

Abstract
Traumatic brain injury (TBI) represents a major cause of death and disability in developed countries. Brain injuries are highly heterogeneous and can also trigger other neurological complications, including epilepsy, depression and dementia. The initial injury often leads to the development of secondary sequelae; cellular hyperexcitability, vasogenic and cytotoxic oedema, hypoxia‐ischaemia, oxidative stress and inflammation, all of which influence expansion of the primary lesion. It is widely known that inflammatory events in the brain following TBI contribute to the widespread cell death and chronic tissue degeneration. Neuroinflammation is a multifaceted response involving a number of cell types, both within the CNS and in the peripheral circulation. Astrocytes and microglia, cells of the CNS, are considered key players in initiating an inflammatory response after injury. These cells are capable of secreting various cytokines, chemokines and growth factors, and following injury to the CNS, undergo changes in morphology. Ultimately, these changes can influence the local microenvironment and thus determine the extent of damage and subsequent repair. This review will focus on the roles of microglia and astrocytes following TBI, highlighting some of the key processes, pathways and mediators involved in this response. Additionally, both the beneficial and the detrimental aspects of these cellular responses will be examined using evidence from animal models and human post‐mortem TBI studies.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- BBB
blood–brain barrier
- CCI
controlled cortical impact
- DAMP
danger‐associated molecular pattern
- GFAP
glial fibrillary acidic protein
- Iba‐1
ionized calcium binding adapter molecule 1
- MHC
major histocompatibility complex
- PRR
pathogen recognition receptor
- TBI
traumatic brain injury
- TLR
Toll‐like receptor
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| CCR2 |
| CX3CR1 |
| P2Y receptors |
| Enzymes b |
| Arg‐1, arginase‐1 |
| iNOS |
| Transporters c |
| EAAT1 |
| EAAT2 |
| Catalytic receptors d |
| TLR, Toll‐like receptors |
| LIGANDS |
|---|
| CCL2 |
| CCL3 |
| FGF |
| IFN‐γ |
| IGF‐1, insulin‐like growth factor 1 |
| IL‐1β |
| IL‐4 |
| IL‐6 |
| IL‐10 |
| IL‐13 |
| NO |
| TGF‐β1 |
| TNF‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a, 2013b, 2013c, 2013d).
Introduction
The pathology of traumatic brain injury (TBI) is complex and multifactorial, with TBIs commonly categorized into primary and secondary injuries. Primary injury results from mechanical disruption of brain tissue, often resulting in axonal shearing and can lead to the formation of contusions and haemorrhage (Werner and Engelhard, 2007). As a result of these events, a secondary cascade of molecular and biochemical changes is initiated within minutes of the initial impact, termed secondary injury. Secondary injury processes, such as excitotoxicity, ischaemia, apoptosis, necrosis and inflammation, are critical in determining the extent of injury expansion and damage to brain tissue following the primary insult (Greve and Zink, 2009). Much of the research and studies performed in the field of TBI are focused on targeting various aspects of the secondary injury cascade in order to control cell death and tissue degeneration post‐injury; however, most have demonstrated limited translational success (Menon, 2009).
Neuroinflammation is as a major pathological process in the secondary response after TBI (Cederberg and Siesjo, 2010). This inflammatory response is designated the term ‘sterile inflammation’ or inflammation in the absence of a pathogenic stimulus and involves multiple cell types within the CNS (Rock et al., 2010). Furthermore, brain injury compromises the integrity of the blood–brain barrier (BBB), a physical barrier separating the brain parenchyma from the body's circulation (Rodriguez‐Baeza et al., 2003; Shlosberg et al., 2010). Injury‐induced BBB damage allows infiltration of blood‐borne cells, adding a layer of complexity to the neuroinflammatory response (Beschorner et al., 2002; Jin et al., 2012). Moreover, increased permeability of the BBB after injury allows passage of both small and large molecules into the brain (Habgood et al., 2007).
In addition to the peripheral response, dying or damaged cells within the lesional and peri‐lesional areas in the brain release cellular debris into the microenvironment, priming local microglia and astrocytes. These resident cells express a variety of pattern recognition receptors (PRRs) at their cell surface and intracellularly, allowing them to mount an immune response (Gorina et al., 2011; Holm et al., 2012; de Rivero Vaccari et al., 2012; Fellner et al., 2013). Toll‐like receptors (TLRs) are a class of membrane‐bound PRRs, which are activated by a variety of endogenous pathogen‐associ ated molecular patterns or danger‐associated molecular patterns (DAMPs), including dsDNA and RNA, CpG motifs and chaperone proteins. These molecules are released by pathogens, or in the case of brain injury, dying cells (Anderson, 2000). Signalling through TLRs can also induce the production of inflammatory cytokines and chemokines, allowing them to signal to cells within damaged tissue, and potentially exacerbate the neuroinflammatory cascade (Lafon et al., 2006; Goodall et al., 2014; Li et al., 2014). Importantly, neurons also express TLRs, allowing them to respond to, and mount, a TLR‐driven inflammatory response (Lafon et al., 2006).
The brain's innate response to injury is crucial; resident astrocytes and microglia are often the primary cell types to initiate an inflammatory cascade upon sensing danger, and proteins associated with the activation of these cells are often used as biomarkers in TBI (Hernandez‐Ontiveros et al., 2013; Diaz‐Arrastia et al., 2014). Their responses include, but are not limited to, secretion of pro‐ and anti‐inflammatory cytokines, chemokines and growth factors, barrier formation around lesional areas, phagocytosis of dying cells and cellular debris, and modulation of cellular responses.
Reactive astrocytes, for example, can influence the responses of other cell types after TBI, both within the brain and in the periphery. Ablation of reactive astrocytes has resulted in an increase in leukocyte infiltration in a stab wound injury model, leading to neurodegeneration (Bush et al., 1999). Contrasting evidence suggest that the glial cell‐mediated invasion of peripheral cells can also be considered a mechanism of protection in a hippocampal entorhinodentate lesion model (Babcock et al., 2003).
Pleiotropic responses of glial cells have been shown to facilitate both inflammation resolution and exacerbation (Johnson et al., 2013; Roth et al., 2014). Because of this, a better understanding of the nature of the inflammatory response generated by astrocytes and microglia will aid in developing therapies to combat cell death and degeneration, and protect viable brain tissue after TBI. The inflammatory responses of both astrocytes and microglia will be discussed in this review, with an examination of the dual nature of these responses, and how these cells modulate the surrounding environment after brain injury.
Astrocytes
Astrocytes form part of the macroglia, cell types comprising oligodendrocytes, radial glia and ependymal cells (Kimelberg and Nedergaard, 2010). They are critical in maintaining physiological homeostasis within the CNS, with important roles in supporting neuronal function, glial transmission and signalling via Ca2+ release and uptake (Chen and Swanson, 2003). Studies have reported that human cortical astrocytes displayed a greater degree of heterogeneity and complexity than their rodent counterparts (Oberheim et al., 2009; Sosunov et al., 2014). Furthermore, astrocytes isolated from human temporal neocortex were larger than rodent astrocytes and capable of transmitting Ca2+ waves much faster than rodent astrocytes (Oberheim et al., 2009). These studies indicate that human astrocytes appear to be much more complex than those in the rodent brain. However, the study of astrocytes after injuries such as TBI is most commonly performed in rodents due to the ease of manipulating pathways and mediators in these models.
Astrocytes also play a role in maintaining BBB integrity, by forming astrocytic end feet around endothelial cells (Risau and Wolburg, 1990; Abbott et al., 2006). Astrocytic interactions with endothelial cells are a critical component of the induction and maintenance of the BBB (see Abbott et al., 2006) and involve processes such as inter‐ and intra‐cellular communication. Disruption of the BBB is associated with high levels of secreted factors from damaged tissue, such as S100B protein (used as a marker of BBB leakage) and MMP (Vajtr et al., 2009). Additionally, other mediators can influence the integrity of the BBB. For example, bradykinin‐induced astrocytic secretion of IL‐6 leads to opening of the BBB (Schwaninger et al., 1999). It is suggested that BBB breakdown follows a biphasic pattern after injury, with an initial increase in permeability hours after TBI, and declining thereafter, and a secondary delayed phase 3–7 days following injury, as found in controlled cortical impact (CCI) and closed head injury models (Shapira et al., 1993; Baskaya et al., 1997). Damage to the BBB after injury can also cause infiltration of peripheral immune cells (Jin et al., 2012). These cells are thought to play roles in repair but can also exacerbate neuroinflammation in the secondary phase of injury. The role of peripheral cells in injury is extensive and is outside the scope of this review. Therefore, this review will focus specifically on astrocytes and microglia in TBI.
Astrocytes and brain injury
An increase in astrocyte reactivity in response to injury is termed astrogliosis (Sofroniew and Vinters, 2010). This response involves changes in morphology, increased expression of the intermediate filament proteins, glial fibrillary acidic protein (GFAP) and vimentin, heightened proliferation and secretion of inflammatory mediators and growth factors (Pekny et al., 1999; Gorina et al., 2011; Zamanian et al., 2012; Paintlia et al., 2013). Many of these factors act in an autocrine and paracrine fashion to facilitate astrocytic reactivity of the cells surrounding them. Interestingly, it has been demonstrated that FGF serves as an inhibitory molecule in rendering astrocytes reactive, both in resting states and after stab wound injury (Kang et al., 2014). This body of work indicates the delicate balance between reactivity and suppression of function in astrocytes after injury.
Reactive astrocytes can acquire a hypertrophic morphology after injury, involving extension of processes and swelling of cell bodies. A recent study conducted in a mouse CCI model reported hypertrophic astrocytes in the lesional and peri‐lesional area 3 days after TBI (Villapol et al., 2014). Further changes in morphology were evident 7 days after injury, with glial scar formation. Reactive gliosis was maintained up to 60 days after injury in this model, demonstrating an ongoing response of astrocytes to brain injury.
Excitotoxicity is another common mechanism of secondary brain injury after TBI (Palmer et al., 1993). It is appreciated that while neurons are highly vulnerable to excitotoxicity, astrocytes have key roles in the re‐uptake of glutamate from synapses, preventing excessive extracellular glutamate accumulation (Chen and Swanson, 2003). Glutamate transporters, such as EAAT1 and EAAT2, are essential for glutamate re‐uptake. It has been demonstrated that blocking these transporters on astrocytes by administering antisense oligonucleotides in rats resulted in increased extracellular glutamate concentration, leading to excitotoxicity and neurodegeneration (Rothstein et al., 1996). Human TBI studies have observed decreases in glial expression of EAAT1 and EAAT2 after TBI in lesional and peri‐lesional areas, suggesting down‐regulation of these transporters after TBI (van Landeghem et al., 2006; Beschorner et al., 2007). This suggests that astrocytic down‐regulation in humans after TBI promotes accumulation of glutamate in extracellular areas and may contribute to excessive excitotoxicity leading to neurodegeneration.
Glial scar formation is commonly seen post‐injury and consists largely of astrocytes, along with microglia, endothelial cells and fibroblasts and extracellular matrix (Silver and Miller, 2004). Several mediators have been implicated in inducing glial scar formation, including TGF‐β1 and TGF‐2, IFN‐γ, FGF and fibrinogen (DiProspero et al., 1997; Moon and Fawcett, 2001; Schachtrup et al., 2010). Knock‐out mice studies have also demonstrated a role for GFAP and vimentin in proper glial scar formation (Pekny et al., 1999; Wilhelmsson et al., 2004). Scarring is thought to act as a physical barrier to encapsulate damaged tissue in order to prevent toxic molecules and DAMPs from leaking out into healthy tissue and to prevent access to invading cell types after injury. It has however also been shown to have an inhibitory effect on axonal regrowth and regeneration (Ribotta et al., 2004; Silver and Miller, 2004).
Another prominent aspect of astrogliosis is the ability of astrocytes to migrate or proliferate towards lesional or damaged tissue. The proliferative capacity of astrocytes as part of the injury‐induced response has been extensively studied in a stab wound model of injury, where the lesion is induced in the somatosensory cortex (Bardehle et al., 2013). Using two‐photon laser scanning microscopy, GFP‐labelled astrocytes were found to up‐regulate GFAP expression and display elongated processes and enlarged cell bodies, indicative of hypertrophy at 7 days post‐injury. Interestingly, proliferation rather than migration of astrocytes was detected at 5–7 days post‐injury. In a mouse CCI model, GFAP‐positive astrocytes were found to proliferate at 1, 3 and 7 days post‐injury, with numbers of proliferating astrocytes peaking 3 days post‐injury (Susarla et al., 2014). These astrocytes were located in close proximity to the lesion and were hypertrophic with extended processes. These studies demonstrate a predominant proliferative response in astrocytes, which peaks in the acute phase after experimental TBI. Collectively, these studies point to a critical role of astrocytes in modulating barrier formation, secretion of inflammatory factors and glial scar formation after injury. Additionally, astrocytes can elicit both protective and deleterious actions, which can influence the extent of brain damage or repair after injury.
Damage versus repair: dual roles of astrocytes following brain injury
Astrogliosis has been defined in the context of both neuroprotection and neurodegeneration (Myer et al., 2006; Zamanian et al., 2012). Reactive astrocytes are capable of producing pro‐inflammatory cytokines, chemokines and MMP that degrade the extracellular matrix and cause further BBB disruption (Carpentier et al., 2005; Kim et al., 2005). However, astrocytes are also capable of producing factors to support repair and regeneration after CNS damage (Kim et al., 2010; Madathil et al., 2013).
Specific roles of reactive astrocytes have been studied in models of moderate and severe CCI TBI (Myer et al., 2006). Selective ablation of proliferating reactive astrocytes allows researchers to elucidate their roles (Bush et al., 1999; Myer et al., 2006). In mice, the removal of proliferating reactive astrocytes resulted in neuronal degeneration and inflammation, and thus, has confirmed their essential role in preserving neuronal tissue after moderate, but not severe TBI (Myer et al., 2006). Furthermore, reactive astrocytes were shown to have a critical role in preventing the infiltration of inflammatory cells in regions containing intact cortical neurons. However, in direct contrast, blocking astrocytic proliferation using agents that disrupt various stages of the cell cycle leads to reduced neuronal cell death after fluid percussion injury in rats (Di Giovanni et al., 2005). Reduced astrocytic proliferation was also accompanied by reduced glial scar formation, microglial activation and improved histological and cognitive outcome after TBI, suggesting that the presence of reactive astrocytes can create an environment permissive to degeneration.
Factors secreted from reactive astrocytes can influence their actions and thus could explain the discrepancy between studies observing favourable roles versus those finding damaging roles for reactive astrocytes. For instance, astrocyte‐specific overexpression of the growth factor IGF‐1 resulted in increased gliosis accompanied by reduced hippocampal neurodegeneration after CCI injury (Madathil et al., 2013). Interestingly, astrocyte‐specific overexpression of the pro‐inflammatory cytokine IL‐6 also resulted in increased wound healing concurrently with increased reactive gliosis (Penkowa et al., 2003; Quintana et al., 2008). In contrast, astrocyte‐specific deletion of a Ca2+‐dependent N cadherin demonstrated a crucial role for this protein in mediating astrogliosis and consequent neurodegeneration (Kanemaru et al., 2013). These studies highlight the diversity of reactive gliosis and how the loss or gain of a single factor in reactive astrocytes can influence inflammatory responses and neuronal outcome.
Additionally, responses of astrocytes can be dependent upon their ability to assume different morphologies and phenotypes. Astrocytic heterogeneity was observed in a study examining the role of astrocytes after LPS injury and mid‐cerebral artery occlusion (Zamanian et al., 2012). LPS‐injured astrocytes assumed deleterious phenotypes, while astrocytes in the brains of mice subjected to mid‐cerebral artery occlusion assume reparative phenotypes. It is possible that after TBI, heterogeneous groups of astrocytes also emerge. Accumulating evidence from morphological and genetic studies reveals that astrocytes can acquire different morphologies and up‐regulate activation markers in varying levels after brain injury (Hill et al., 1996; Bardehle et al., 2013; Martin‐Lopez et al., 2013). Astrocytic responses are influenced by their location with respect to the injury, the signals they receive from their environment and also factors during their development (Martin‐Lopez et al., 2013).
Astrocytes have also been found to affect microglial responses. In a stab wound TBI model, impaired astrocyte recruitment after depletion of the RhoGTPase Cdc42 resulted in increased microglial activation (Robel et al., 2011). Additionally, increased astrocyte activation in an in vitro oxygen‐glucose deprivation model influenced the production of anti‐inflammatory mediators and suppressed microglial activation (Kim et al., 2010). These responses imply that microglia can heighten their activity as a compensatory mechanism for depleted astrocytic numbers, and conversely, excessive activation of astrocytes can dampen microglial responses. It is evident from these studies that astrocytes can affect the local environment after TBI, either in concert with, or by affecting neighbouring cells. Indeed, like astrocytes, microglial cells play instrumental roles in shaping the microenvironment after TBI.
Microglia
Microglia are specialized immune cells of the brain with phagocytic and antigen‐presenting capabilities (Hickey and Kimura, 1988). Originating from mesodermal cells of the yolk sac, microglia are derived from erythromyeloid precursor cells, and recently, their development has been shown to be dependent upon the transcription factors Pu.1 and interferon regulatory factor‐8 (Ginhoux et al., 2010; Kierdorf et al., 2013). There is a large degree of heterogeneity in microglial structure and shape, depending upon their activation state. Broadly, microglia are described as ‘ramified’ when in a resting or quiescent state (Glenn et al., 1992). Upon activation, microglia can transform to a hypertrophic or bushy morphology (Tambuyzer et al., 2009). Additionally, microglia are described as acquiring an amoeboid morphology during early stages of development or when actively phagocytosing cellular debris. In the healthy brain, microglia are often described as ‘resting’, although they are constantly surveying their environment in preparation for insult or injury (Nimmerjahn et al., 2005). Additionally, microglia are also capable of pruning synapses on neighbouring neurons during development by direct engulfment, thus serving a homeostatic function (Paolicelli et al., 2011).
Microglia express a variety of molecules at their cell surface, thus allowing their identification, and in certain cases, distinction from peripheral macrophages. Microglia are Mac 1/CD11b+ CD45low‐expressing cells, whereas macrophages are Mac‐1/CD11b+ CD45high‐expressing cells (D'Mello et al., 2009). The recent development of CCR2‐RFP knock‐in mice crossed with CX3CR1‐GFP mice has allowed the differentiation of monocytes from microglia (Saederup et al., 2010). Additionally, microglia express high levels of the chemokine receptor, CX3CR1, and the intracellular Ca2+‐binding protein, ionized calcium binding adapter molecule 1 (Iba‐1) (Fukuda et al., 1996; Nishiyori et al., 1998). Microglia in activated states up‐regulate the expression of Iba‐1, which can occur in cases of infection or injury to the CNS. Additionally, CD68 (ED1) is also used as a marker to confirm microglial activation (Graeber et al., 1990), and CD68 immunoreactivity is observed after both mouse and human TBI (Frugier et al., 2010; Loane et al., 2014). Following CNS trauma, microglia undergo morphological changes and can secrete a variety of factors, which either exacerbate or limit tissue damage.
Microglia in TBI
Microglia are instrumental in mounting an immune response after TBI. A study investigating pathological changes in injured post‐mortem human brains showed a prolonged and persistent activation of microglia, which was present even years after injury (Johnson et al., 2013). With chronic trauma, patients displayed increased levels of the major histocompatibility complex (MHC) Class II molecule CR3/43 and CD68 on activated microglia compared with acutely injured and non‐injured controls. In addition, increased white matter degeneration and disruption of myelinated fibres were associated with the chronic cohort of patients, clearly demonstrating an association between chronic microglial activation after injury and neurodegeneration.
Similarly, in a mouse CCI model, neuropathological and inflammatory changes were monitored up to 1 year after experimental TBI (Loane et al., 2014). Microglia were chronically activated, with increased levels of Iba‐1, CD68 and the MHC Class II molecule CR3/43 compared with controls. Morphology was assessed as being hypertrophic or bushy. This was accompanied with lesion expansion, loss of hippocampal neurons, white matter damage and loss of myelin. This study is corroborated by earlier work that reported the presence of mononuclear phagocytes in injured rat brains 3 months after weight‐drop injury, along with an up‐regulation in MHC Class II and the release of IL‐1β and TNF‐α (Holmin and Mathiesen, 1999). These smaller scale studies in rodents confirm the results found in the longitudinal study in humans, demonstrating the presence of chronically activated microglia and tissue degeneration after injury.
Evidence from alternative TBI models suggests that microglia have various roles after injury, adding to the complexity of microglial responses in the context of brain injury. In a novel closed head injury model, it was revealed that skull thinning and compression in mice resulted in cell death of the meningeal layers (Roth et al., 2014). This was associated with an extension of microglial processes surrounding the lesion and microglia surrounding dying astrocytes in the glial limitans. The microglial response was found to be dependent upon purinergic signalling, and blocking this signalling resulted in increased permeability of the glial limitans and subsequent cell death around the meninges. Similarly, in a laser‐induced focal injury model, the microglial response was determined to be dependent upon ATP release and activation of P2Y receptors (Davalos et al., 2005). In response to injury, it was established that ATP mediated extension of microglial processes to surround the injury site, allowing a barrier to be formed between damaged and intact tissues. Together, these significant studies emphasize that the microglial response in injury can be dependent upon context and injury severity (highlighted through the use of different injury models), and therefore, a better understanding of the pleiotropic nature of these responses is needed to advance our understanding of microglial contribution in injury.
The dual nature of the microglial response; M1 and M2 phenotypes
The microglial response in TBI is dualistic; highly dependent upon timing and the nature of the injury itself. Macrophages/microglia can acquire heterogeneous phenotypes following CNS insult in response to varying environmental cues. These cells are ascribed the nomenclature of ‘M1’ or ‘M2’, with varying classes of M2 cells (Mantovani and Locati, 2009; Sica and Mantovani, 2012). Additionally, the terms ‘classical activation’ for M1, ‘alternative activation (M2a)’ and ‘acquired deactivation (M2c)’ are commonly used to group microglia (Cao et al., 2012; Kumar et al., 2013). Cells polarize from a ‘resting’ or ‘M0’ phenotype into an M1 phenotype after LPS or IFN‐γ exposure and are considered neurotoxic following CNS injury (Kigerl et al., 2009). It has been suggested that M1 microglia exhibit reduced phagocytosing capability compared with M0 microglia in response to oxygen‐glucose deprivation in vitro, and M2 exhibit increased activity (Hu et al., 2012; Wang et al., 2013). In contrast, an M2 phenotype is acquired following exposure to IL‐4 or IL‐13, and these cells are considered neuroprotective (Kigerl et al., 2009). M1 microglia are known to produce mediators such as pro‐inflammatory cytokines and inducible NOS to elicit their deleterious effects, and M2 microglia produce scavenger receptors, growth factors such as TGF‐β and the anti‐inflammatory cytokine IL‐10 (Figure 1). As such, these mediators or cell surface receptors for corresponding M1 and M2 cells are used as markers to broadly differentiate the two cell types in injury states. Table 1 lists the different markers used to study M1/M2 polarization following TBI.
Figure 1.
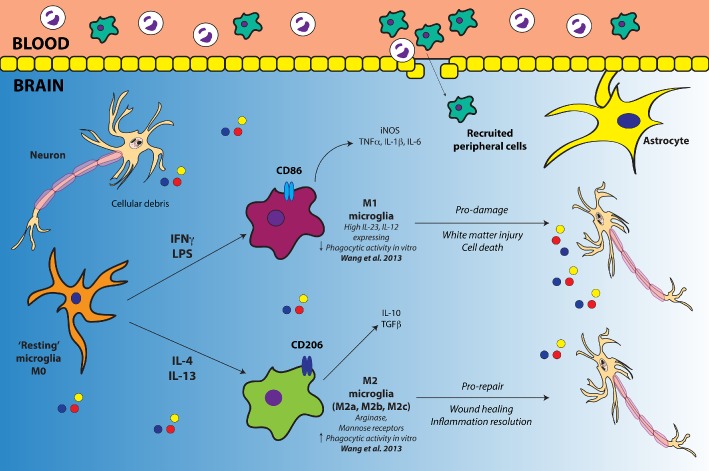
Diagrammatic representation of M1/M2 polarization in microglia. For simplicity, M2 microglia have been represented as just the one phenotype, as opposed to illustrating the various classes. Cellular debris and other mediators released by dying neurons after injury prime microglia. An environment rich with the classical pro‐inflammatory stimuli, such as IFN‐γ and LPS, promotes the polarization of resting microglia into an M1 phenotype. M1 microglia release pro‐inflammatory cytokines, chemokines and iNOS. An M1 environment is neurotoxic, facilitating white matter injury and cell death. In contrast, a neuroinflammatory environment rich in anti‐inflammatory IL‐4 or IL‐13 drives the development of an M2 phenotype. M2 microglia release IL‐10 and TGF‐β, while promoting repair and inflammation resolution. In addition to microglia, peripherally infiltrating macrophages can also undergo polarization into M1 and M2 phenotypes, and recently, this has shown to be true of neutrophils in a murine ischaemic stroke model (Cuartero et al., 2013).
Table 1.
Markers to classify M1 and M2 microglia commonly used in studies of TBI
| Study | Microglial polarization | Other microglial activation markers used | |
|---|---|---|---|
| M1 | M2 | ||
| Jin et al. (2012) | CD86 | CD206 | Iba‐1, CD11b+ CD45low‐expressing cells characterized as microglia |
| Wang et al. (2013) | CD32, CD16, iNOS, CD11b, CD86 | CD206, IL‐10, Ym1/2, TGF‐β, Arg‐1, CCL22 | Iba‐1 |
| Kumar et al. (2013) | IL‐1β, TNF‐α, CD86, iNOS, CCL2, CCL3 (classical activation) | Arg‐1, Ym1, Mrc, Fizz‐1 (alternative activation; M2a) and IL‐4rα, SOCS3, TGF‐β (acquired deactivation; M2c) | Iba‐1, CD11b, ED1, MHC II |
| Cao et al. (2012) | TNF‐α, CD45 (classical activation) | Arg‐1 (alternative activation), TGF‐βΙ, TGF‐βRII (acquired deactivation) | Iba‐1, MHC I, MHC II, TSPO |
| Bachstetter et al. (2013) | IL‐1β, IL‐6, TNF‐α, CCL2, CCL3 (classical activation) | Arg‐1, Ym1 (alternative activation) | Iba‐1, CD68, CD45, MHC II |
| Dohi et al. (2010) | iNOS, NO, TNF‐α (classical activation) | Arg‐1, Ym1 (alternative activation) | CD11B |
Some studies use the nomenclature M2a and M2c (‘alternative activation’ and ‘acquired deactivation’) to describe M2 microglia and ‘classical activation’ for M1 microglia.
Arg‐1, arginase‐1; iNOS, inducible NOS.
There is still some debate over the use of specific markers to distinguish M1 and M2 cells. Furthermore, the grouping of cells into ‘M1’ and ‘M2’ categories is not a unanimous view; there are reports suggesting that macrophage/microglial polarization fits onto a spectrum rather than into two distinct groups (reviewed in Mosser and Edwards, 2008). These conflicting reports may be a reflection of the studies, with changes observed in whole tissue specimens rather than in specific cell types (Kumar et al., 2013; Wang et al., 2013). Many studies therefore describe microglial polarization as being skewed towards either an ‘M1‐like’ or ‘M2‐like’ state. Additionally, it is possible that that M1/M2 polarization may be reversible, as previously polarized cells may be able to transform to different phenotypes depending upon the presence of additional environmental stimuli (Butovsky et al., 2005; Schwartz et al., 2006). Furthermore, cells may be undergoing phenotypic switches when isolated, influencing levels of receptors/marker proteins. A recent review has addressed complexities regarding macrophage nomenclature (Murray et al., 2014). It is suggested that researchers use more standardized methods of isolation and use a wider range of markers to designate categories for polarized macrophages. Recently, using a direct RNA sequencing technique, a unique set of genes, termed the ‘microglial sensome’, was identified in microglia isolated from aged mice (Hickman et al., 2013). Furthermore, microglial‐specific genes that were up‐regulated during classical and alternative microglial priming were identified in both young and aged mice, allowing a greater understanding of microglial polarization states. Evidently, similar such studies in microglia after TBI are warranted and will increase our knowledge of microglial‐specific polarization following injury. As such, the M1/M2 paradigm is no doubt an evolving one; and as new evidence comes to light, these categories and definitions will be subject to re‐evaluation.
Studies performed in CCI models indicate that the onset of activation varies significantly between M1 and M2 microglia (Jin et al., 2012; Wang et al., 2013). By qPCR and immunohistochemistry, Wang et al. demonstrated an immediately increased and transient M2‐like environment after TBI, in contrast to the delayed but slightly more prolonged M1‐like environment (Wang et al., 2013). However, in the absence of cell‐specific markers, it was suggested that this environment could be attributed to both microglial and macrophage populations. However, in distinguishing resident microglia from peripheral macrophages by assessing CD45/CD11B and Iba‐1 expression, Jin et al. confirmed a similar pattern of M1/M2 activation with an initial and transient M2 peak, followed by a predominant M1 response at 21 days post‐injury (Jin et al., 2012). Specifically, microglial numbers followed a multiphasic pattern, with increased levels 7 days post‐injury, a decrease in microglial numbers 14 days post‐injury and, finally, a gradual increase after 21 days. The exact period of activation differed slightly between these studies, perhaps reflecting the different methods of analysis of M1/M2 populations.
Evidence from stroke models also suggests that there is a propensity for an M2‐favoured environment early after injury, followed by a delayed onset of M1 microglia/macrophages in vivo (Hu et al., 2012). These results are also observed with astrocyte populations after ischaemic stroke, suggesting the presence of beneficial and detrimental subsets of astrocyte, with a protective astrocytic phenotype observed in a murine stroke model (Zamanian et al., 2012). Collectively, this body of evidence suggests the need to further characterize the polarization properties of microglia specifically rather than in whole brain specimens. The studies reviewed here provide evidence for M1/M2 environments in the brain following injury, and when combined with information about the specific phenotypic changes of microglia exclusively, a greater understanding of M1/M2 dynamics following brain injury will be achieved. Additionally, studies that selectively investigate microglial properties following injury will aid our understanding of the exact nature and timing of both beneficial and detrimental microglial responses following injury.
Discussion
It is clear that the diverse roles of astrocytes and microglia in TBI arise as a result of divergent stimuli they receive from the surrounding cells and the local microenvironment. Substrates produced in injury, such as inflammatory mediators, proteases, complement factors and DAMPs, trigger complex cascades, promoting a variety of cellular responses. Importantly, responses of astrocytes and microglia in experimental TBI studies can be largely dependent upon the nature and severity of injury; focal models, such as stab wound models, can induce different responses in these cells compared with diffuse models, such as weight‐drop injury. This is highlighted nicely in the suite of microglial studies, which demonstrated both reparative and deleterious roles for these cells in various models of experimental TBI (Davalos et al., 2005; Loane et al., 2014; Roth et al., 2014). Studies investigating responses of reactive glial cells must therefore take this into account and ideally incorporate results from more than just one TBI model. Additionally, the outcomes of glial responses depend upon both environmental cues and other cell types, emphasizing the complex nature of reactive gliosis in injury.
Studies of reactive astro‐ and microgliosis have identified somewhat opposing roles in injury: those promoting neurotoxicity and degeneration and those promoting repair and regeneration. Microglia are not exclusive in their ability to polarize into different phenotypes; recently, it has been shown that astrocytes can assume both pro‐repair and pro‐damage phenotypes depending upon their stimulus, and accordingly, these astrocytes up‐regulate phenotype‐specific markers (Zamanian et al., 2012). The identification of glial polarization in the context of brain injuries is crucial in advancing our understanding of glial heterogeneity, thereby allowing us to manipulate these properties to limit damage and support regeneration following TBI.
It is important to note that these cellular responses may not occur in isolation. Although not extensively discussed within this review, cells within the brain can act in concert with recruited haematopoietic cells after injury to exert their biological function. Signals released by microglia, for example, have been shown to recruit monocytes into the brain in response to peripheral organ inflammation (D'Mello et al., 2009). Neuroimmune crosstalk is emerging as a critical concept in studies of brain injury and stroke, and the use of chimeric mice and the selective depletion of brain‐derived or peripheral cells is allowing investigators to tease apart the roles of these different cell types (Gliem et al., 2012; Downes et al., 2013; Low et al., 2014).
Therapies targeting CNS injuries must take into account the multifaceted nature of cellular responses if they are going to be effective in limiting neuronal damage after TBI. Inflammatory responses of astrocytes and microglia in TBI represent an interesting therapeutic opportunity. By harnessing the protective or reparative effects of these responses while simultaneously dampening their deleterious effects, we may be able to control the progression and exacerbation of inflammation and protect viable brain tissue. Therefore, a greater understanding of the mechanisms governing reactive gliosis and how gliosis affects other cell types will aid in the development of better therapeutics for TBI.
Conflict of interest
The authors declare no conflicts of interest.
Karve, I. P. , Taylor, J. M. , and Crack, P. J. (2016) The contribution of astrocytes and microglia to traumatic brain injury. British Journal of Pharmacology, 173: 692–702. doi: 10.1111/bph.13125.
References
- Abbott NJ, Ronnback L, Hansson E (2006). Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci 7: 41–53. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol, 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol, 170: 1706–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL Spedding M et al (2013d). The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol, 170, 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KV (2000). Toll signaling pathways in the innate immune response. Curr Opin Immunol 12: 13–19. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T (2003). Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 23: 7922–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ (2013). The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci 33: 6143–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H et al (2013). Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16: 580–586. [DOI] [PubMed] [Google Scholar]
- Baskaya MK, Rao AM, Dogan A, Donaldson D, Dempsey RJ (1997). The biphasic opening of the blood‐brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett 226: 33–36. [DOI] [PubMed] [Google Scholar]
- Beschorner R, Nguyen TD, Gozalan F, Pedal I, Mattern R, Schluesener HJ et al (2002). CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol 103: 541–549. [DOI] [PubMed] [Google Scholar]
- Beschorner R, Dietz K, Schauer N, Mittelbronn M, Schluesener HJ, Trautmann K et al (2007). Expression of EAAT1 reflects a possible neuroprotective function of reactive astrocytes and activated microglia following human traumatic brain injury. Histol Histopathol 22: 515–526. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN et al (1999). Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar‐forming, reactive astrocytes in adult transgenic mice. Neuron 23: 297–308. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben‐Yaakov K, Schwartz M (2005). Activation of microglia by aggregated beta‐amyloid or lipopolysaccharide impairs MHC‐II expression and renders them cytotoxic whereas IFN‐gamma and IL‐4 render them protective. Mol Cell Neurosci 29: 381–393. [DOI] [PubMed] [Google Scholar]
- Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J (2012). Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 225: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD (2005). Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49: 360–374. [DOI] [PubMed] [Google Scholar]
- Cederberg D, Siesjo P (2010). What has inflammation to do with traumatic brain injury? Childs Nerv Syst 26: 221–226. [DOI] [PubMed] [Google Scholar]
- Chen Y, Swanson RA (2003). Astrocytes and brain injury. J Cereb Blood Flow Metab 23: 137–149. [DOI] [PubMed] [Google Scholar]
- Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA et al (2013). N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist rosiglitazone. Stroke 44: 3498–3508. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005). ATP mediates rapid microglial response to local brain injury in vivo . Nat Neurosci 8: 752–758. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B et al (2005). Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A 102: 8333–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Arrastia R, Wang KK, Papa L, Sorani MD, Yue JK, Puccio AM et al (2014). Acute biomarkers of traumatic brain injury: relationship between plasma levels of ubiquitin C‐terminal hydrolase‐L1 and glial fibrillary acidic protein. J Neurotrauma 31: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiProspero NA, Meiners S, Geller HM (1997). Inflammatory cytokines interact to modulate extracellular matrix and astrocytic support of neurite outgrowth. Exp Neurol 148: 628–639. [DOI] [PubMed] [Google Scholar]
- D'Mello C, Le T, Swain MG (2009). Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K et al (2010). Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes CE, Wong CH, Henley KJ, Guio‐Aguilar PL, Zhang M, Ates R et al (2013). MyD88 is a critical regulator of hematopoietic cell‐mediated neuroprotection seen after stroke. PLoS ONE 8: e57948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W et al (2013). Toll‐like receptor 4 is required for α‐synuclein dependent activation of microglia and astroglia. Glia 61: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier T, Morganti‐Kossmann C, O'Reilly D, McLean C (2010). In situ detection of inflammatory mediators in post‐mortem human brain tissue following traumatic injury. J Neurotrauma 27: 497–507. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Tomatsu S, Masuno M, Ogawa T, Yamagishi A, Rezvi GM et al (1996). Mucopolysaccharidosis IVA: submicroscopic deletion of 16q24.3 and a novel R386C mutation of N‐acetylgalactosamine‐6‐sulfate sulfatase gene in a classical Morquio disease. Hum Mutat 7: 123–134. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn JA, Ward SA, Stone CR, Booth PL, Thomas WE (1992). Characterisation of ramified microglial cells: detailed morphology, morphological plasticity and proliferative capability. J Anat 180 (Pt 1): 109–118. [PMC free article] [PubMed] [Google Scholar]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP et al (2012). Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 71: 743–752. [DOI] [PubMed] [Google Scholar]
- Goodall KJ, Poon IK, Phipps S, Hulett MD (2014). Soluble heparan sulfate fragments generated by heparanase trigger the release of pro‐inflammatory cytokines through TLR‐4. PLoS ONE 9: e109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina R, Font‐Nieves M, Marquez‐Kisinousky L, Santalucia T, Planas AM (2011). Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88‐dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59: 242–255. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kiefer R, Schoen SW, Kreutzberg GW (1990). New expression of myelomonocytic antigens by microglia and perivascular cells following lethal motor neuron injury. J Neuroimmunol 27: 121–132. [DOI] [PubMed] [Google Scholar]
- Greve MW, Zink BJ (2009). Pathophysiology of traumatic brain injury. Mt Sinai J Med 76: 97–104. [DOI] [PubMed] [Google Scholar]
- Habgood MD, Bye N, Dziegielewska KM, Ek CJ, Lane MA, Potter A et al (2007). Changes in blood‐brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci 25: 231–238. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV (2013). Microglia activation as a biomarker for traumatic brain injury. Front Neurol 4: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF, Kimura H (1988). Perivascular microglial cells of the CNS are bone marrow‐derived and present antigen in vivo . Science 239: 290–292. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK et al (2013). The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Barbarese E, McIntosh TK (1996). Regional heterogeneity in the response of astrocytes following traumatic brain injury in the adult rat. J Neuropathol Exp Neurol 55: 1221–1229. [DOI] [PubMed] [Google Scholar]
- Holm TH, Draeby D, Owens T (2012). Microglia are required for astroglial Toll‐like receptor 4 response and for optimal TLR2 and TLR3 response. Glia 60: 630–638. [DOI] [PubMed] [Google Scholar]
- Holmin S, Mathiesen T (1999). Long‐term intracerebral inflammatory response after experimental focal brain injury in rat. Neuroreport 10: 1889–1891. [DOI] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S et al (2012). Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43: 3063–3070. [DOI] [PubMed] [Google Scholar]
- Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T (2012). Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS ONE 7: e41892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136 (Pt 1): 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemaru K, Kubota J, Sekiya H, Hirose K, Okubo Y, Iino M (2013). Calcium‐dependent N‐cadherin up‐regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc Natl Acad Sci U S A 110: 11612–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Balordi F, Su N, Chen L, Fishell G, Hebert JM (2014). Astrocyte activation is suppressed in both normal and injured brain by FGF signaling. Proc Natl Acad Sci U S A 111: E2987–E2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG et al (2013). Microglia emerge from erythromyeloid precursors via Pu.1‐ and Irf8‐dependent pathways. Nat Neurosci 16: 273–280. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29: 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Fillmore HL, Reeves TM, Phillips LL (2005). Elevation of hippocampal MMP‐3 expression and activity during trauma‐induced synaptogenesis. Exp Neurol 192: 60–72. [DOI] [PubMed] [Google Scholar]
- Kim JH, Min KJ, Seol W, Jou I, Joe EH (2010). Astrocytes in injury states rapidly produce anti‐inflammatory factors and attenuate microglial inflammatory responses. J Neurochem 115: 1161–1171. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Nedergaard M (2010). Functions of astrocytes and their potential as therapeutic targets. Neurother 7: 338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ (2013). Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging 34: 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon M, Megret F, Lafage M, Prehaud C (2006). The innate immune facet of brain: human neurons express TLR‐3 and sense viral dsRNA. J Mol Neurosci 29: 185–194. [DOI] [PubMed] [Google Scholar]
- van Landeghem FK, Weiss T, Oehmichen M, von Deimling A (2006). Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma 23: 1518–1528. [DOI] [PubMed] [Google Scholar]
- Li XQ, Wang J, Fang B, Tan WF, Ma H (2014). Intrathecal antagonism of microglial TLR4 reduces inflammatory damage to blood‐spinal cord barrier following ischemia/reperfusion injury in rats. Mol Brain 7: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014). Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73: 14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PC, Manzanero S, Mohannak N, Narayana VK, Nguyen TH, Kvaskoff D et al (2014). PI3Kδ inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nat Commun 5: 3450. [DOI] [PubMed] [Google Scholar]
- Madathil SK, Carlson SW, Brelsfoard JM, Ye P, D'Ercole AJ, Saatman KE (2013). Astrocyte‐specific overexpression of insulin‐like growth factor‐1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS ONE 8: e67204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Locati M (2009). Orchestration of macrophage polarization. Blood 114: 3135–3136. [DOI] [PubMed] [Google Scholar]
- Martin‐Lopez E, Garcia‐Marques J, Nunez‐Llaves R, Lopez‐Mascaraque L (2013). Clonal astrocytic response to cortical injury. PLoS ONE 8: e74039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon DK (2009). Unique challenges in clinical trials in traumatic brain injury. Crit Care Med 37 (1 Suppl.): S129–S135. [DOI] [PubMed] [Google Scholar]
- Moon LD, Fawcett JW (2001). Reduction in CNS scar formation without concomitant increase in axon regeneration following treatment of adult rat brain with a combination of antibodies to TGFbeta1 and beta2. Eur J Neurosci 14: 1667–1677. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP (2008). Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S et al (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV (2006). Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129 (Pt 10): 2761–2772. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N et al (1998). Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett 429: 167–172. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F et al (2009). Uniquely hominid features of adult human astrocytes. J Neurosci 29: 3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Singh AK, Singh I (2013). S‐nitrosoglutathione induces ciliary neurotrophic factor expression in astrocytes that has implication to protect CNS under pathological conditions. J Biol Chem 288: 3831–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, DeKosky ST (1993). Traumatic brain injury‐induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem 61: 2015–2024. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T et al (1999). Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol 145: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernandez J et al (2003). Astrocyte‐targeted expression of IL‐6 protects the CNS against a focal brain injury. Exp Neurol 181: 130–148. [DOI] [PubMed] [Google Scholar]
- Quintana A, Molinero A, Borup R, Nielsen FC, Campbell IL, Penkowa M et al (2008). Effect of astrocyte‐targeted production of IL‐6 on traumatic brain injury and its impact on the cortical transcriptome. Dev Neurobiol 68: 195–208. [DOI] [PubMed] [Google Scholar]
- Ribotta MG, Menet V, Privat A (2004). Glial scar and axonal regeneration in the CNS: lessons from GFAP and vimentin transgenic mice. Acta Neurochir Suppl 89: 87–92. [DOI] [PubMed] [Google Scholar]
- Risau W, Wolburg H (1990). Development of the blood‐brain barrier. Trends Neurosci 13: 174–178. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Minkiewicz J, Wang X, De Rivero Vaccari JC, German R, Marcillo AE et al (2012). Astrogliosis involves activation of retinoic acid‐inducible gene‐like signaling in the innate immune response after spinal cord injury. Glia 60: 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Bardehle S, Lepier A, Brakebusch C, Gotz M (2011). Genetic deletion of cdc42 reveals a crucial role for astrocyte recruitment to the injury site in vitro and in vivo . J Neurosci 31: 12471–12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Latz E, Ontiveros F, Kono H (2010). The sterile inflammatory response. Annu Rev Immunol 28: 321–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Baeza A, Reina‐de la Torre F, Poca A, Marti M, Garnacho A (2003). Morphological features in human cortical brain microvessels after head injury: a three‐dimensional and immunocytochemical study. Anat Rec A Discov Mol Cell Evol Biol 273: 583–593. [DOI] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014). Transcranial amelioration of inflammation and cell death after brain injury. Nature 505: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes‐Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW et al (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686. [DOI] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL et al (2010). Selective chemokine receptor usage by central nervous system myeloid cells in CCR2‐red fluorescent protein knock‐in mice. PLoS ONE 5: e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL et al (2010). Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF‐beta after vascular damage. J Neurosci 30: 5843–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaninger M, Sallmann S, Petersen N, Schneider A, Prinz S, Libermann TA et al (1999). Bradykinin induces interleukin‐6 expression in astrocytes through activation of nuclear factor‐kappaB. J Neurochem 73: 1461–1466. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK (2006). Microglial phenotype: is the commitment reversible? Trends Neurosci 29: 68–74. [DOI] [PubMed] [Google Scholar]
- Shapira Y, Setton D, Artru AA, Shohami E (1993). Blood‐brain barrier permeability, cerebral edema, and neurologic function after closed head injury in rats. Anesth Analg 77: 141–148. [DOI] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D, Friedman A (2010). Blood‐brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A (2012). Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH (2004). Regeneration beyond the glial scar. Nat Rev Neurosci 5: 146–156. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV (2010). Astrocytes: biology and pathology. Acta Neuropathol 119: 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov AA, Wu X, Tsankova NM, Guilfoyle E, McKhann GM 2nd, Goldman JE (2014). Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J Neurosci 34: 2285–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susarla BT, Villapol S, Yi JH, Geller HM, Symes AJ (2014). Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice. ASN Neuro 6: 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambuyzer BR, Ponsaerts P, Nouwen EJ (2009). Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 85: 352–370. [DOI] [PubMed] [Google Scholar]
- Vajtr D, Benada O, Kukacka J, Prusa R, Houstava L, Toupalik P et al (2009). Correlation of ultrastructural changes of endothelial cells and astrocytes occurring during blood brain barrier damage after traumatic brain injury with biochemical markers of BBB leakage and inflammatory response. Physiol Res 58: 263–268. [DOI] [PubMed] [Google Scholar]
- Villapol S, Byrnes KR, Symes AJ (2014). Temporal dynamics of cerebral blood flow, cortical damage. Apoptosis 5: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X et al (2013). Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab 33: 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Engelhard K (2007). Pathophysiology of traumatic brain injury. Br J Anaesth 99: 4–9. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C et al (2004). Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post‐traumatic regeneration. J Neurosci 24: 5016–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG et al (2012). Genomic analysis of reactive astrogliosis. J Neurosci 32: 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]