

Abstract
Cigarette smoking has reached epidemic proportions within many regions of the world and remains the highest risk factor for chronic obstructive pulmonary disease (COPD) and lung cancer. Squamous cell lung cancer is commonly detected in heavy smokers, where the risk of developing lung cancer is not solely defined by tobacco consumption. Although therapies that target common driver mutations in adenocarcinomas are showing some promise, they are proving ineffective in smoking‐related squamous cell lung cancer. Since COPD is characterized by an excessive inflammatory and oxidative stress response, this review details how aberrant innate, adaptive and systemic inflammatory processes can contribute to lung cancer susceptibility in COPD. Activated leukocytes release increasing levels of proteases and free radicals as COPD progresses and tertiary lymphoid aggregates accumulate with increasing severity. Reactive oxygen species promote formation of reactive carbonyls that are not only tumourigenic through initiating DNA damage, but can directly alter the function of regulatory proteins involved in host immunity and tumour suppressor functions. Systemic inflammation is also markedly increased during infective exacerbations in COPD and the interplay between tumour‐promoting serum amyloid A (SAA) and IL‐17A is discussed. SAA is also an endogenous allosteric modifier of FPR2 expressed on immune and epithelial cells, and the therapeutic potential of targeting this receptor is proposed as a novel strategy for COPD–lung cancer overlap.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- COPD
chronic obstructive pulmonary disease
- FGFR1
fibroblast growth factor‐1
- FPR2
formyl peptide receptor 2
- LOH
loss of heterozygosity
- LXA4
lipoxinA4
- NSCLC
non‐small cell lung cancer
- NFE2L2
nuclear factor, erythroid 2‐like 2
- PIK3CA
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha
- PTEN
phosphatase and tensin homologue
- SAA
serum amyloid A
- SCC
squamous cell cancer
- RvD1
resolvinD1
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a, 2013b, 2013c).
Squamous cell carcinomas are non‐responsive to current therapies tailored for adenocarcinomas
Globally, lung cancer is the most commonly diagnosed form of cancer and is currently the leading cause of cancer‐related deaths (Jemal et al., 2011). Conventional therapy for lung cancer includes surgical resection, cytotoxic agents, radiation and in some cases targeted molecular therapies. Despite the advances in new molecular therapies, the prognosis remains poor for lung cancer, with an overall 5 year survival rate of around 15% (Jemal et al., 2011). Lung cancers are broadly subdivided into histological types: small‐cell lung cancers and non‐small‐cell lung cancers (NSCLCs), where NSCLCs represent 80–85% of all lung cancers. NSCLCs are further classified into different subtypes where squamous‐cell carcinoma (SCC) and adenocarcinomas account for the majority of NSCLC cases. SCC accounts for approximately 20–30% of NSCLC cases and adenocarcinomas account for about 40–50% of NSCLC cases. As many lung cancers present in advanced stages, most patients are unresectable and the 5 year survival rates for advanced histological subtype stage IV NSCLC falls to below 2% for SCC (Cetin et al., 2011). Adenocarcinomas can occur in non‐smokers, particularly in Asian populations, whereas SCC is strongly associated with a history of cigarette smoking. Histological classification of tumours is increasingly being supplanted by molecular typing into subsets based upon dominant mutational drivers and the use of selective tyrosine kinase such as gefitinib, which targets EGFR mutations and improves progression‐free survival (Fukuoka et al., 2011). The outlook for smoking‐related SCC is much less optimistic with respect to targeting of dominant mutations that drive tumour progression. This, in part, reflects a complex and differential driver mutation profile for SCC, as dominant mutations in adenocarcinomas, such as EGFR, ALK and ROS1 fusions and KRAS mutations, are relatively infrequent in SCC (<5% frequency rate) as reviewed in Pao and Girard (2011) and summarized in Table 1.
Table 1.
Prevalence of abnormalities detected in NSCLC subsets and in COPD
| Adenocarcinoma | SCC | COPD | |
|---|---|---|---|
| EGF receptor mutations | 5–15% frequency | Infrequent | EGF receptor amplification occurs |
| ELM4/ALK translocations | 5–15% frequency | Infrequent | Not detected |
| KRAS mutations | >15% frequency | Infrequent | Not detected |
| FGFR family amplification/overexpression | Infrequent | 20% frequency in FGFR1 amplification | Overexpression of FGF1/2 and FGFR1 detected |
| PI3KCA amplification | Infrequent | >30% frequency | Increased PI3K pathway activation |
| Loss of PTEN status | Infrequent | Mutations and methylation detected (10–40% frequency) | PTEN pathway down‐regulated |
Cigarette smoking is also the predominant risk factor for chronic obstructive pulmonary disease (COPD). Globally, COPD is predicted to become the third leading cause of death in the world by 2020 (Lopez and Murray, 1998). Cigarette smoking accounts for more than 95% of cases in industrialized countries (Barnes et al., 2003), although environmental pollutants are recognized as important causes in developing countries (Dennis et al., 1996). COPD is characterized by progressive airflow obstruction and is associated with an abnormal and chronic inflammatory response of the lungs to noxious particles and gases (Pauwels et al., 2001). The pathology of COPD is heterogeneous encompassing (i) airways disease, chronic obstructive bronchiolitis with fibrosis and obstruction of small airways and/or mucous metaplasia and mucus gland hypertrophy leading to plugging of the larger airways, and (ii) emphysema, enlargement of airspaces and destruction of lung parenchyma. Of significance, approximately 30% of patients with mild to moderate COPD have been reported to die from lung cancer (Anthonisen et al., 1994), which has traditionally been linked to a common aetiological exposure, namely tobacco smoke. There are however multiple studies to show that both airways disease and emphysema are significant risk factors for lung cancer, even when adjustments for smoking history are made (Skillrud et al., 1986; Mannino et al., 2003; Ueda et al., 2006; Purdue et al., 2007). In particular, the presence of COPD was found to increase the risk for squamous cell histological subtypes by more than fourfold (Papi et al., 2004).
Cytogenetic studies have demonstrated that the lung epithelium of heavy smokers transforms into a squamous metaplasia phenotype that is correlated with the severity of airway obstruction (Cosio et al., 1978). Hence, the chronic injurious state of this lung microenvironment may facilitate tumour development progressing from metaplasia, dysplasia, carcinoma in situ and subsequent malignant transformation. Morphological changes in the bronchial epithelium are accompanied by an increase in loss of heterozygosity (LOH) and field cancerization involving the accumulation of mutations that eventually predispose the lung to cancer (Franklin et al., 1997; Minna et al., 2002; Wistuba et al., 2002). The widespread presence of TP53 mutations in smoker epithelium exhibiting squamous metaplasia occurs early during this transformation, which can expand from a single progenitor clone to populate broad areas of the injured bronchial mucosa (Franklin et al., 1997). Microsatellite instability (MSI) is frequent in the non‐malignant bronchial epithelium of COPD and is associated with EGFR amplification (Romeo et al., 2003). Increased EGFR expression has been observed in COPD epithelium (de Boer et al., 2006) and EGFR transactivation augments inflammatory responses initiated by viral and bacterial infection in the bronchial epithelial cells (Liu et al., 2008a, 2008b). A significant increase in severe exacerbations was observed in COPD patients exhibiting MSI (Makris et al., 2008). The acquisition of early somatic mutations required for tumourigenesis may also worsen inflammation in COPD (Anderson and Bozinovski, 2003).
Smoke‐mediated epithelial transformation can also drive a fibrotic response contributing to small airway wall thickening in COPD through TGFβ‐dependent mechanisms (Araya et al., 2007). Increased bronchial expression of fibroblast growth factors (FGF‐1/2) that target FGFR1 have been implicated in the remodelling of bronchial airways in COPD (Kranenburg et al., 2005). Focal FGFR1 amplification has been detected in 22% of SCC where this receptor pathway is associated with tumour growth and survival (Weiss et al., 2010). Signature genes activated by the phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha (PIK3CA) pathway are also overexpressed in the bronchial airway of smokers with dysplastic lesions, suggesting that PIK3CA is activated early during carcinogenesis (Gustafson et al., 2010). Dysregulated PIK3CA/PTEN/Akt/mTOR signalling coordinates tumour‐promoting survival, metabolism, migration and angiogenesis. Activation of Akt leads to inhibition of downstream signalling proteins, including glycogen synthase kinase 3, forkhead box O transcription factors and BAD, thereby suppressing apoptotic signals. In addition, Akt indirectly activates mammalian target of rapamycin (mTOR), a master regulator of cell growth and metabolism, through its regulation of anabolic processes, including lipid and protein biosynthesis. This pathway is activated by multiple tyrosine kinase receptors, including EGF, IGF1, VEGF and PDGF receptors, and mutations are frequently detected along this pathway. Amplification of PIK3CA has been detected in high frequency in SCC, in contrast to gain‐of‐function mutations that are much less frequent (Drilon et al., 2012).
Loss of PTEN protein expression has been characterized as an independent poor prognostic factor for patients with NSCLC associated with a more aggressive subset of lung tumours (Tang et al., 2006; Lim et al., 2007). Somatic mutation or deletion of PTEN has been reported in a variety of tumour types and genetic analysis of SCC demonstrates a mutational frequency around 10% (Drilon et al., 2012). An increased frequency of PTEN promoter methylation has been proposed as an alternative method for loss of PTEN expression in NSCLC (Soria et al., 2002). The microRNA miR‐29b has been shown to regulate PTEN gene expression through inducing hypomethylation in the PTEN promoter (Li et al., 2012). However, mutations of PTEN and methylation of its promoter in NSCLC are infrequent relative to PTEN protein expression, which is reported to be reduced or lost in 74% NSCLC tumours (Marsit et al., 2005). In this study, neither methylation of the PTEN promoter nor loss of LOH at MSI surrounding and intragenic to the PTEN locus was a significant predictor of PTEN protein expression (Marsit et al., 2005). PTEN expression is also dysregulated in COPD; however, the implications of this observation are not well characterized. Microarray analysis of primary airway epithelial cells revealed down‐regulation of PTEN expression in chronic smokers that progressively decreased with development of COPD (Shaykhiev et al., 2011). Cigarette smoke extract has been shown to down‐regulate PTEN expression, which was associated with an increase in EGFR transactivation required for increased mucin production (Lee et al., 2006); however, the epigenetic mechanism behind reduced PTEN expression has yet to be determined.
Oxidative stress, COPD and squamous cell carcinoma
Another important pathogenic feature of COPD that can initiate lung tumourigenesis is excessive and uncompensated oxidative stress, where oxidative DNA damage has been shown to be prominent in COPD lungs (Pastukh et al., 2011). Moreover, oxidative stress can induce oncogenic lipid peroxides, inactivate defensive mechanisms and permissively alter the extracellular matrix. A lifetime of cigarette smoking exposes lung cells to over 4700 different chemical compounds and more than 1015 oxidants/free radicals per puff (Church and Pryor, 1985; Nakayama et al., 1989; Pryor and Stone, 1993; Rahman, 2012). At least 69 of these are carcinogenic compounds, including polycyclic aromatic hydrocarbons, tobacco‐specific nitrosamines, aromatic amines and volatile carcinogens such as formaldehyde and benzene (Hoffmann et al., 2001). In addition, free radicals in cigarette smoke are known to initiate conversion of procarcinogens into their active state to promote formation of DNA adducts (Pryor, 1997). Highly reactive molecules are also generated enzymatically by inflammatory and epithelial cells within the lung in response to repeated exposure to smoke constituents and inhaled pathogens. Activation of leukocytes by cigarette smoke generates superoxide radicals (O2 ●−), which can then either react with NO to form reactive peroxynitrite (ONOO−) or alternatively be rapidly converted to hydrogen peroxide (H2O2) under the influence of superoxide dismutase. Non‐enzymatic production of the more damaging hydroxyl radical (●OH) from H2O2 through Fenton reactions catalysed by free iron can also proceed (Vlahos and Bozinovski, 2013). ROS activity as assessed by measuring exhaled H2O2 in current smokers and patients with COPD is significantly higher than non‐smokers (Nowak et al., 1996), and levels are further increased during exacerbations of COPD due to increased release of O2 ●− (Dekhuijzen et al., 1996).
Thus, while ROS are required for host defence against invading pathogens, increased levels of ROS have been implicated in sustaining a damaging cycle of inflammation in COPD through redox‐dependent activation of inflammatory transcription factors such as NF‐κB and AP‐1 (Rahman and Adcock, 2006; Rahman, 2012). Increased production of ROS in COPD is also confounded by the down‐regulation of nuclear factor, erythroid 2‐like 2 (NFE2L2 or NRF2) in COPD (Malhotra et al., 2008), which is a transcription factor that promotes expression of a suite of cytoprotective and antioxidant genes that contain an antioxidant response element in their promoter. Agents that increase NFE2L2 expression have shown promise in pre‐clinical models of lung cancer when administered during the initiation phase of carcinogenesis, and consequently, there has been interest in their use as cancer chemopreventative agents (reviewed in Sporn and Liby, 2012). However, emerging reports have led to safety concerns relating to the long‐term use of such agents. It has been shown that oncogenes can increase NFE2L2, a master regulator of antioxidant defence levels, and this facilitates a cytoprotective advantage in the tumour microenvironment (DeNicola et al., 2011), which may have significant implications during chemotherapy. Dietary supplementation with antioxidants can also increase tumour progression and reduce survival in mouse models of lung cancer (Sayin et al., 2014). In this model, the oncogenic drivers KRAS or B‐Raf are already introduced at initiation of antioxidant therapy and NAC provided a survival advantage in the advanced tumours. The opposing effects of antioxidants in various cancer models may suggest that timing of antioxidant therapy will be critical, where early intervention can prevent accumulation of cytotoxic DNA damage, whereas late intervention may facilitate tumour survival.
Oxidative stresses can also directly influence the activity of the tumour suppressor gene, PTEN. PTEN is reported to be very susceptible to oxidative modification, which inactivates its enzymatic activity leading to increased PIK3CA/Akt signalling (Leslie et al., 2003). Smoking and COPD are associated with ROS‐dependent peroxidation of polyunsaturated fatty acids and generation of reactive carbonyl species, including acrolein, 4‐hydroxynonenal and malondialdehyde. Reactive carbonyls lead to the oxidation of proteins, lipids, carbohydrates and DNA and can target approximately 10% of the entire proteome during ageing, starvation or disease (Maisonneuve et al., 2009). Hence, protein carbonylation represents a major pathway to protein oxidation (Stadtman, 1993) where reactive carbonyls target susceptible arginine, histidine, lysine, proline or threonine residues (Dalle‐Donne et al., 2006; 2009). Once proteins are carbonylated, they are rapidly ubiquinated and degraded within proteosomal complexes as a primary mechanism for removal of carbonylated proteins (Bernhard et al., 2005). Reactive carbonyls are increased in COPD (Rahman et al., 2002; Kirkham et al., 2011) and carbonyl stress caused by cigarette smoke has been associated with impairment of macrophage function required for bacterial clearance (Bozinovski et al., 2011). Reactive carbonyls also covalently modify and inactivate cellular PTEN, leading to activation of PIK3CA/Akt signalling (Covey et al., 2010). Hence, loss of PTEN function by post‐translational modification caused by reactive carbonyls occurs independently of genetic PTEN alterations that are typically screened for and represents an alternative pathway to maintaining tumour‐promoting PIK3CA/Akt/mTOR signalling.
Inflammatory responses in COPD support tumour initiation and progression
Hanahan and Weinberg comprehensively described emerging hallmarks and enabling characteristics of cancer (Hanahan and Weinberg, 2011). Enabling characteristics that promote acquired functional capabilities allow cancer cells to survive, proliferate and disseminate. The two enabling characteristics include (i) the development of genomic instability in cancer cells and (ii) the initiation of tumour‐promoting inflammation driven by cells of the immune system. They also highlighted a core hallmark involving the active evasion by cancer cells from attack and elimination by immune cells (Hanahan and Weinberg, 2011). Hence, inflammation and immunity play a key role in tumour initiation and progression, and as COPD is a chronic inflammatory condition, aberrant immunity in COPD will be central to increased risk of lung cancer, independently of tobacco exposure. Immunological processes in COPD are complex but involve disruption of both innate and adaptive immunity, and a greater understanding of this distinct microenvironment is needed to develop therapeutic strategies to combat tumour‐promoting inflammation and tumour‐evading immunity in COPD. A prominent hallmark of the tumour microenvironment is the emergence of tumour‐associated macrophages (TAMs), which accumulate within hypoxic regions of tumours. TAMs promote pro‐angiogenic programmes through production of angiogenic factors, such as VEGF and PDGF, and facilitate tumour progression by secreting matrix‐degrading enzymes (Allavena et al., 2008). In an analogous manner, neutrophils have been shown to accumulate in certain human tumours and their presence can either be associated with better or worse outcomes. Like macrophages, tumour‐associated neutrophils are likely to be transformed by their microenvironment, leading to divergent phenotypes (N1 vs. N2) that can either support tumour growth through expression of pro‐angiogenic factors [VEGF, matrix metalloproteinase‐9 (MMP‐9)] or suppress tumour growth.
Innate immunity
Macrophages and neutrophils are considered to be central in the pathophysiology of COPD, where they accumulate in the airways and lung parenchyma (Keatings et al., 1996; Pesci et al., 1998). In particular, macrophage numbers in the parenchyma of patients with emphysema markedly increase (Retamales et al., 2001) and their selective depletion conferred protection against the development of emphysema in an experimental model of COPD (Beckett et al., 2013). In response to cigarette smoke, irritants and infection, macrophages release inflammatory mediators and secrete elastolytic enzymes including MMP‐9 (Barnes et al., 2003). There is an increase in lung parenchymal MMP‐9 activity in emphysematic patients (Ohnishi et al., 1998) and proteinases released in COPD are well known for their ability to promote tumour growth and invasion (Houghton, 2013). MMP‐9 has been shown to promote angiogenesis through proteolytic activation of ECM‐bound VEGF (Bergers et al., 2000) and tumour progression has been shown to be reduced in mice deficient in MMP‐9 (Itoh et al., 1998).
The molecular profiling of disease‐associated macrophages in COPD demonstrates that they do not conform to the classic M1/M2 dichotomy and it is likely that the inflammatory environment of the COPD airways drives the development of both M1 and M2 macrophages (Mosser and Edwards, 2008; Hodge et al., 2011). The emergence of alveolar macrophages expressing an M2 gene profile in heavy smokers with normal lung function is progressive in patients with COPD (Shaykhiev et al., 2009). The accumulation of M2 airway macrophages may reflect a deficiency in processes that normally resolve inflammation and restore lung homeostasis, where oxidative stress is known to impair phagocytic and efferocytic programmes required to clear infected/damaged tissue (Vlahos and Bozinovski, 2014). In addition, impaired macrophage efferocytosis is observed in lung cancer and this deficiency is associated with tumour‐derived arachidonic acid products including PGE2 (Dehle et al., 2013). The accumulation of macrophages in the tumour stroma in NSCLC is associated with a worse prognosis, in contrast to accumulation within the tumour inlet, which conferred a significant survival advantage (Welsh et al., 2005). Distinct macrophage phenotypes have been characterized in NSCLC tissue, where M1 macrophages found in the tumour islet are associated with survival extension, whereas M2 macrophages that express CD163 and VEGF were not related to increased survival (Ohri et al., 2009). CD163‐positive macrophages are also found in ex‐smokers with COPD (Kunz et al., 2011). Hence, there is a need to better define the relative contribution of M1 versus M2‐skewed macrophages in COPD and lung cancer overlap as both populations do concurrently exist and their relative ratio and localization will be very important in the progression of lung tumours.
Neutrophilic inflammation is also prominent in COPD, where cigarette smoke promotes the recruitment of neutrophils (Vlahos et al., 2006) and smoking cessation fails to fully resolve inflammation (Stanescu et al., 1996; Rutgers et al., 2000; Willemse et al., 2005). Since neutrophils are relatively short‐lived, their persistence is indicative of continual recruitment even when the primary insult of smoke exposure is removed. Neutrophils are also a potent source of ROS in COPD, which are released in response to inhaled irritants and respiratory microbes that infect the airways. During neutrophil respiratory burst, myeloperoxidase can catalyse the formation of the potent and very damaging oxidants hypochlorous acid (HOCl) and hypobromous acid (HOBr) from H2O2 in the presence of chloride (Cl−) and bromide (Br−) ions respectively. Persistent activation of neutrophils can therefore contribute to the accumulation of DNA damage through generation of potent free radicals. The insufficiency to clear exhausted neutrophils can also lead to indiscriminate degranulation and subsequent release of proteases, including neutrophil elastase from azurophilic granules, and neutrophil elastase activity increases with COPD severity in the presence of inhaled glucocorticosteroids (Vlahos et al., 2012). Colonizing pathogens and viral infections can directly cause neutrophil necrosis and release of azurophilic granular content in COPD (Naylor et al., 2007; Mallia et al., 2012).
Anti‐proteinases such as α1‐antitrypsin (α1‐AT) that normally provide an anti‐proteinase screen become overwhelmed in COPD. Neutrophil elastase degrades extracellular matrix components, including elastin and the degree of elastase localized to lung elastic fibres correlates with the degree of emphysema (Damiano et al., 1986). Neutrophil elastase can also directly activate TLR4 signalling that promotes CXCL8 expression in the bronchial epithelial cells (Walsh et al., 2001; Kuwahara et al., 2006) and promote mucin production via EGFR transactivation (Shao and Nadel, 2005). α1‐AT deficiency remains the only heritable genetic defect that leads to accelerated emphysematic phenotype, and of significance, carriers are also at increased risk for developing lung cancer (Yang et al., 2008). In a murine KRAS mutant lung cancer model, ablation of neutrophil recruitment significantly reduced the number of lung tumours, where neutrophil elastase activity was required for tumour‐promoting proliferation and angiogenesis (Gong et al., 2013). Neutrophil elastase was also shown to accelerate lung tumour growth by regulating the activity of the PIK3CA/Akt pathway through the degradation of its binding partner, IRS1 (Houghton et al., 2010). Hence, excessive neutrophil degranulation cannot only contribute to tumour initiation by facilitating DNA damage through release of ROS, but its proteolytic content can induce proliferation, angiogenesis and migration required for tumour progression.
Adaptive immunity
COPD is also characterized by the accumulation of adaptive immune cells. As COPD progresses, organized tertiary lymphoid follicles emerge with increasing disease severity (Hogg et al., 2004). These organized structures consist of B‐cells, T‐cells and dendritic cells that are maintained by up‐regulation of homeostatic chemokines, where B‐cell recruitment is dependent upon CXCL13 (Bracke et al., 2013). B‐cells may contribute to deleterious autoantibody production against self‐antigens, leading to local complement fixation and tissue damage; however, a causative role has yet to be established. Neutralizing CXCL13 activity effectively reduced B‐cell tissue accumulation and partially reduced alveolar tissue destruction in a chronic smoke model but did not alter other features of emphysema, inflammation and airway wall remodelling (Bracke et al., 2013). There may also be a case for a protective role for lymphoid follicles in COPD. Inducible bronchus‐associated lymphoid tissues are increasingly recognized for their ability to enhance protective immunity and maintain memory cells in the lungs against respiratory pathogens (Foo and Phipps, 2010). As infectious exacerbations become more frequent with increasing severity of COPD, the emergence of lymphoid follicles may play a role in protective immunity in this setting.
The role for lymphoid follicles in lung cancer is also intriguing, where the combination of follicular B‐cell and mature dendritic cell densities was predictive of longer survival in early and advanced stage NSCLC (Germain et al., 2014). In addition, intra‐tumoural follicles are associated with the development of antigen‐specific humoral responses, with the emergence of plasma cells secreting tumour antigen‐specific immunoglobulins (Germain et al., 2014). Since the risk of developing lung cancer decreases in severe COPD relative to mild‐moderate COPD, the potential anti‐tumourigenic role for lymphoid aggregates warrants further investigation. This robust humoral response may further contribute to antigen‐specific CD8 T‐cell generation and expansion central to protective immunity. Dendritic cells also play a key role in tumour eradication by ingesting tumour debris, homing back to the draining lymph nodes and facilitating the development of tumour‐specific CD4 and CD8 T‐cells. Tumour‐specific T‐cells can then home to the tumour site where their cytolytic function effectively destroys the remaining antigen‐bearing cancer cells. In COPD, several dendritic cell subsets are present in the respiratory mucosa and their relative abundance and activation state appear to be influenced by smoking status and severity of COPD (Brusselle et al., 2011). Dendritic cells will regulate activation of cytotoxic T‐cells in COPD that contribute to development of emphysema through promoting apoptosis of structural cells (Brusselle et al., 2011). Further investigation into the interactions between dendritic and cytotoxic T‐cell function in COPD will reveal their role in lung cancer susceptibility.
The concept of cancer immunoediting now recognizes that the immune system can play a dual role in cancer that not only suppresses tumour growth by destroying cancer cells but can also promote tumour progression by either selecting poorly immunogenic tumours or establishing a microenvironment that facilitate tumour outgrowth (Dunn et al., 2002). Destruction of cancer cells by tumour infiltrating cytotoxic CD8 and NK cells recognize tumour antigen and produce mediators such as IFN‐γ, TNF‐α, granzymes and perforin to promote tumour control, and their levels have been shown to be decreased in lung cancer (Hodge et al., 2014). Reduced granzyme B release from CD8 T‐cells has been associated with increased expression of its inhibitor, PI‐9, by the lung tumour cells (Soriano et al., 2012). In COPD, there is an expansion of cytotoxic CD8 T‐cells that increase in the airway and alveolar compartment with disease severity (Saetta et al., 1998), where they are thought to promote emphysema through release of perforin and granzymes that promote apoptosis of the structural cells (Urbanowicz et al., 2010). Although the emergence of cytotoxic CD8 T‐cells in COPD may theoretically provide a protective role in reducing lung cancer risk, it has also been reported that cigarette smoke can induce a state of T‐cell anergy that is dependent upon the degree of smoke exposure (Stampfli and Anderson, 2009). Consistent with this concept, oxidative stress has been shown to alter the survival, activation, differentiation and migration of cytotoxic effector cells directly through oxidative modification including carbonylation and nitrosylation (Klemke and Samstag, 2009). The expression of the CD3 zeta chain of T‐cell receptor complex is particularly vulnerable to ROS, where circulating T‐cells produce less cytokines in the presence of activated granulocytes (Schmielau and Finn, 2001). In addition to suppression of inflammatory cytokine release by epithelial cells (Laan et al., 2004), ROS has also been shown to decrease cytokine production by blocking NF‐κB signalling in T‐cells (Malmberg et al., 2001). In addition, peroxynitrite can enhance nitration of tyrosine residues on TCR and CD8 molecules on CD8 T‐cells, thereby blocking their ability to bind antigen presented by MHC‐I (Nagaraj et al., 2007). Oxidation of the actin cytoskeleton in T‐cells can also promote a state of T‐cell hyporesponsiveness (Klemke et al., 2008), which ultimately compromises immune surveillance required for eradication of cancer cells.
Checkpoint inhibitors are increasingly been evaluated for their ability to modulate cytotoxic T‐cell function and improve immunological eradication of cancer cells, including ipilimumab, a monoclonal blocking antibody against cytotoxic T lymphocyte associated protein 4 (CTLA‐4), and pembrolizumab, a monoclonal antibody against programmed cell death protein 1 (PD‐1). Since immune checkpoint pathways normally prevent excessive effector activity by cytotoxic T‐cells, their inhibition has shown promise in increasing T‐cell activation and killing of tumour cells. An increase in CTLA‐4 expression on the cell surface of lymphocytes from patients with NSCLC has been observed (Erfani et al., 2012), which may contribute to an anergic phenotype that fails to eradicate tumour cells. There are multiple clinical trials that are currently evaluating the efficacy of ipilimumab in lung cancer. A phase II trial has shown that phased ipilimumab plus paclitaxel and carboplatin improved immune‐related progression‐free survival, and importantly, this appeared to be greater in patients with squamous histology then non‐squamous histology (Lynch et al., 2012). Like CTLA‐4, PD‐1 is a surface receptor that is expressed on many cell types including activated T‐cells and interaction with its ligand (PD‐L1) induces T‐cell tolerance by reducing proliferation and cytokine production. PD‐L1 has been shown to be expressed by NSCLC cells and was associated with poor survival (Mu et al., 2011), which may contribute to immune evasion in this setting. The anti‐tumour activity of PD‐1 inhibitors such as pembrolizumab is currently being evaluated for advanced NSCLC expressing PD‐1. In addition, the activity and safety of nivolumab, an anti‐PD‐1 immune checkpoint inhibitor, has been assessed in patients with advanced, refractory SCC, and 14.5% subjects had an objective response with a manageable safety profile in this phase II trial (Rizvi et al., 2015). The Food and Drug Administration has now approved the use of nivolumab in SCC. In COPD, circulating levels of PD‐1 + exhausted effector T‐cells were found to be increased (Kalathil et al., 2014), which may not only mitigate deficient antiviral and antibacterial effector functions in COPD but may also compromise tumour immune surveillance.
IL‐17A, COPD and lung cancer
IL‐17A is increasingly recognized as a fundamentally important regulator of cellular immunity and is conventionally considered to arise predominantly from the ‘Th17’ specific subset of CD4 cells (Park et al., 2005). IL‐17A is uniquely positioned at the interface of innate and adaptive immunity (Ouyang et al., 2008) and can regulate lung inflammation by promoting recruitment of leukocytes, release of myeloperoxidase, neutrophil elastase and MMP‐9 (Prause et al., 2004; Ivanov et al., 2007). There is also evidence for an emerging role for IL‐17A in COPD, where IL‐17A+ cells are increased in bronchial submucosa of chronic smokers and stable COPD subjects (Di Stefano et al., 2009; Doe et al., 2010). Genetic ablation of IL‐17 receptors also prevented the development of experimental emphysema (Chen et al., 2011) and inhibition of IL‐17A reduced neutrophilic inflammation induced by cigarette smoke (Shen et al., 2011). Of significance, IL‐17A expression was induced in NOD.SCID mice, which reveals that alternate sources of IL‐17A including NK cells are activated by smoke exposure (S. Bozinovski et al., unpubl. data). Consistent with this concept, both IL‐17A and NK cells were shown to be induced by cigarette smoke exposure and were persistently up‐regulated following prolonged smoking cessation (Hansen et al., 2014). NK cell function has not been extensively characterized in COPD; however, chronic cigarette smoke exposure primed NK cells to release inflammatory mediators including IL‐12 and IL‐18 in mice (Motz et al., 2010). The NK cell group 2D (NKG2D) ligand is also elevated in CS‐exposed pulmonary epithelial cells, which may sustain deleterious activation of cytotoxic T‐cells including NK cells involving release of granzymes and perforin (Borchers et al., 2009).
IL‐17A is also increasingly recognized as a key cytokine in lung cancer where its levels are inversely correlated with patient survival and is implicated in metastasis of lung cancer by promoting lymphangiogenesis (Chen et al., 2010). IL‐17A transcript levels were increased in NSCLC biopsies and inhibition of IL‐17A reduced tumour growth by inducing activation of tumour‐infiltrating CD4 T‐cells in mice injected with L1C2 tumour cells (Reppert et al., 2011). Suppression of IL‐17A in the KRASG12D mouse model of lung cancer also resulted in the reduction of tumour cell proliferation and angiogenesis and decreased the expression of pro‐inflammatory mediators (Chang et al., 2014). In this model, IL‐17A promoted the recruitment of myeloid cells that were required for tumour growth (Chang et al., 2014). Hence, there is a compelling case for targeting IL‐17A in both COPD and lung cancer, where both conventional TH17 and alternate innate sources will contribute to disease progression and poor prognosis.
SAA and FPR2 in lung cancer and COPD
COPD is associated with systemic inflammation as characterized by an increase in circulating cytokines, acute phase proteins and abnormalities in circulating cells, where the degree of inflammation increases with disease severity (Agusti et al., 2012). Systemic inflammation also markedly rises during infectious exacerbations of COPD, and the acute phase reactant termed serum amyloid A (SAA) has been shown to be predictive of exacerbation severity (Bozinovski et al., 2008). Systemically, SAA is secreted by the liver and associates with HDLs (Coetzee et al., 1986), where this complex is involved in mobilizing and recycling macrophage cholesterol during tissue injury. There is also evidence of extrahepatic SAA production, as de novo transcript synthesis is increased in the lung tissue from mice exposed to cigarette smoke, LPS and influenza infection (Bozinovski et al., 2012). In addition, SAA immunoreactvity was detected in lung resection samples obtained from COPD subjects with lung cancer (Bozinovski et al., 2012). SAA is considered to be pro‐inflammatory as it is a potent chemotactic factor that mediates migration of leukocytes (Su et al., 1999) and can also stimulate expression of pro‐inflammatory mediators under in vitro (He et al., 2003) and in vivo conditions (Bozinovski et al., 2012). More recently, SAA was shown to promote the differentiation of human monocyte‐derived macrophages into a pro‐inflammatory phenotype that expressed higher levels of the TH17 polarizing cytokines, IL‐6 and IL‐1β (Anthony et al., 2014). Inhibition of IL‐17A signalling in this model also markedly reduced the recruitment of neutrophils into the airways in response SAA (Anthony et al., 2013). Hence, increased expression of SAA in the lungs of COPD subjects with lung cancer may contribute to tumour growth by stimulating an M2‐like alternative macrophage phenotype and inducing IL‐17A‐mediated inflammation.
Like COPD, SAA has been identified as a severity biomarker and a potential therapeutic candidate in lung cancer. Proteomic screening of sera from lung cancer patients have demonstrate that when SAA is combined with other circulating inflammatory markers, high sensitivity and specificity (>90%) are achieved with respect to differentiating healthy subjects from lung cancer (Liu et al., 2011; Dowling et al., 2012). Furthermore, when SCC patients are stratified on the basis of pre‐surgery SAA levels into rapid recurrence group versus no evidence of recurrence group following resection of tumour, high SAA levels (cut‐off of 17 μg·mL−1) were predictive of rapid recurrence and poor survival outcomes (Liu et al., 2012). Hence, the application of SAA in combination with other markers are not only powerful in prognosis determination but can also inform on treatment optimization and patient‐tailored therapies based upon serum signatures. Like COPD, there is also emerging literature that SAA function extends beyond its clinical utility as a disease biomarker and may transition into an important pathogenic mediator of disease progression.
There is now evidence for production of SAA within the tumour microenvironment in melanoma (De Santo et al., 2010) and lung cancer (Bozinovski et al., 2012) in close proximity to tumour‐infiltrating macrophages. In addition, there was a positive association between the number of neutrophils and the expression of SAA in lung resection samples obtained from subjects with COPD and lung cancer (Anthony et al., 2013). This is an important observation, as SAA has been shown to promote the recruitment and differentiation of an IL‐10+ expressing neutrophil population that has been shown to be immunosuppressive towards cytotoxic T‐cells (De Santo et al., 2010). The expansion of immunosuppressive neutrophils by SAA was shown to be dependent upon the activation of MAPK and PIK3CA through its interaction with the FPR2 receptor (De Santo et al., 2010). Immunosuppressive neutrophils are normally counteracted by crosstalk with invariant or CD1‐restricted NKT cells, which reverse this suppressive phenotype by decreasing IL‐10 and promoting IL‐12 production, and restoring proliferation of antigen‐specific CD8+ T‐cells (De Santo et al., 2010). The functional capabilities of NKT cells have not been determined in COPD; however, possible defects in this mucosal cell type have been implicated in recurrent infections (Stampfli and Anderson, 2009). Furthermore, SAA has been reported to promote immune evasion of cancer cells by enhancing the suppressive capacity of myeloid‐derived suppressor cells (Lee et al., 2014) via TLR2‐dependent mechanisms, which have been proposed as an alternative receptor for SAA. In another study, a mouse‐specific isoform of SAA was shown to stimulate inflammation associated with the accelerated migration of primary tumour cells to the lungs, where blocking SAA function in the pre‐metastatic phase prevented pulmonary metastasis in mice (Hiratsuka et al., 2008).
SAA has also been shown to modulate the differentiation of monocyte‐derived macrophages, including the induction of IL‐10 and TH17 inducing cytokines (Anthony et al., 2014). In addition, SAA has been shown to promote an M2‐like polarization state that exacerbated hepatocellular carcinoma cell invasion (Li et al., 2011). In this study, the polarization towards a tumour‐promoting phenotype was dependent upon FPR2 signalling, and alternative ligands to this receptor involved in the resolution of inflammation skewed macrophages towards an anti‐tumourigenic phenotype (Li et al., 2011). FPR2 is a GPCR superfamily member characterized by seven putative TM domains that display diverse ligand affinities that extend beyond its interaction with SAA to include pro‐resolving mediators (PRM) such as lipoxins, series D‐resolvins and annexin A1. The diverse conformation of endogenous and synthetic ligands that bind to alternate regions of the FPR2 can promote ligand‐biased signalling, leading to differential biological actions (Cooray et al., 2013). Lipoxin A4 (LXA4) is synthesized in response to cell–cell interactions (reviewed in Serhan, 2005; Chiang et al., 2006), where it opposes leukocyte migration and activation (Papayianni et al., 1996). LXA4 can also suppress inflammatory cytokine production in mucosal epithelial cells (Bonnans et al., 2006) and reduce lung inflammation initiated by SAA (Bozinovski et al., 2012). Other important roles for LXA4 include the stimulation of macrophage‐mediated efferocytosis (Godson et al., 2000; El Kebir et al., 2009) and the promotion of an alternative macrophage phenotype associated with anti‐tumourigenic properties (Li et al., 2011). The D series resolvins (RvD1) derived from the omega‐3 fatty acid, docosahexaenoic acid also engage FPR2 and have been shown to reduce neutrophilic lung inflammation, inflammatory cytokine production and promote phagocytosis in an acute cigarette smoke exposure model (Hsiao et al., 2013). There are also emerging data that FPR2 and its murine homologues are critical in mediating homeostasis, inflammation and epithelial repair processes and any perturbation of this mechanism may contribute to tumour development (Bonnans et al., 2006; Khau et al., 2011; Chen et al., 2013). Further work is needed to characterize what effect different FPR2 ligands have on lung epithelial survival and proliferation.
Conclusions
COPD is a chronic inflammatory condition where aberrant immunity not only contributes to excessive oxidative stress and deleterious lung remodelling but will also contribute to lung cancer susceptibility and progression (summarized in Figure 1). COPD is associated with persistent activation of innate immune cells including neutrophils, where the uncontrolled release of powerful free radicals and proteases will not only cause DNA damage but also promote tumour migration. Airway macrophages in COPD are skewed towards an M2 state that will release supportive factors within the tumour microenvironment. Adaptive immunity in COPD is also compromised, which is potentially associated with the development of exhausted T‐cell populations that fail to effectively respond to respiratory infections. T‐cell anergy in COPD may also lead to tumour evasion through suppressed cytotoxic T‐cell clearance of cancer cells. The stimulation of T‐cell function using CTLA‐4 and PD‐1 inhibitors may provide an opportunity to stimulate T‐cell function in COPD in order to reduce exacerbation and lung cancer risk; however, activation of cytotoxic T‐cells are also implicated in development of emphysema through apoptosis of structural cells. Non‐conventional T‐cells may also be important sources of IL‐17A in COPD, which is a pivotal immunological cytokine that is increasingly recognized as a mediator of tumour growth. Systemic inflammation is common in COPD and lung cancer, and SAA has predictive utility in both diseases with respect to severity indices. There is also emerging evidence to suggest that SAA is actively modulating inflammation and immunity in cancer by promoting pro‐tumourigenic inflammation and facilitating tumour evasion. Since the treatment of SCC remains a major therapeutic challenge, targeted inhibition of FPR2 expressed on immune cells provides a therapeutic opportunity to combat pro‐tumourigenic inflammation and immunity caused by underlying COPD. Epimers of LXA4 and RvD1 can be produced following aspirin treatment (Serhan et al., 2008) and since aspirin may reduce the risk of developing lung cancer (Jonsson et al., 2013), the relative contribution of these known FPR2 ligands warrants further investigation.
Figure 1.
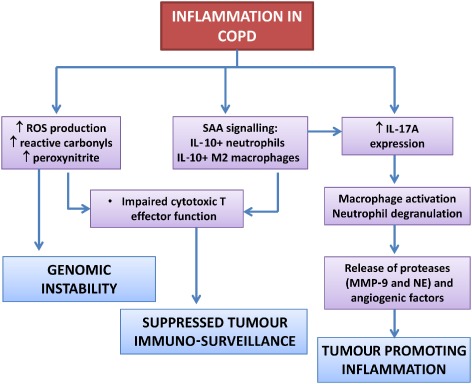
Inflammatory and oxidative mechanisms prominent in COPD that can promote lung cancer by causing genomic instability, suppressing tumour immuno‐surveillance mechanisms and promoting inflammation that is beneficial to tumour growth and migration.
Bozinovski, S. , Vlahos, R. , Anthony, D. , McQualter, J. , Anderson, G. , Irving, L. , and Steinfort, D. (2016) COPD and squamous cell lung cancer: aberrant inflammation and immunity is the common link. British Journal of Pharmacology, 173: 635–648. doi: 10.1111/bph.13198.
References
- Agusti A, Edwards LD, Rennard SI, MacNee W, Tal‐Singer R, Miller BE et al (2012). Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS ONE 7: e37483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A (2008). The inflammatory micro‐environment in tumor progression: the role of tumor‐associated macrophages. Crit Rev Oncol Hematol 66: 1–9. [DOI] [PubMed] [Google Scholar]
- Anderson GP, Bozinovski S (2003). Acquired somatic mutations in the molecular pathogenesis of COPD. Trends Pharmacol Sci 24: 71–76. [DOI] [PubMed] [Google Scholar]
- Anthonisen NR, Connett JE, Kiley JP, Altose MD, Bailey WC, Buist AS et al (1994). Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA 272: 1497–1505. [PubMed] [Google Scholar]
- Anthony D, Seow HJ, Uddin M, Thompson M, Dousha L, Vlahos R et al (2013). Serum amyloid A promotes lung neutrophilia by increasing IL‐17A levels in the mucosa and gammadelta T cells. Am J Respir Crit Care Med 188: 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony D, McQualter JL, Bishara M, Lim EX, Yatmaz S, Seow HJ et al (2014). SAA drives proinflammatory heterotypic macrophage differentiation in the lung via CSF‐1R‐dependent signaling. FASEB J 28: 3867–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A et al (2007). Squamous metaplasia amplifies pathologic epithelial‐mesenchymal interactions in COPD patients. J Clin Invest 117: 3551–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Shapiro SD, Pauwels RA (2003). Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 22: 672–688. [DOI] [PubMed] [Google Scholar]
- Beckett EL, Stevens RL, Jarnicki AG, Kim RY, Hanish I, Hansbro NG et al (2013). A new short‐term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol 131: 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K et al (2000). Matrix metalloproteinase‐9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2: 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard D, Csordas A, Henderson B, Rossmann A, Kind M, Wick G (2005). Cigarette smoke metal‐catalyzed protein oxidation leads to vascular endothelial cell contraction by depolymerization of microtubules. FASEB J 19: 1096–1107. [DOI] [PubMed] [Google Scholar]
- de Boer WI, Hau CM, van Schadewijk A, Stolk J, van Krieken JH, Hiemstra PS (2006). Expression of epidermal growth factors and their receptors in the bronchial epithelium of subjects with chronic obstructive pulmonary disease. Am J Clin Pathol 125: 184–192. [DOI] [PubMed] [Google Scholar]
- Bonnans C, Fukunaga K, Levy MA, Levy BD (2006). Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am J Pathol 168: 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchers MT, Wesselkamper SC, Curull V, Ramirez‐Sarmiento A, Sanchez‐Font A, Garcia‐Aymerich J et al (2009). Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD‐like disease. J Clin Invest 119: 636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozinovski S, Hutchinson A, Thompson M, Macgregor L, Black J, Giannakis E et al (2008). Serum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177: 269–278. [DOI] [PubMed] [Google Scholar]
- Bozinovski S, Vlahos R, Zhang Y, Lah LC, Seow HJ, Mansell A et al (2011). Carbonylation caused by cigarette smoke extract is associated with defective macrophage immunity. Am J Respir Cell Mol Biol 45: 229–236. [DOI] [PubMed] [Google Scholar]
- Bozinovski S, Uddin M, Vlahos R, Thompson M, McQualter JL, Merritt AS et al (2012). Serum amyloid A opposes lipoxin A(4) to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A 109: 935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracke KR, Verhamme FM, Seys LJ, Bantsimba‐Malanda C, Cunoosamy DM, Herbst R et al (2013). Role of CXCL13 in cigarette smoke‐induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 188: 343–355. [DOI] [PubMed] [Google Scholar]
- Brusselle GG, Joos GF, Bracke KR (2011). New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378: 1015–1026. [DOI] [PubMed] [Google Scholar]
- Cetin K, Ettinger DS, Hei YJ, O'Malley CD (2011). Survival by histologic subtype in stage IV nonsmall cell lung cancer based on data from the Surveillance, Epidemiology and End Results Program. Clin Epidemiol 3: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS et al (2014). T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci U S A 111: 5664–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, Kreindler JL et al (2011). IL‐17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS ONE 6: e20333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Liu M, Liu Y, Yoshimura T, Shen W, Le Y et al (2013). Formylpeptide receptor‐2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J Clin Invest 123: 1694–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wan J, Liu J, Xie W, Diao X, Xu J et al (2010). Increased IL‐17‐producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer 69: 348–354. [DOI] [PubMed] [Google Scholar]
- Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE et al (2006). The lipoxin receptor ALX: potent ligand‐specific and stereoselective actions in vivo. Pharmacol Rev 58: 463–487. [DOI] [PubMed] [Google Scholar]
- Church DF, Pryor WA (1985). Free‐radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 64: 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC (1986). Serum amyloid A‐containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem 261: 9644–9651. [PubMed] [Google Scholar]
- Cooray SN, Gobbetti T, Montero‐Melendez T, McArthur S, Thompson D, Clark AJ et al (2013). Ligand‐specific conformational change of the G‐protein‐coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci U S A 110: 18232–18237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosio M, Ghezzo H, Hogg JC, Corbin R, Loveland M, Dosman J et al (1978). The relations between structural changes in small airways and pulmonary‐function tests. N Engl J Med 298: 1277–1281. [DOI] [PubMed] [Google Scholar]
- Covey TM, Edes K, Coombs GS, Virshup DM, Fitzpatrick FA (2010). Alkylation of the tumor suppressor PTEN activates Akt and beta‐catenin signaling: a mechanism linking inflammation and oxidative stress with cancer. PLoS ONE 5: e13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle‐Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A (2006). Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med 10: 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle‐Donne I, Carini M, Orioli M, Vistoli G, Regazzoni L, Colombo G et al (2009). Protein carbonylation: 2,4‐dinitrophenylhydrazine reacts with both aldehydes/ketones and sulfenic acids. Free Radic Biol Med 46: 1411–1419. [DOI] [PubMed] [Google Scholar]
- Damiano VV, Tsang A, Kucich U, Abrams WR, Rosenbloom J, Kimbel P et al (1986). Immunolocalization of elastase in human emphysematous lungs. J Clin Invest 78: 482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R et al (2010). Invariant NKT cells modulate the suppressive activity of IL‐10‐secreting neutrophils differentiated with serum amyloid A. Nat Immunol 11: 1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehle FC, Mukaro VR, Jurisevic C, Moffat D, Ahern J, Hodge G et al (2013). Defective lung macrophage function in lung cancer ± chronic obstructive pulmonary disease (COPD/emphysema)‐mediated by cancer cell production of PGE2? PLoS ONE 8: e61573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL et al (1996). Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 154: 813–816. [DOI] [PubMed] [Google Scholar]
- Dennis RJ, Maldonado D, Norman S, Baena E, Martinez G (1996). Woodsmoke exposure and risk for obstructive airways disease among women. Chest 109: 115–119. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K et al (2011). Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A et al (2009). T helper type 17‐related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol 157: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P et al (2010). Expression of the T helper 17‐associated cytokines IL‐17A and IL‐17F in asthma and COPD. Chest 138: 1140–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling P, Clarke C, Hennessy K, Torralbo‐Lopez B, Ballot J, Crown J et al (2012). Analysis of acute‐phase proteins, AHSG, C3, CLI, HP and SAA, reveals distinctive expression patterns associated with breast, colorectal and lung cancer. Int J Cancer 131: 911–923. [DOI] [PubMed] [Google Scholar]
- Drilon A, Rekhtman N, Ladanyi M, Paik P (2012). Squamous‐cell carcinomas of the lung: emerging biology, controversies, and the promise of targeted therapy. Lancet Oncol 13: e418–e426. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3: 991–998. [DOI] [PubMed] [Google Scholar]
- El Kebir D, Jozsef L, Pan W, Wang L, Petasis NA, Serhan CN et al (2009). 15‐epi‐lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med 180: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erfani N, Mehrabadi SM, Ghayumi MA, Haghshenas MR, Mojtahedi Z, Ghaderi A et al (2012). Increase of regulatory T cells in metastatic stage and CTLA‐4 over expression in lymphocytes of patients with non‐small cell lung cancer (NSCLC). Lung Cancer 77: 306–311. [DOI] [PubMed] [Google Scholar]
- Foo SY, Phipps S (2010). Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol 3: 537–544. [DOI] [PubMed] [Google Scholar]
- Franklin WA, Gazdar AF, Haney J, Wistuba II, La Rosa FG, Kennedy T et al (1997). Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest 100: 2133–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V et al (2011). Biomarker analyses and final overall survival results from a phase III, randomized, open‐label, first‐line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non‐small‐cell lung cancer in Asia (IPASS). J Clin Oncol 29: 2866–2874. [DOI] [PubMed] [Google Scholar]
- Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J et al (2014). Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 189: 832–844. [DOI] [PubMed] [Google Scholar]
- Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte‐derived macrophages. J Immunol 164: 1663–1667. [DOI] [PubMed] [Google Scholar]
- Gong L, Cumpian AM, Caetano MS, Ochoa CE, De la Garza MM, Lapid DJ et al (2013). Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer 12: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson AM, Soldi R, Anderlind C, Scholand MB, Qian J, Zhang X et al (2010). Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci. Transl. Med. 2: 26ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Hansen MJ, Chan SP, Langenbach SY, Dousha LF, Jones JE, Yatmaz S et al (2014). IL‐17A and serum amyloid A are elevated in a cigarette smoke cessation model associated with the persistence of pigmented macrophages, neutrophils and activated NK cells. PLoS ONE 9: e113180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Sang H, Ye RD (2003). Serum amyloid A induces IL‐8 secretion through a G protein‐coupled receptor, FPRL1/LXA4R. Blood 101: 1572–1581. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Watanabe A, Sakurai Y, Akashi‐Takamura S, Ishibashi S, Miyake K et al (2008). The S100A8‐serum amyloid A3‐TLR4 paracrine cascade establishes a pre‐metastatic phase. Nat Cell Biol 10: 1349–1355. [DOI] [PubMed] [Google Scholar]
- Hodge G, Barnawi J, Jurisevic C, Moffat D, Holmes M, Reynolds PN et al (2014). Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)‐gamma by infiltrating lung tissue T cells, natural killer (NK) T‐like and NK cells. Clin Exp Immunol 178: 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge S, Matthews G, Mukaro V, Ahern J, Shivam A, Hodge G et al (2011). Cigarette smoke‐induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol 44: 673–681. [DOI] [PubMed] [Google Scholar]
- Hoffmann D, Hoffmann I, El‐Bayoumy K (2001). The less harmful cigarette: a controversial issue. A tribute to Ernst L. Wynder. Chem Res Toxicol 14: 767–790. [DOI] [PubMed] [Google Scholar]
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L et al (2004). The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350: 2645–2653. [DOI] [PubMed] [Google Scholar]
- Houghton AM (2013). Mechanistic links between COPD and lung cancer. Nat Rev Cancer 13: 233–245. [DOI] [PubMed] [Google Scholar]
- Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE et al (2010). Neutrophil elastase‐mediated degradation of IRS‐1 accelerates lung tumor growth. Nat Med 16: 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA et al (2013). A novel anti‐inflammatory and pro‐resolving role for resolvin D1 in acute cigarette smoke‐induced lung inflammation. PLoS ONE 8: e58258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S (1998). Reduced angiogenesis and tumor progression in gelatinase A‐deficient mice. Cancer Res 58: 1048–1051. [PubMed] [Google Scholar]
- Ivanov S, Bozinovski S, Bossios A, Valadi H, Vlahos R, Malmhall C et al (2007). Functional relevance of the IL‐23‐IL‐17 axis in lungs in vivo. Am J Respir Cell Mol Biol 36: 442–451. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011). Global cancer statistics. CA Cancer J Clin 61: 69–90. [DOI] [PubMed] [Google Scholar]
- Jonsson F, Yin L, Lundholm C, Smedby KE, Czene K, Pawitan Y (2013). Low‐dose aspirin use and cancer characteristics: a population‐based cohort study. Br J Cancer 109: 1921–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathil SG, Lugade AA, Pradhan V, Miller A, Parameswaran GI, Sethi S et al (2014). T‐regulatory cells and programmed death 1 + T cells contribute to effector T‐cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 190: 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatings VM, Collins PD, Scott DM, Barnes PJ (1996). Differences in interleukin‐8 and tumor necrosis factor‐alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 153: 530–534. [DOI] [PubMed] [Google Scholar]
- Khau T, Langenbach SY, Schuliga M, Harris T, Johnstone CN, Anderson RL et al (2011). Annexin‐1 signals mitogen‐stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J 25: 483–496. [DOI] [PubMed] [Google Scholar]
- Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B et al (2011). Oxidative stress‐induced antibodies to carbonyl‐modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184: 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke M, Samstag Y (2009). Molecular mechanisms mediating oxidative stress‐induced T‐cell suppression in cancer. Adv Enzyme Regul 49: 107–112. [DOI] [PubMed] [Google Scholar]
- Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y (2008). Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity 29: 404–413. [DOI] [PubMed] [Google Scholar]
- Kranenburg AR, Willems‐Widyastuti A, Mooi WJ, Saxena PR, Sterk PJ, de Boer WI et al (2005). Chronic obstructive pulmonary disease is associated with enhanced bronchial expression of FGF‐1, FGF‐2, and FGFR‐1. J Pathol 206: 28–38. [DOI] [PubMed] [Google Scholar]
- Kunz LI, Lapperre TS, Snoeck‐Stroband JB, Budulac SE, Timens W, van Wijngaarden S et al (2011). Smoking status and anti‐inflammatory macrophages in bronchoalveolar lavage and induced sputum in COPD. Respir Res 12: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara I, Lillehoj EP, Lu W, Singh IS, Isohama Y, Miyata T et al (2006). Neutrophil elastase induces IL‐8 gene transcription and protein release through p38/NF‐{kappa}B activation via EGFR transactivation in a lung epithelial cell line. Am J Physiol Lung Cell Mol Physiol 291: L407–L416. [DOI] [PubMed] [Google Scholar]
- Laan M, Bozinovski S, Anderson GP (2004). Cigarette smoke inhibits lipopolysaccharide‐induced production of inflammatory cytokines by suppressing the activation of activator protein‐1 in bronchial epithelial cells. J Immunol 173: 4164–4170. [DOI] [PubMed] [Google Scholar]
- Lee JM, Kim EK, Seo H, Jeon I, Chae MJ, Park YJ et al (2014). Serum amyloid A3 exacerbates cancer by enhancing the suppressive capacity of myeloid‐derived suppressor cells via TLR2‐dependent STAT3 activation. Eur J Immunol 44: 1672–1684. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kang EJ, Hur GY, Jung KH, Jung HC, Lee SY et al (2006). Peroxisome proliferator‐activated receptor‐gamma inhibits cigarette smoke solution‐induced mucin production in human airway epithelial (NCI‐H292) cells. Am J Physiol Lung Cell Mol Physiol 291: L84–L90. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP (2003). Redox regulation of PI 3‐kinase signalling via inactivation of PTEN. EMBO J 22: 5501–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Zhao J, Peng X, Liang J, Deng X, Chen Y (2012). The mechanism involved in the loss of PTEN expression in NSCLC tumor cells. Biochem Biophys Res Commun 418: 547–552. [DOI] [PubMed] [Google Scholar]
- Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y et al (2011). Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor‐2. Oncogene 30: 3887–3899. [DOI] [PubMed] [Google Scholar]
- Lim WT, Zhang WH, Miller CR, Watters JW, Gao F, Viswanathan A et al (2007). PTEN and phosphorylated AKT expression and prognosis in early‐ and late‐stage non‐small cell lung cancer. Oncol Rep 17: 853–857. [PubMed] [Google Scholar]
- Liu K, Anderson GP, Bozinovski S (2008a). DNA vector augments inflammation in epithelial cells via EGFR‐dependent regulation of TLR4 and TLR2. Am J Respir Cell Mol Biol 39: 305–311. [DOI] [PubMed] [Google Scholar]
- Liu K, Gualano RC, Hibbs ML, Anderson GP, Bozinovski S (2008b). Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem 283: 9977–9985. [DOI] [PubMed] [Google Scholar]
- Liu L, Liu J, Wang Y, Dai S, Wang X, Wu S et al (2011). A combined biomarker pattern improves the discrimination of lung cancer. Biomarkers 16: 20–30. [DOI] [PubMed] [Google Scholar]
- Liu YS, Luo XY, Li QR, Li H, Li C, Ni H et al (2012). Shotgun and targeted proteomics reveal that pre‐surgery serum levels of LRG1, SAA, and C4BP may refine prognosis of resected squamous cell lung cancer. J Mol Cell Biol 4: 344–347. [DOI] [PubMed] [Google Scholar]
- Lopez AD, Murray CC (1998). The global burden of disease, 1990–2020. Nat Med 4: 1241–1243. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R et al (2012). Ipilimumab in combination with paclitaxel and carboplatin as first‐line treatment in stage IIIB/IV non‐small‐cell lung cancer: results from a randomized, double‐blind, multicenter phase II study. J Clin Oncol 30: 2046–2054. [DOI] [PubMed] [Google Scholar]
- Maisonneuve E, Ducret A, Khoueiry P, Lignon S, Longhi S, Talla E et al (2009). Rules governing selective protein carbonylation. PLoS ONE 4: e7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris D, Tzanakis N, Damianaki A, Ntaoukakis E, Neofytou E, Zervou M et al (2008). Microsatellite DNA instability and COPD exacerbations. Eur Respir J 32: 612–618. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Thimmulappa R, Navas‐Acien A, Sandford A, Elliott M, Singh A et al (2008). Decline in NRF2‐regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ‐1. Am J Respir Crit Care Med 178: 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mallia P, Footitt J, Sotero R, Jepson A, Contoli M, Trujillo‐Torralbo MB et al (2012). Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 186: 1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg KJ, Arulampalam V, Ichihara F, Petersson M, Seki K, Andersson T et al (2001). Inhibition of activated/memory (CD45RO(+)) T cells by oxidative stress associated with block of NF‐kappaB activation. J Immunol 167: 2595–2601. [DOI] [PubMed] [Google Scholar]
- Mannino DM, Aguayo SM, Petty TL, Redd SC (2003). Low lung function and incident lung cancer in the United States: data from the first National Health and Nutrition Examination Survey follow‐up. Arch Intern Med 163: 1475–1480. [DOI] [PubMed] [Google Scholar]
- Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK et al (2005). PTEN expression in non‐small‐cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol 36: 768–776. [DOI] [PubMed] [Google Scholar]
- Minna JD, Fong K, Zochbauer‐Muller S, Gazdar AF (2002). Molecular pathogenesis of lung cancer and potential translational applications. Cancer J 8 (Suppl. 1): S41–S46. [PubMed] [Google Scholar]
- Mosser DM, Edwards JP (2008). Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz GT, Eppert BL, Wortham BW, Amos‐Kroohs RM, Flury JL, Wesselkamper SC et al (2010). Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease. J Immunol 184: 4460–4469. [DOI] [PubMed] [Google Scholar]
- Mu CY, Huang JA, Chen Y, Chen C, Zhang XG (2011). High expression of PD‐L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol 28: 682–688. [DOI] [PubMed] [Google Scholar]
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L et al (2007). Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13: 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Church DF, Pryor WA (1989). Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic Biol Med 7: 9–15. [DOI] [PubMed] [Google Scholar]
- Naylor EJ, Bakstad D, Biffen M, Thong B, Calverley P, Scott S et al (2007). Haemophilus influenzae induces neutrophil necrosis: a role in chronic obstructive pulmonary disease? Am J Respir Cell Mol Biol 37: 135–143. [DOI] [PubMed] [Google Scholar]
- Nowak D, Antczak A, Krol M, Pietras T, Shariati B, Bialasiewicz P et al (1996). Increased content of hydrogen peroxide in the expired breath of cigarette smokers. Eur Respir J 9: 652–657. [DOI] [PubMed] [Google Scholar]
- Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT (1998). Matrix metalloproteinase‐mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest 78: 1077–1087. [PubMed] [Google Scholar]
- Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P (2009). Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J 33: 118–126. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Kolls JK, Zheng Y (2008). The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28: 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Girard N (2011). New driver mutations in non‐small‐cell lung cancer. Lancet Oncol 12: 175–180. [DOI] [PubMed] [Google Scholar]
- Papayianni A, Serhan CN, Brady HR (1996). Lipoxin A4 and B4 inhibit leukotriene‐stimulated interactions of human neutrophils and endothelial cells. J Immunol 156: 2264–2272. [PubMed] [Google Scholar]
- Papi A, Casoni G, Caramori G, Guzzinati I, Boschetto P, Ravenna F et al (2004). COPD increases the risk of squamous histological subtype in smokers who develop non‐small cell lung carcinoma. Thorax 59: 679–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH et al (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastukh VM, Zhang L, Ruchko MV, Gorodnya O, Bardwell GC, Tuder RM et al (2011). Oxidative DNA damage in lung tissue from patients with COPD is clustered in functionally significant sequences. Int J Chron Obstruct Pulmon Dis 6: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS, Committee GS (2001). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 163: 1256–1276. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR . (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci A, Balbi B, Majori M, Cacciani G, Bertacco S, Alciato P et al (1998). Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J 12: 380–386. [DOI] [PubMed] [Google Scholar]
- Prause O, Bozinovski S, Anderson GP, Linden A (2004). Increased matrix metalloproteinase‐9 concentration and activity after stimulation with interleukin‐17 in mouse airways. Thorax 59: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor WA (1997). Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect 105 (Suppl. 4): 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor WA, Stone K (1993). Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 686: 12–27, discussion 27–18. [DOI] [PubMed] [Google Scholar]
- Purdue MP, Gold L, Jarvholm B, Alavanja MC, Ward MH, Vermeulen R (2007). Impaired lung function and lung cancer incidence in a cohort of Swedish construction workers. Thorax 62: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I (2012). Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim Biophys Acta 1822: 714–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, Adcock IM (2006). Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J 28: 219–242. [DOI] [PubMed] [Google Scholar]
- Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W et al (2002). 4‐Hydroxy‐2‐nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 166: 490–495. [DOI] [PubMed] [Google Scholar]
- Reppert S, Boross I, Koslowski M, Tureci O, Koch S, Lehr HA et al (2011). A role for T‐bet‐mediated tumour immune surveillance in anti‐IL‐17A treatment of lung cancer. Nat Commun 2: 600. [DOI] [PubMed] [Google Scholar]
- Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC et al (2001). Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 164: 469–473. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ et al (2015). Activity and safety of nivolumab, an anti‐PD‐1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non‐small‐cell lung cancer (CheckMate 063): a phase 2, single‐arm trial. Lancet Oncol 16: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo MS, Sokolova IA, Morrison LE, Zeng C, Baron AE, Hirsch FR et al (2003). Chromosomal abnormalities in non‐small cell lung carcinomas and in bronchial epithelia of high‐risk smokers detected by multi‐target interphase fluorescence in situ hybridization. J Mol Diagn 5: 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutgers SR, Postma DS, ten Hacken NH, Kauffman HF, van Der Mark TW, Koeter GH et al (2000). Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 55: 12–18. [DOI] [PubMed] [Google Scholar]
- Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE et al (1998). CD8+ T‐lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 157: 822–826. [DOI] [PubMed] [Google Scholar]
- Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO (2014). Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6: 221ra215. [DOI] [PubMed] [Google Scholar]
- Schmielau J, Finn OJ (2001). Activated granulocytes and granulocyte‐derived hydrogen peroxide are the underlying mechanism of suppression of t‐cell function in advanced cancer patients. Cancer Res 61: 4756–4760. [PubMed] [Google Scholar]
- Serhan CN (2005). Lipoxins and aspirin‐triggered 15‐epi‐lipoxins are the first lipid mediators of endogenous anti‐inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 73: 141–162. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE (2008). Resolving inflammation: dual anti‐inflammatory and pro‐resolution lipid mediators. Nat Rev Immunol 8: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao MX, Nadel JA (2005). Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF‐alpha‐converting enzyme. J Immunol 175: 4009–4016. [DOI] [PubMed] [Google Scholar]
- Shaykhiev R, Krause A, Salit J, Strulovici‐Barel Y, Harvey BG, O'Connor TP et al (2009). Smoking‐dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 183: 2867–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici‐Barel Y et al (2011). Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci 68: 877–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen N, Wang J, Zhao M, Pei F, He B (2011). Anti‐interleukin‐17 antibodies attenuate airway inflammation in tobacco‐smoke‐exposed mice. Inhal Toxicol 23: 212–218. [DOI] [PubMed] [Google Scholar]
- Skillrud DM, Offord KP, Miller RD (1986). Higher risk of lung cancer in chronic obstructive pulmonary disease. A prospective, matched, controlled study. Ann Intern Med 105: 503–507. [DOI] [PubMed] [Google Scholar]
- Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL et al (2002). Lack of PTEN expression in non‐small cell lung cancer could be related to promoter methylation. Clin Cancer Res 8: 1178–1184. [PubMed] [Google Scholar]
- Soriano C, Mukaro V, Hodge G, Ahern J, Holmes M, Jersmann H et al (2012). Increased proteinase inhibitor‐9 (PI‐9) and reduced granzyme B in lung cancer: mechanism for immune evasion? Lung Cancer 77: 38–45. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Liby KT (2012). NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 12: 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER (1993). Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal‐catalyzed reactions. Annu Rev Biochem 62: 797–821. [DOI] [PubMed] [Google Scholar]
- Stampfli MR, Anderson GP (2009). How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 9: 377–384. [DOI] [PubMed] [Google Scholar]
- Stanescu D, Sanna A, Veriter C, Kostianev S, Calcagni PG, Fabbri LM et al (1996). Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 51: 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ et al (1999). A seven‐transmembrane, G protein‐coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med 189: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JM, He QY, Guo RX, Chang XJ (2006). Phosphorylated Akt overexpression and loss of PTEN expression in non‐small cell lung cancer confers poor prognosis. Lung Cancer 51: 181–191. [DOI] [PubMed] [Google Scholar]
- Ueda K, Jinbo M, Li TS, Yagi T, Suga K, Hamano K (2006). Computed tomography‐diagnosed emphysema, not airway obstruction, is associated with the prognostic outcome of early‐stage lung cancer. Clin Cancer Res 12: 6730–6736. [DOI] [PubMed] [Google Scholar]
- Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC (2010). Enhanced effector function of cytotoxic cells in the induced sputum of COPD patients. Respir Res 11: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S (2013). Glutathione peroxidase‐1 as a novel therapeutic target for COPD. Redox Rep 18: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S (2014). Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol 5: 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A et al (2006). Differential protease, innate immunity, and NF‐kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol 290: L931–L945. [DOI] [PubMed] [Google Scholar]
- Vlahos R, Wark PA, Anderson GP, Bozinovski S (2012). Glucocorticosteroids differentially regulate MMP‐9 and neutrophil elastase in COPD. PLoS ONE 7: e33277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DE, Greene CM, Carroll TP, Taggart CC, Gallagher PM, O'Neill SJ et al (2001). Interleukin‐8 up‐regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF‐6 in human bronchial epithelium. J Biol Chem 276: 35494–35499. [DOI] [PubMed] [Google Scholar]
- Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM et al (2010). Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Science translational medicine 2: 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh TJ, Green RH, Richardson D, Waller DA, O'Byrne KJ, Bradding P (2005). Macrophage and mast‐cell invasion of tumor cell islets confers a marked survival advantage in non‐small‐cell lung cancer. J Clin Oncol 23: 8959–8967. [DOI] [PubMed] [Google Scholar]
- Willemse BW, ten Hacken NH, Rutgers B, Lesman‐Leegte IG, Postma DS, Timens W (2005). Effect of 1‐year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J 26: 835–845. [DOI] [PubMed] [Google Scholar]
- Wistuba II, Mao L, Gazdar AF (2002). Smoking molecular damage in bronchial epithelium. Oncogene 21: 7298–7306. [DOI] [PubMed] [Google Scholar]
- Yang P, Sun Z, Krowka MJ, Aubry MC, Bamlet WR, Wampfler JA et al (2008). Alpha1‐antitrypsin deficiency carriers, tobacco smoke, chronic obstructive pulmonary disease, and lung cancer risk. Arch Intern Med 168: 1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]