: 1618–1631. [DOI] [PubMed] [Google Scholar]
- Zhou J, Perelman JM, Kolosov VP, Zhou X (2013). Neutrophil elastase induces MUC5AC secretion via protease‐activated receptor 2. Mol Cell Biochem 377: 75–85. [DOI] [PubMed] [Google Scholar]