

Abstract
Microglia are critical nervous system‐specific immune cells serving as tissue‐resident macrophages influencing brain development, maintenance of the neural environment, response to injury and repair. As influenced by their environment, microglia assume a diversity of phenotypes and retain the capability to shift functions to maintain tissue homeostasis. In comparison with peripheral macrophages, microglia demonstrate similar and unique features with regards to phenotype polarization, allowing for innate immunological functions. Microglia can be stimulated by LPS or IFN‐γ to an M1 phenotype for expression of pro‐inflammatory cytokines or by IL‐4/IL‐13 to an M2 phenotype for resolution of inflammation and tissue repair. Increasing evidence suggests a role of metabolic reprogramming in the regulation of the innate inflammatory response. Studies using peripheral immune cells demonstrate that polarization to an M1 phenotype is often accompanied by a shift in cells from oxidative phosphorylation to aerobic glycolysis for energy production. More recently, the link between polarization and mitochondrial energy metabolism has been considered in microglia. Under these conditions, energy demands would be associated with functional activities and cell survival and thus, may serve to influence the contribution of microglia activation to various neurodegenerative conditions. This review examines the polarization states of microglia and their relationship to mitochondrial metabolism. Additional supporting experimental data are provided to demonstrate mitochondrial metabolic shifts in primary microglia and the BV‐2 microglia cell line induced under LPS (M1) and IL‐4/IL‐13 (M2) polarization.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- 2‐DG
2‐deoxy‐glucose
- AMPK
AMP‐activated PK
- BBB
blood–brain barrier
- CD172 (SIRP1A)
signal‐regulatory protein
- CD206
mannose receptor
- EAE
experimental autoimmune encephalomyelitis
- FA
fatty acid
- Fizz1
found in inflammatory zone 1
- HK
hexokinase
- iNOS
inducible NOS
- MHC‐II
major histocompatibility complex‐II
- NLRP
nucleotide‐binding oligomerization domain‐like receptor family pyrin domain‐containing
- NODs
nucleotide‐binding oligomerization domains
- PPP
pentose phosphate pathway
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SR
scavenger receptor
- TCA
tricarboxylic acid cycle
- TLR
Toll‐like receptor
Tables of Links
| TARGETS |
|---|
| Enzymes a |
| Akt |
| AMPK |
| Arg‐1, arginase 1 |
| Caspase‐1 |
| Histone demethylase |
| HK, hexokinase |
| iNOS |
| PI3K |
| GPCRs b |
| CCR2 |
| Catalytic receptors c |
| NLRC4 (IPAF) |
| NLRP1 |
| NLRP3 |
| TLR |
| LIGANDS | |
|---|---|
| CCL2 | IL‐6 |
| CCL17 | IL‐10 |
| CCL20 | IL‐13 |
| CCL22 | IL‐18 |
| CCL24 | Il‐21 |
| CX3CL1 | IL‐23 |
| CXCL13 | IL‐33 |
| IFN‐γ | LPS |
| IL‐1β | NO |
| IL‐3 | TGF‐β |
| IL‐4 | TNF‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a, 2013b, 2013c).
Introduction
The innate immune response of the body recruits a number of different cells to initiate a response to a novel stimulus such as a pathogen. These various cells of the immune system communicate and cooperate in a complex fashion to successfully complete their assigned tasks to clear the invading factor and return the system back to homeostasis. Such cells include circulating lymphocytes (T‐cells, B‐cells, NK cells) and monocytes that can develop into either dendritic cells or macrophages. They also include tissue associated bone marrow‐derived mast cells (effector cells of allergic reactions) and tissue‐specific macrophages. Within this arsenal, cells that phagocytose the initiating factor serve in a workhorse‐type capacity. In the periphery, these actions are primarily the function of bone marrow‐derived polymorphonuclear leucocytes that constantly circulate in the blood and in tissue‐specific macrophages that originate as monocytes from stem cells in the bone marrow. Once the macrophage has phagocytosed the material, it initiates intracellular processes that ensure the destruction of such engulfed material. As this function relates to the clearance of invading microbes, the cell can accomplish this task in two main ways, aerobically or anaerobically. The phagocyte can produce oxygen‐based chemicals that, by being reactive, can disrupt the microbe. This is often considered as an oxidative burst or respiratory burst. Alternatively, the cell can kill the microbe without oxygen by either increasing the acidity of the internal environment or by depriving the microbe of iron to inhibit metabolism.
Tissue‐specific macrophages can be found in virtually all tissues of the body and are representative of distinct classes (Gautier et al., 2012). In the CNS, microglia function as resident mononuclear phagocytes. In comparison with peripheral tissue macrophages and antigen‐presenting dendritic cells that originate from bone marrow‐derived monocytes (Parwaresch and Wacker, 1984; Fogg et al., 2006), microglia are derived from primitive yolk sac myeloid progenitors that actively seed the brain parenchyma during mid‐embryonic development (Alliot et al., 1999; Ginhoux et al., 2010). Similar to other tissue‐specific resident macrophages, microglia represent 10–15% of the total cell population within the brain parenchyma (Carson et al., 2006) and display a level of morphological heterogeneity across regions (Lawson et al., 1990; Mittelbronn et al., 2001; Harry and Kraft, 2012). As resident cells of the brain, microglia are involved in regulatory processes critical for tissue development, architectural refinement, maintenance of the neural environment, response to injury and subsequent remodelling/repair. Similar to macrophages, microglia mount an immune response to pathogens, monitor for tissue changes and maintain tissue homeostasis by clearing pathogens, dying cells, debris or aberrant proteins (Gehrmann et al., 1995; Bruce‐Keller, 1999; Stevens et al., 2007; Wake et al., 2009; Nagamoto‐Combs et al., 2010; Sierra et al., 2010; Tremblay et al., 2010; Olah et al., 2011; Paolicelli et al., 2011). It is thought that through such capacities, microglia play a role in brain development and in various neurological and neurodegenerative disorders (Kettenmann et al., 2011).
Under normal conditions, microglia assume a neural‐specific phenotype (Schmid et al., 2009) and retain a relative quiescent surveillance phenotype for constant monitoring of the brain parenchyma (Davalos et al., 2005; Nimmerjahn et al., 2005). Maintaining microglia in a relatively quiescent state is, in part, due to signals derived from neuronal‐ and astrocyte‐derived factors (Cardona et al., 2006; Neumann et al., 2009; Ransohoff and Cardona, 2010). This role is attributed to expression of several receptors on microglia including triggering receptor expressed on myeloid cells‐2, signal‐regulatory protein CD172 (SIRP1A) the chemokine CX3CL1, colony‐stimulating factor 1 receptor and CD200R (Wright et al., 2003; Kierdorf and Prinz, 2013). Healthy neurons accomplish regulatory tasks via secreted and membrane bound signals including CX3CL1 (Hoek et al., 2000; Barclay et al., 2002; Sunnemark et al., 2005; Lyons et al., 2007), CD200 (Hoek et al., 2000; Frank et al., 2006), neurotransmitters (Pocock and Kettenmann, 2007), neurotrophins and CD22 (Mott et al., 2004), which acts on the CD45R for negative regulation of microglia via inhibition of p44/45 MAPK (Tan et al., 2000). In addition, within the normal CNS environment, microglia express high levels of microRNA‐124, reducing expression of CD46, major histocompatibility complex‐II (MHC‐II) and CD11b serving to maintain the cells in a quiescent yet, surveillance state (Conrad and Dittel, 2011).
M1 polarization state of macrophages and microglia
Macrophages respond to endogenous stimuli generated following infection or injury and demonstrate both pathogenic and protective roles (Mills, 2012; Boche et al., 2013; Wynn et al., 2013). Upon appropriate stimulation, classically activated, pro‐inflammatory (M1) macrophages serve in the first line of defence of the innate immune system occurring often within the first few hours or days. Microglia share phenotypic characteristics with peripheral monocytes. This allows for innate immunological functions. They recognize harmful stimuli using a full array of immune receptors, such as toll‐like receptors (TLRs), nucleotide‐binding oligomerization domains (NODs), NOD‐like receptors and many scavenger receptors (SRs; Ransohoff and Perry, 2009; Ransohoff and Brown, 2012). Within injured tissue, microglia exist in various states of activation and retain the capability to shift their functional phenotype during the inflammatory response (Stout et al., 2005; Graeber, 2010). With injury, resident microglia or macrophages infiltrating from the circulation become polarized towards a pro‐inflammatory (M1) phenotype upon exposure to pro‐inflammatory cytokines IFN‐γ, TNF‐α and cellular or bacterial debris. These cells then produce pro‐inflammatory cytokines (TNF‐α, interleukin (IL)‐1β, IL‐12), present antigen, and express high levels of inducible NO (iNOS) for NO production (Gordon and Taylor, 2005; Villalta et al., 2009). This action is geared to kill the offending foreign pathogen and polarize T‐cells to mount an adaptive immune response. In many experimental models, the M1 response is characterized following exposure to bacterial‐derived products such as, LPS or signals associated with infection such as IFN‐γ (Martinez and Gordon, 2014). In the absence of microorganisms, a similar, but sterile inflammatory response occurs often as a result of trauma, ischaemia‐reperfusion injury or chemical exposure (Chen and Nunez, 2010; Shechter and Schwartz, 2013; McPherson et al., 2014). Like peripheral macrophages, microglia respond by producing M1 associated factors such as, pro‐inflammatory cytokines (IL‐1α, IL‐1β, IL‐6, IL‐12, IL‐23, TNF‐α), chemokines, redox molecules (NADPH oxidase, phagocytic oxidase, iNOS), SRs (macrophage receptor with collagenous structure), co‐stimulatory proteins (CD40) and MHC‐II (Hanisch and Kettenmann, 2007; Henkel et al., 2009; Ransohoff and Perry, 2009; Colton and Wilcock, 2010; Varnum and Ikezu, 2012; Boche et al., 2013). Early work on the MMGT12 murine microglia cell line (Briers et al., 1994) and primary microglia provided a wide‐ranging transcription and functional profile using the M1/M2 differentiation spectrum (Michelucci et al., 2009). An extensive transcription profile was examined including LPS or IFN‐γ‐induced M1 markers, such as IL‐1β, IL‐6, TNF‐α, NOS2, COX‐2, C‐C chemokines CCL2 and CCL20, and the receptor CCR2. Functional aspects of the polarization showed that phagocytosis was inhibited with M1 polarization. While the majority of work is in rodent microglia, primary microglia obtained from adult human brain can be induced to an M1 phenotype with LPS + IFN‐γ (Durafourt et al., 2012).
The outcome of a M1 polarizing event is dependent upon a number of features, not the least of which is whether the response includes a production of iNOS, reactive oxygen species (ROS) or activation of NOD‐like receptor family pyrin domain‐containing 3 (NLRP3) inflammasome complex (Bordt and Polster, 2014; de Rivero Vaccari et al., 2014). The NLRP3 inflammasome protein complex facilitates production of active caspase‐1 for generation of IL‐1β and IL‐18 from precursor proteins (Netea et al., 2014). NLRP1, NLRP3 and NLRC4 are primary complexes (Martinon et al., 2009) governing caspase‐1 activation for proteolytic processing and secretion of pro‐inflammatory cytokines (Labbe and Saleh, 2008; Salminen et al., 2008) to activate the full cytokine cascade. Limited data are starting to become available regarding the role of inflammasome activation in CNS injury (de Rivero Vaccari et al., 2014). A role for NLRP3 inflammasomes in IL‐1β release has been reported from LPS‐primed prion‐infected microglia (Shi et al., 2012). In lentivirus‐infected microglia, NLRP3 inflammasome activation is an early aspect of infection (Walsh et al., 2014). Chronic exposure to exogenous glucocorticoids primes microglia towards an exacerbated pro‐inflammatory response and induces NLRP3 within the hippocampus (Frank et al., 2013).
M2 polarization of macrophages and microglia
While the initial response of macrophages to injury has been known for some time, positive influences on tissue remodelling have been recognized more recently (Longbrake et al., 2007; Ruffell et al., 2009; Deng et al., 2012; Novak and Koh, 2013; Shechter and Schwartz, 2013; Shechter et al., 2013). In the early 1990s, the concept of macrophage alternative activation was developed largely based on work showing a role for IL‐4 in the induction of an alternative (M2) activation state (Stein et al., 1992) inducing expression of the anti‐inflammatory cytokines (Il4, Il10, Il13 and TGF‐β) as well as, arginase‐1 (Arg1), CD206 and Chitinase‐3‐like‐3 (Ym1 in rodents) (Colton, 2009; Henkel et al., 2009). M2 macrophages play a role in allergy response, parasite clearance, inflammatory dampening, tissue remodelling, angiogenesis, immunoregulation and tumour promotion (Sica and Mantovani, 2012). Upon further study, subclasses of M2 activation have been identified. The M2a activation state is induced by parasitic products or associated signals (IL‐4 and IL‐13) with a longer‐term function for resolution and repair (Rutschman et al., 2001; Gordon, 2003; Lawrence and Natoli, 2011; Mills, 2012; Wynn et al., 2013). In this case, signalling occurs through IL‐4 receptor α leading to inhibition of NF‐κB signalling induced by M1 activation. M2b polarization is observed with triggering of Fcγ receptors, TLRs and immune complexes (Martinez and Gordon, 2014). M2c polarization occurs in response to specific anti‐inflammatory factors such as, IL‐10, TGF‐β and glucocorticoids (Vodovoz et al., 1993; Gordon, 2003; Martinez et al., 2008; Morris, 2009; Mills, 2012). Other cytokines induce M2 polarization such as IL‐3 (Kuroda et al., 2009), IL‐21 (Pesce et al., 2006) and IL‐33, (Kurowska‐Stolarska et al., 2009) as well as the chemokines, CCL2 and CXCL4 (Roca et al., 2009; Gleissner et al., 2010). In addition, cells can shift from an M2b phenotype to a mixture of M1 and M2a/b (Lisi et al., 2014). M2 polarization of microglia is similar to peripheral macrophages (Fenn et al., 2012; Liu et al., 2012; Chhor et al., 2013; Freilich et al., 2013), generating different mRNA profiles for IL‐4 and IL‐10 stimulation including Arg1, Mmr, Ym1, found in inflammatory zone 1 (Fizz1) and Ppar (Michelucci et al., 2009). While these associations have been demonstrated in vitro, M2 is induced in vivo in sterile wounds in the absence IL‐4 or IL‐13 (Crane et al., 2014) suggesting an alternative stimulus. In this model, M2 macrophages were derived from M1 macrophages that matured into repair macrophages within the tissue after recruitment from the circulation (Italiani et al., 2014). Thus, the inherent phenotype of the cells may differ as a function of source and environment.
M2 macrophages facilitate resolution of inflammation through anti‐inflammatory factors (e.g. IL‐10, IL‐13, TGF‐β), VEGF, EGF, Arg1) to deactivate pro‐inflammatory cell phenotypes and re‐establish homeostasis (Gordon, 2003; Gordon and Martinez, 2010; Ortega‐Gómez et al., 2013). This includes production of IL‐10 to down‐regulate inflammatory cells, extracellular matrix protecting proteins like Ym1/2, ornithine, polyamines for wound repair and higher levels of receptors associated with phagocytosis (Martinez et al., 2009). IL‐10 induces STAT3 and downstream genes including Il10, Tgfb1, macrophage mannose receptor Mrc1 (Lang et al., 2002; Gordon, 2003). Upon STAT6 activation, induction occurred in Ym1, Mrc1 and Fizz1. Functional changes associated with M2 activation include increased engulfment of apoptotic cells by cells stimulated with IL‐10 (Ghigo et al., 2001; 2004; Benoit et al., 2008b; Michelucci et al., 2009). In contrast, macrophages from IL‐10 over expressing mice do not show enhanced phagocytosis, but are unable to clear Coxiella burnetii infection. They exhibit an M2‐type transcriptional programme with increased mRNA levels for Arg1, mannose receptor (Mr) and Ym1/2 and down‐modulated inflammatory markers (Meghari et al., 2008). Activation of the PPAR‐γ characterizes M2 polarization (Benoit et al., 2008a; Rajaram et al., 2010). M2 cells express chemokines (CCL17, CCL22 and CCL24) (Mantovani et al., 2004) and co‐express macrophage activation factor with CD68 or CD163 (Barros et al., 2013). The specificity of these markers remains in question. For example, CD163 is considered a M2‐specific marker (Buechler et al., 2000); yet, differential expression of CD163 has not been observed in human disease (Barros et al., 2013) or polarized macrophages (Kittan et al., 2013). In addition, the primary ‘marker’ for M2, Arg1, is also induced in M1 macrophages and expressed in some resident and mycobacteria‐infected macrophages (El Kasmi et al., 2008).
In a few studies, the effects , particularly on neuroprotection and repair, of M2 microglia have been demonstrated (see Cherry et al., 2014a). Butovsky et al. (2006) suggested that glatiramer acetate could induce microglia to express insulin‐like growth factor 1 for neuroprotective action. In a murine experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis, Ponomarev et al. (2007) reported a regulatory role for CNS‐derived IL‐4 in the induction of Ym1 protein and mRNA. This elevation occurred in the absence of NO production. Clinical signs of EAE were exacerbated in chimera mice deficient in IL‐4 suggesting an association with a diminished M2 microglia phenotype (Ponomarev et al., 2007). Based upon the temporal expression of M1‐ and M2‐related factors in a mouse spinal cord injury model, Kigerl et al. (2009) examined the effect of conditioned media from bone marrow‐derived macrophages on dorsal root ganglion cell survival and neurite outgrowth. Conditioned media from LPS + IFN‐γ induced macrophages damaged neurons while media from IL‐4‐induced macrophages‐stimulated neurite outgrowth. Additional studies are now being reported linking the M2 microglia phenotype to diminished injury and potential repair; however, the strongest data still remain from spinal cord injury models.
Polarization transitions
Injury‐induced inflammatory processes are dynamic, demonstrating spatial and temporal heterogeneity, with the possibility that individual cells express transitional phenotypes. It has been suggested that macrophages transition from a M1 phenotype to a more regulatory or anti‐inflammatory M2 phenotypes to promote positive functional outcomes and minimize scar tissue formation. Alternatively, subpopulations of macrophages within an injury environment may express specific phenotypes resulting in concurrent expression of M1‐ and M2‐related factors or mixed M1/M2 phenotypes (Ziegler‐Heitbrock et al., 2010; Pettersen et al., 2011; Wong et al., 2011; Vogel et al., 2013). In vitro studies indicate that human monocytes can polarize to a M1 phenotype then mature into a M2 phenotype as a function of sequential changes in culture conditions (Italiani et al., 2014). Following exposure to classic M1 signals, TLR ligands or IFN‐γ, M2 macrophages can be reprogrammed to express M1 genes (Stout et al., 2005; Mylonas et al., 2009). Recent work suggests that histone H3K27me3 demethylase Jumonji domain containing three was essential for microglial M2 polarization and M1 down‐regulation (Tang et al., 2014). While a shift to an M1 phenotype would be a relatively standard transition, it is considered rare that once activated, peripheral M1 cells would switch to an M2 phenotype. Rather, for peripheral immune cells, it is thought that M1 stage cells become terminal and die during the inflammatory response (Albina et al., 1989). However, it has also been shown that inflammatory monocyte‐derived M1 macrophages can undergo phenotype conversion and become tissue‐resident macrophages (Hashimoto et al., 2013; Yona et al., 2013). Alternatively, any apparent increase in M2 phenotype cells may be associated with the loss of NO‐producing cells and an increase in TGF‐β for amplification of M2 polarization. Thus, the question remains as to whether M1 and M2 macrophages are phenotypically distinct subpopulations that, within different stages of an inflammatory response, would perform different functions (Auffray et al., 2007; Nahrendorf et al., 2007) or rather, shift between functional phenotypes depending upon environmental signals (Arnold et al., 2007; Crane et al., 2014).
Resident microglia versus peripheral macrophages
Evaluation of the M1/M2 paradigm in the CNS becomes complex as compared with other tissues due to the presence of the blood–brain barrier (BBB) that prevents the infiltration of blood‐borne monocytes/macrophages. Physical injury or late‐stage disease states can lead to a disruption of the BBB allowing monocyte‐derived macrophages to infiltrate and influence the injury and repair process. However, a directed response of resident microglia alone can occur in the absence of infiltrating monocytes (Peng et al., 2008; Funk et al., 2011), which may be more reflective of early stages of neurodegenerative disorders. Distinguishing between in vivo signals of resident microglia and infiltrating cells that assume a brain macrophage phenotype remains a confounding factor in identifying signals unique to the CNS. It has been suggested that infiltrating cells would be more involved in severe inflammatory injuries, while resident microglia would focus on maintaining tissue homeostasis (Ginhoux and Jung, 2014). Thus, characterization of the stage of an inflammatory response would depend upon the contributing cell type for example microglia versus infiltrating blood‐borne monocytes.
While microglia and peripheral macrophages maintain many similar features, they remain uniquely different. Comparison of non‐activated microglia to peritoneal macrophages identified a significant number of similarities in gene transcript expression yet, also distinct differences. Of the genes highly enriched in microglia, several were classified as ‘sensome’ genes allowing cells to sense and interact with the local environment (Hickman et al., 2013). These included putative P2ry12, P2ry13, Tmem119, GPCR 34 (Gpr34), the 1‐type lectin receptor Siglec‐h, Trem2 and Cx3cr1. Additional unique transcripts included the enzyme hexosaminidase B (Hexb) and the antimicrobial peptides cathelicidin antimicrobial peptide (Camp) and neutrophilic granule proteins (Ngp). While macrophages expressed a number of ‘sensome’ genes, the enriched genes included those encoding for fibronectin, CXCL13 and the endothelin B receptor in contrast to microglia. In an elegant study set out to directly compare M1 and M2 polarization capabilities of human microglia and blood‐derived macrophages gene expression (PCR array; 26 M1 and 11 M2 genes), microglia were observed to be more restricted in their capacity to adopt an M2 phenotype and cytokine profile, compared with macrophages (Durafourt et al., 2012). Both macrophages and microglia showed a greater induction of gene expression in response to M1, compared with M2 polarization. The majority of genes differentially regulated in M1‐polarized macrophages were also observed in M1 microglia compared with their M2 counterparts. Some differences were observed when comparing the M1‐ and M2‐polarized cell populations. Comparison of M1 macrophages with M1 microglia demonstrated that macrophages over‐expressed antigen presentation markers, CD1A, 1B and 1C. HLA‐DM expression was also increased in M2 macrophages compared with M2 microglia. Upon M2 polarization, microglia and macrophages were found to express similar genes with the exception of CD64 that was observed in both M1 and M2 microglia.
Metabolism under polarization states
Increasing evidence suggests a role of metabolic reprogramming in the regulation of the innate inflammatory response. Modification of metabolic functions from a growth‐promoting capacity (M2) to a killing/inhibitory capacity (M1) allows macrophages to respond with appropriate functions in distinct contexts (Mills et al., 2000; Rodríguez‐Prados et al., 2010; Odegaard and Chawla, 2011; Biswas and Mantovani, 2012; Mills, 2012). Under normal oxygen conditions, cells obtain energy via two different mechanisms. In the first, glucose is converted to pyruvate via glycolysis, entering the mitochondrial tricarboxylic acid cycle (TCA) to produce ATP through oxidative phosphorylation (Dashty, 2013). Under hypoxic conditions, anaerobic glycolysis converts pyruvate into lactate. This metabolic switch is promoted by PI3K/Akt signalling and inhibited by AMP‐activated PK (AMPK) (Hardie, 2007) and IL‐10 (Murray, 2006). Recent evidence suggests that immune cells have the ability to switch from oxidative phosphorylation to aerobic glycolysis; not dissimilar to the Warburg effect seen in tumour cells (Warburg, 1956; Vander Heiden et al., 2009). In this shift, cells preferentially use glycolysis rather than catabolic mitochondrial pathways to conserve and generate metabolic resources that are necessary to meet demands of cellular proliferation and activation while, still producing a sufficient supply of ATP.
M1 polarization
In classically activated M1 macrophages and dendritic cells, metabolism is shifted towards glycolysis and NO and citrulline production. This switch increases glucose uptake and lactate production (Krawczyk et al., 2010; Rodríguez‐Prados et al., 2010) with activation of the pentose phosphate pathway (PPP) and decreased mitochondrial oxygen consumption (Haschemi et al., 2012). In M1 macrophages, the Krebs cycle intermediate, succinate, regulates hypoxia‐inducible factor 1α to drive a sustained production of IL‐1β (Galvan‐Peña and O'Neill, 2014). Increased glycolysis is permissive to quickly trigger microbiocidal activity and allows cells to survive in a hypoxic environment.
A key feature of M1 macrophages is associated with their production of ROS to facilitate killing of phagocytosed bacteria (West et al., 2011). Intracellular damage from ROS is limited due to the increased generation of NADPH required for maintenance of reduced glutathione (Kletzien et al., 1994; Salvemini et al., 1999) and also NO production (Bredt and Snyder, 1990; Knowles and Moncada, 1994). NO is synthesized by oxidation of L‐arginine by inducible NOS (iNOS) using the electrons supplied by NADPH. At high concentrations NO reversibly inhibits mitochondrial respiration by competing with O2 in cytochrome c oxidase. With reduced mitochondrial respiration ROS production, in the form of superoxide anion (O2 −), is increased and converted into H2O2 by superoxide dismutase (SOD)3. This then diffuses into the cytoplasm (Fukai et al., 2002). With prolonged production, NO can react with O2 − to produce peroxynitrite (ONOO−), irreversibly inhibiting the electron transport chain (Bolaños et al., 2004). NO inhibits the enzyme pyruvate dehydrogenase that converts pyruvate into acetyl CoA before entering the Krebs cycle (Klimaszewska‐Łata et al., 2014). Functionally, M1‐produced NO serves as an effector molecule with microbiocidal activity and the capacity to inhibit cell proliferation (MacMicking et al., 1997). Using a screening strategy in macrophages, it was shown that the specific modulation of glycolytic energy flux is critical to macrophage activation and is likely to define cell polarization (Haschemi et al., 2012). In this study, a number of non‐protein nutrient kinases were reported to have a contributory role including CARKL, a sedoheptulose kinase of the PPP, for repression of LPS‐induced macrophage activation. In primary murine macrophages, a decrease in CARKL expression accompanied M1 polarization with only a minimal increase in expression observed following IL‐4 or IL‐13 (Haschemi et al., 2012) .
The majority of the published data on the bioenergetics of polarization states has been generated in peripheral immune cells (Biswas and Mantovani, 2012; O'Neill and Hardie, 2013; Pearce and Pearce, 2013) with only a few studies examining microglia (Moss and Bates, 2001; Chenais et al., 2002; Bernhart et al., 2010; Sohn et al., 2012; Voloboueva et al., 2013; Gimeno‐Bayon et al., 2014). The majority of microglia studies were related to the generation of ROS (Innamorato et al., 2009; Ferger et al., 2010; Bordt and Polster, 2014; Chen et al., 2014). As in macrophages, microglia in the surveillance state are likely to rely on oxidative phosphorylation metabolism (Cherry et al., 2014b). Similar to other immune cells, when stimulated with TLR agonists (e.g. LPS), microglia switch from oxidative metabolism towards glycolytic metabolism (Voloboueva et al., 2013; Gimeno‐Bayon et al., 2014). Voloboueva et al. (2013) showed that upon stimulation by LPS (1 μg·mL−1, 3 h), BV‐2 cells increased lactate production concurrent with a decrease in mitochondrial oxygen consumption and ATP production as measured using an extracellular flux analyzer (Seahorse Bioscience, Billerica, MA, USA). This shift was modulated by the mitochondrial glucose‐regulated protein 75/mortalin. Over expression of 75/mortalin attenuated LPS‐induced oxidative and metabolic responses, as well as, suppressed pro‐inflammatory activation. From these findings the authors proposed that LPS‐induced elevations in glycolytic activity and lactate levels contributed to the associated pro‐inflammatory response. When the LPS (100 μg·mL−1) stimulus was augmented by IFN‐γ (0.5 μg·mL−1) a 24‐h exposure resulted in an increase in NO formation and a metabolic reprogramming of BV‐2 cells based on increased glucose consumption, hexokinase (HK) activity, glucose‐6‐phosphate dehydrogenase activity, phosphofructokinase‐1 activity, lactate dehydrogenase activity and lactate release, suggesting a potentiated glycolysis (Gimeno‐Bayon et al., 2014). Time‐lapsed confocal imaging of the inner membrane potential by the mitochondrial membrane potential‐sensitive dye TMRE showed that the cells maintained their electron transport chain usage. In comparison, stimulation with IL‐4 (0.5 μg·mL−1) for 24 h resulted in a reduction of glucose consumption and lactate production. The authors suggested that this shift is association with phagocytic actions of the cells and the reduction in the need for anabolic reactions. These data suggest that different metabolic programming is associated with the different phenotype states of microglia.
To provide specific experimental data on the bioenergetics profile of microglia following polarization, we examined specific features of the profile under different polarization states using a XF24 extracellular flux analyzer (Seahorse Bioscience; see Supporting Information for detailed methods). In murine BV‐2 microglia cells, LPS (100 μg·mL−1; 24 h) exposure resulted in an elevation in M1‐related pro‐inflammatory genes (Figure 1A). Under this M1 stimulatory condition, the cells shifted from a primary oxidative metabolic state towards glycolytic metabolism (Figure 1B–D) and showed no evidence of cell death. LPS‐polarized BV‐2 cells were unresponsive to the mitochondrial stressors oligomycin, FCCP and rotenone, highlighting the loss of mitochondrial function (Figure 1B). Similar to findings in the work of Voloboueva et al. (2013) shifting of BV‐2 cells to a glycolytic metabolism decreased mitochondrial oxygen consumption (OCR) (Figure 1C) and, as a consequence of lactate release, the cells increased their extracellular acidification rate (ECAR) (Figure 1D). Under the stimulatory conditions induced by LPS, the data suggest that in BV‐2 cells, a switch to glycolysis appears to serve as a survival response to maintain ATP levels, after inhibition of oxidative phosphorylation by NO. This finding is similar to that obtained by Everts et al., (2012) with dendritic cells (DCs).
Figure 1.
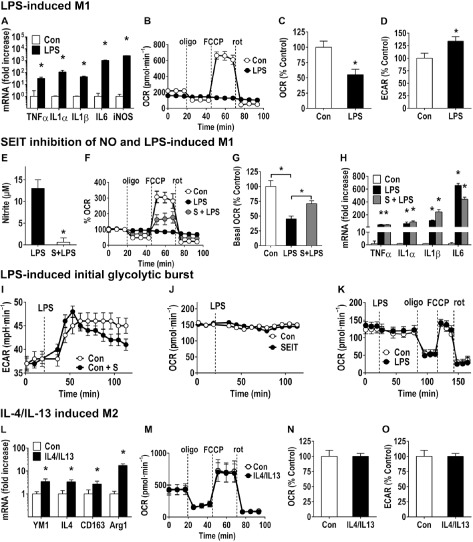
LPS and IL‐4/IL‐13 stimulation of BV‐2 cells. (A–D) LPS‐induced M1 phenotype of BV‐2 cells. BV‐2 cells were plated in six‐well tissue culture plates for mRNA (2 × 105 cells per well) or in 24‐well Seahorse plates (2.5 × 104 per well). 24 h post‐plating, cells were exposed to LPS (100 ng·mL−1 final concentration; 24 h) or media (Con). (A) Total RNA was isolated by Trizol and mRNA levels for M1‐related genes determined by qRT‐PCR (Supporting Information). Threshold cycle values were determined, GAPDH was used for normalization, and the mean fold changes over saline controls were calculated according to the 2−ΔΔC T method. Data represent mean ± SEM (n = 6). (B) Representative example of a bioenergetics profile (Seahorse Bioscience; Supporting Information) shows a normal response pattern for control BV‐2 cells (2.5 × 104 cells per well) for basal respiration (first three readings), and following addition of the mitochondrial stressors oligomycin (oligo; 0.75 μM), FCCP (0.75 μM) and rotenone (rot; 1 nM). LPS‐exposed cells showed a decrease in basal respiration and were unresponsive to the different mitochondrial stressors, suggesting an impairment of mitochondrial function. Calculation of (C) OCR and (D) ECAR as a percentage of control demonstrated a significant difference between controls and LPS‐exposed cells suggesting an increased extracellular acidification of the media. Data represent mean ± SEM calculated as a percentage of control (six to seven individual wells across three independent experiments) (n = 3). *P < 0.05, significantly different from control; Student's t‐test. (E–H) Response of BV‐2 cells to LPS following inhibition of iNOS with SEIT. BV‐2 cells (nitrite release; mRNA: 2 × 105 cells per well per six‐well; 24‐well Seahorse plate (2.5 × 104 per well) were pre‐exposed to the iNOS‐inhibitor, SEIT (S, 200 μM) for 1 h followed by LPS (100 ng·mL−1, 24 h) exposure. (E) LPS‐induced nitrite release into the media, as determined by Griess reaction, was significantly inhibited by SEIT (S). *P < 0.05, significantly different from LPS alone; Student's t‐test. (F) Seahorse bioenergetics profile showed that inhibition of iNOS partially blunted the mitochondrial impairment induced by LPS as demonstrated by the cellular response following FCCP. (G) Calculation of OCR indicated a significant decrease with LPS exposure and a blunting of this effect with SEIT. (H) mRNA levels for M1‐related genes as determined by qRT‐PCR showed no significant difference between cells exposed to LPS and those exposed to SEIT + LPS. Data represent mean ± SEM (n = 6). *P < 0.05, significantly different from control; anova with Bonferroni's test. (I–K) LPS‐induced polarization provokes a glycolytic burst in BV‐2 cells. (I–J) BV‐2 cells (2.5 × 104 per well Seahorse plate) were exposed to the iNOS‐inhibitor, SEIT (S; 1 h, 200 μM) or media (Con) followed by LPS (100 ng·mL−1 final concentration; 24 h). (I) Cells showed an increase in ECAR following LPS. SEIT dosed cells showed a decrease in ECAR over time as compared with controls suggesting a role for iNOS in maintaining elevated ECAR. (J) SEIT alone showed no effect on basal respiration (OCR). (K) To examine mitochondrial function during the initial LPS‐induced glycolytic burst, BV‐2 cells were administered LPS (100 μg·mL−1 final concentration) or media (Con) at the 20 min time point after recording basal respiration (line indicating dosing). No significant differences were observed in response to oligomycin, FCCP or rotenone, indicating that cells maintained normal mitochondrial function during the initial glycolytic burst. Data represent mean ± SEM calculated as a percentage of control (six to seven individual wells across three independent experiments) (n = 3). (L–N) IL4/IL13 induction of M2 phenotype. 24 h post‐plating, BV‐2 cells (mRNA: 2 × 105 cells per well per well plate; 2.5 × 104 per well per 24‐well Seahorse plate) were exposed to IL4/IL13 (10 ng·mL−1 final concentration of each) or media (Con) for 24 h. (L) Total RNA was isolated by Trizol and mRNA levels for M2‐related genes determined by qRT‐PCR (Supporting Information). Threshold cycle values were determined, GAPDH was used for normalization, and the mean fold changes over saline controls were calculated according to the 2−ΔΔC T method. Data represent mean ± SEM (n = 6). *P < 0.05, significantly different from control; Student's t‐test. (M) Seahorse bioenergetics profile shows no significant effect of M2 polarization by IL4/IL13 as compared with media controls and (N) OCR and (O) ECAR were not altered with the addition of IL4/IL13. Data represent mean ± SEM calculated as a percentage of control from seven independent wells from duplicate experiments.
The response of dendritic cells to LPS is characterized by a rapid increase in glycolytic flux that occurs within minutes of TLR activation, independent of NO (Everts et al., 2014). This serves to stimulate de novo synthesis of fatty acids (FA) and secretion of proteins critical for cell activation (Everts et al., 2014). In comparison, a similar phenomenon was observed in BV‐2 cells. To examine the role of NO in the metabolic shift of microglia, NO production in LPS‐stimulated BV‐2 cells was blocked by pre‐treatment with the iNOS‐inhibitor S‐ethyl‐isothiourea (SEIT). Pretreatment of cells with SEIT (S, 200 μM) for 1 h prior to LPS (100 μg·mL−1, 24 h) exposure was effective in inhibiting LPS‐induced nitrite production (Figure 1E), partially blunting mitochondrial impairment (Figure 1F and G). The inhibition of iNOS, however, did not alter the LPS‐induced elevation in mRNA levels for M1‐related genes (Figure 1H) and was ineffective in preventing the initial glycolytic burst observed within the first hour following LPS (Figure 1I). Under these conditions, OCR was not altered with the inhibition of NO (Figure 1J) suggesting an increase in glycolytic metabolism while maintaining mitochondrial function. Everts et al. (2014) reported that in DCs, the increase in glycolysis and the stimulation of pyruvate flux into the TCA contributes to an increased spare respiratory capacity. In contrast, while BV‐2 cells demonstrated an initial glycolytic burst, there was no indication of an increase in spare respiratory capacity (Figure 1K). However, similar to peripheral immune cells such as, dendritic cells, the transition from oxidative phosphorylation to glycolysis in microglia appears dependent upon NO.
BV‐2 cells offer significant advantages over primary microglia with regards to their ability to generate sufficient cells to conduct biochemical studies. However, they are limited and do have some significant differences as compared with primary microglia. To confirm if the findings on mitochondrial bioenergetics observed in BV‐2 cells would also be observed in primary microglia, we conducted a focused investigation in primary murine microglia following exposure to LPS (100 μg·mL−1, for 24 h). We demonstrated that under LPS stimulatory conditions, the primary microglia replicated the effects observed for BV‐2 cells (Figure 2) including a loss of mitochondrial function (Figure 2A), decreased basal respiration (Figure 2C) and increased ECAR (Figure 2D).
Figure 2.
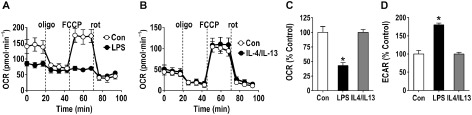
Representative mitochondrial function analysis of polarized primary microglia cells. Primary microglia cells were seeded in a Seahorse XF plate (Seahorse Bioscience, Supporting Information) at a density of 1.25 × 105 cells per well and polarized in situ for 24 h with LPS (100 ng·mL−1) or a combination of IL‐4 and IL‐13 (10 ng·mL−1 each). After signal stabilization (three measures) the cells were sequentially exposed to the mitochondrial stressors oligomycin (0.75 μM), FCCP (0.75 μM) and rotenone (rot, 1 μM). (A) Representative bioenergetics profile demonstrates a normal response pattern for control primary microglia. LPS‐exposed cells showed a decrease in basal respiration and were unresponsive to the different mitochondrial stressors, suggesting an impairment of mitochondrial function. (B) Following exposure to IL‐4/IL‐13, cells maintained a bioenergetics profile similar to controls. Calculation of (C) OCR and (D) ECAR as a percentage of control showed that LPS exposure decreased basal respiration, and increased extracellular acidification of the media, respectively, with no changes observed with IL‐4/IL‐13 exposure. Data represent mean ± SEM calculated as a percentage of control (seven individual wells each condition). *P < 0.05, significantly different from control; Student's t‐test. All studies were conducted under an animal protocol approved by National Institute of Environmental Health Sciences Animal Care and Use Committee.
Integration of the data on peripheral immune cells with the limited data on microglia lead to the proposal of a two‐stage process during activation following TLR signalling (Figure 3). It is proposed that, while in the initial stage, cells are capable of utilizing both oxidative and glycolytic metabolism yet, at the same time, activating the PPP. In the second stage, microglia shift to rely on glycolytic metabolism for survival and activation of the PPP.
Figure 3.
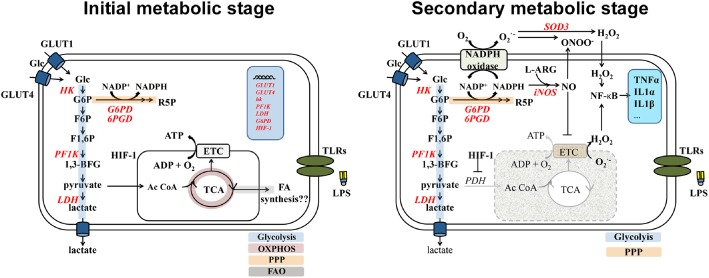
Schematic representation of LPS‐induced BV‐2 polarization. Activation of TLRs by LPS provokes dramatic changes in the metabolism of microglia, inducing a glycolytic switch that decreases mitochondrial O2 consumption and increases extracellular acidification via production of lactate. As observed in other immune cells (Everts et al., 2014), the data suggest that activation of microglia goes through two metabolic steps. In the first step, immediately after LPS stimulation, glycolytic metabolism is enhanced independently of NO production, increasing intracellular glucose (Glc) via glucose transporter (GLUT)‐1 and GLUT‐4 and production of several glycolytic enzymes. The PPP is induced via expression of its rate‐limiting enzyme, 6‐phosphogluconate dehydrogenase (G6PD). The electron transport chain (ETC) remains functional and the cells rely on oxidative phosphorylation and glycolysis for energy production. While LPS provokes a rapid glycolytic burst in microglia, there is little evidence of increased oxidative phosphorylation for synthesis of new molecules, as observed in DCs (Everts et al., 2014). In the second stage, NADPH, generated through the PPP, is used to produce ROS. H2O2 is then used as a bactericidal and also as a second messenger to modulate NF‐κB. In the presence of NADPH, iNOS oxidation of L‐arginine (L‐ARG) produces NO to inhibit Cytochrome c. In addition, HIF‐1α inhibits pyruvate dehydrogenase (PDH) and thus, the conversion of pyruvate into acetyl CoA, forcing a sole reliance on glycolysis for cell survival. As a consequence, mitochondrial dysfunction provokes the generation of additional mitochondrial ROS that are transported to the cytoplasm activating NF‐κB to exacerbate the pro‐inflammatory response. Abbreviations: 1,3‐BFG, 1,3‐biphosphoglycerate; ETC, electronic transport chain; FAO, fatty‐acid oxidation; F6P, fructose‐6‐phosphate; F‐1,6P, fructose‐1,6‐biphosphate; G6P, glucose‐6‐phosphate; NADP +, oxidized form of NADPH; OXPHOS, oxidative phosphorylation; PF1K, phosphofructose‐1‐kinase; R5P, ribose‐5‐phosphate; SOD3, superoxide dismutase 3.
M2 polarization
M2 macrophages use oxidative metabolism for the more long‐term functions involved in tissue repair and wound healing (Biswas and Mantovani, 2012). The M2 phenotype has also been considered as the ‘default’ polarization of resident macrophages (Murray and Wynn, 2011) with the production of ornithine to promote cell proliferation and repair through polyamine and collagen synthesis, fibrosis and tissue remodelling functions (Pesce et al., 2009). In M2 macrophages, glucose consumption is significantly lower as compared with M1 (Rodríguez‐Prados et al., 2010) and the sedoheptulose kinase CARKL is critical for regulating the PPP (Galvan‐Peña and O'Neill, 2014). IL‐4 induced M2 macrophages utilize FA oxidation and oxidative respiration for energy production (Odegaard and Chawla, 2011). Under these conditions, arginine metabolism is shifted to ornithine and polyamines (Mills et al., 2000). This contributes to phagocytosis by regulating energy demands and membrane fluidity.
Currently, there are limited data on the role of mitochondria as associated with M2 activation of microglia. Gimeno‐Bayon et al. (2014) reported that IL‐4‐stimulated (0.5 μg·mL−1; 24 h) BV‐2 cells decreased glucose consumption and lactate production. The authors suggested that this shift was association with phagocytic actions of the cells and the reduced need for anabolic reactions. When we examined the response of BV‐2 cells following stimulation with IL‐4/IL‐13 (10 μg·mL−1, 24 h), mRNA levels for a number of M2‐related genes were found to be elevated (Figure 1L). When the bioenergetic state was examined upon such stimulation, the cells were found to remain within an oxidative metabolic state (Figure 1M) maintaining OCR and ECAR at levels similar to non‐stimulated cells (Figure 1N,O). These observations are in contrast to peripheral macrophages where IL‐4 stimulates glucose uptake in addition to FA metabolism and mitochondrial biogenesis (Vats et al., 2006). When similar dynamics were examined in primary murine microglia, it was found that upon stimulation with IL‐4/IL‐13, cells remained within an oxidative metabolic state (Figure 2B). They showed no alteration in basal respiration and OCR (Figure 2C) and ECAR (Figure 2D) were similar to non‐stimulated cells. In peripheral macrophages, inhibition of mitochondrial respiration inhibits induction of arginase activity and minimizes the anti‐inflammatory effects of IL‐4 on LPS‐induced secretion of IL‐6 and TNF‐α (Vats et al., 2006). Ferger et al. (2010) found that non‐toxic doses of the mitochondrial electron transport chain inhibitors, rotenone or 3‐nitropropionic acid, impaired IL‐4 stimulation of M2‐related genes and inhibited LPS‐stimulated IL‐6 and TNF‐α release. In contrast to peripheral macrophages, primary microglia showed no inhibition of IL‐4 down‐regulation of LPS‐induced secretion of IL‐1β protein (Ferger et al., 2010). In mixed glia cultures, IL‐4 was found to enhance LPS‐induced IL‐1β production suggesting that, under these conditions, IL‐4 could activate the NLR inflammasome (Cao et al., 2007).
Modifications of the innate immune response
The innate immune system is shaped and conditioned to subsequent responses for days or months following activation or immunological signals (Netea et al., 2011; Kleinnijenhuis et al., 2012). This has been associated with increased non‐specific resistance to infectious agents following exposure to microbial agents. An augmented innate immune response occurring upon a secondary infection or challenge has been termed ‘trained innate immunity’ (Netea et al., 2011) and a lymphocyte‐independent shaping of innate immunity has been termed ‘memory’ (Kurtz and Franz, 2003; Kleinnijenhuis et al., 2012). Such a memory can occur in macrophages previously exposed to IFN‐γ with an elevated response to LPS (Nathan et al., 1984; Bosisio et al., 2002), potentially for a protective inflammatory response (Netea, 2013; Quintin et al., 2014). In contrast, preconditioning can occur with the development of endotoxin tolerance or hypo‐responsiveness to a subsequent challenge as a defence strategy to limit damage (Medzhitov et al., 2012). One could consider that stimuli encountered by microglia on a regular basis may serve to provide a pool of memory‐like cells enhancing performance upon a subsequent challenge. LPS‐induced tolerant macrophages express a transcriptional profile similar to M2 polarization with Il10, Arg1, Ccl17 and Ccl22 rather than a diminished M1 response, suggestive of a reorientation of function (Biswas and Lopez‐Collazo, 2009; Porta et al., 2009). While inhibition of mitochondrial function can be influenced by LPS stimulation, the reverse also occurs in that mitochondrial inhibition can influence the level of stimulation induced by LPS (Park et al., 2013). Questions remain as to whether microglia resume a normal functional phenotype once activated. If they conserve a memory of past inflammatory activation, do they shift response to a new challenge or react as naïve cells? Earlier work suggested that oxidative metabolism primed macrophages for a less‐inflammatory mode of activation (Vats et al., 2006). Thus, how this relates to mitochondrial functioning and shifts in metabolism with polarization warrants further examination.
Disruption of mitochondrial function is linked to a range of cellular effects in the nuclear genome including loss of heterozygosity, chromosome instability and epigenetic modifications (Veatch et al., 2009; Seoane et al., 2011). Thus, such shifts in the microglial activation state could lead to a broader cascade of effects as they relate to cell function over the lifespan. Warburg (1956) initially suggested that respiratory insufficiency was irreversible. However, the absence of cell death with microglia M1 polarization and down‐regulation to a quiescent phenotype with return to homeostasis suggests that microglia survive respiratory insufficiency. As an example, 2‐deoxy‐glucose (2‐DG), a glucose analogue that blocks glycolysis by inhibiting HK activity, is capable of blunting TNF‐α and IL‐6 production by inhibiting NF‐κB signalling in primary microglia (Wang et al., 2014). However, inhibition of the glycolytic metabolism in M1 microglia, during which the cell relies solely on glycolysis for survival, could prove fatal. For example, Lyons and Kettenmann (1998) reported that the substitution of glucose by 2‐DG under hypoxic conditions was lethal to 90% of cultured microglia. In a more recent study, Vilalta and Brown (2014) reported that 2‐DG killed microglia when co‐cultured with neurons. Rather than directly inhibiting the glycolytic metabolism, an alternative approach would involve preventing the metabolic switch or in enhancing oxidative metabolism. Something similar has been observed in DCs with the anti‐inflammatory cytokine, IL‐10 (Krawczyk et al., 2010) and may be translated to microglia. A better understanding of the inflammatory‐associated metabolic state and changes that occur with a polarization shift should help identify appropriate targets for modulating and regulating actions of microglia.
Future directions
Translation to chronic conditions
The current review focuses primarily on cellular responses initiated upon acute or short‐interval exposures. It is from such models that experimental data support an association between the activation state and cellular bioenergetics. These events are likely to reflect those that occur in brain or spinal cord injury with traumatic events or stroke. However, gaining a better understanding of the acute response capability of microglia may become critical for understanding associations with neurodegenerative diseases. This will require a better understanding of the complex nature and heterogeneity of cellular responses as they occur within more chronic conditions. The association of neuroinflammation and activation state with various neurodegenerative diseases including Alzheimer's disease (Varnum and Ikezu, 2012; Tang and Le, 2015), Parkinson's disease (Blandini, 2013; Kannarkat et al., 2013; Moehle and West, 2014), Huntington disease (Ellrichmann et al., 2013; Crotti et al., 2014), amyotrophic lateral sclerosis (Evans et al., 2013; Zhao et al., 2013; Hooten et al., 2015), prion disease (Gómez‐Nicola et al., 2014) and multiple sclerosis (Goldmann and Prinz, 2013; Strachan‐Whaley et al., 2014) suggests a contribution to disease progression (Boche et al., 2013). Whether the contribution of microglia actions in progressive neurodegenerative diseases is associated with an elevation in the M1 pro‐inflammatory phenotype or a diminished ability of the cells to differentiate into an M2‐type phenotype remains an issue under current study. Given the diverse pathological patterns of each of these diseases, questions remain as to whether the contribution of microglia and the associated inflammatory response follows a general pattern or a level of specificity as may be influenced by the environmental niche. In addition, it is not clear if responses of individual microglia within such disease states may follow an acute response pattern or a prolonged cellular shift. In human patients, data are limited with regards establishing a temporal progression of cellular changes and inflammatory responses. Restrictions also exist in the various animal models of specific aspects of human neurodegenerative diseases with regards to examination of the temporal aspect of associations with classical pro‐inflammatory activation and alternative or repair associated phenotypes. In addition, within such disease‐oriented conditions there exists the potential confounding factor of a perivascular macrophage or infiltrating blood‐borne immune cell contribution occurring with tissue degeneration (Mildner et al., 2011; Funk et al., 2013). This then brings an entirely different dynamics to the environment as compared with a response limited to resident microglia (Jung and Schwartz, 2012; Yamasaki et al., 2014). Hypotheses have been put forward that (i) under chronic degenerative conditions, microglia maintain a high pro‐inflammatory state leading to an enhanced and prolonged generation of low MW mediators such as NO, ONOO−, and ROS, and (ii) that pro‐inflammatory molecules impair the ability of microglia to clear excess or aberrant proteins, which could then extend the stimulating environment. How either of these two situations would result in an alteration of the bioenergetics of microglia has yet to be examined; however, it is likely that the ability of microglia to shift their bioenergetic profile will significantly influence the final outcome (Urrutia et al., 2014).
Classification for microglia phenotyping
The initial concept of individual macrophages having either a M1 or M2 phenotype has effectively set a framework for experimental examination of inflammation (Figure 4). Currently available data demonstrate a more complex phenotyping, especially in vivo with influence of various cell types comprising the inflammatory niche.
Figure 4.
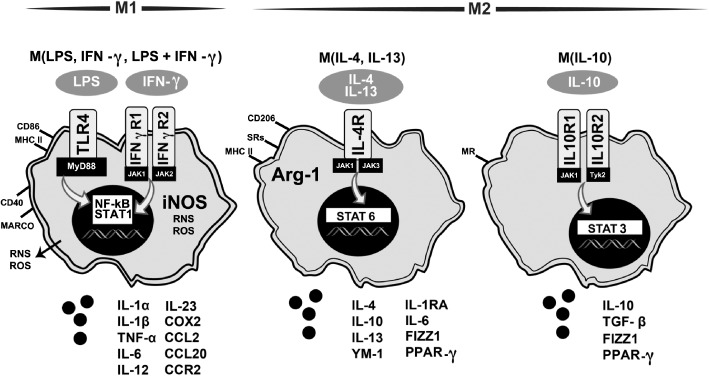
Diagram of activation states of microglia based on inflammatory profile and effector function. Based upon peripheral macrophage nomenclature, M1 and M2 polarization states of microglia have been proposed as a framework to evaluate the heterogeneity of responses (Colton, 2009; Mosser and Edwards, 2008) with recent evaluations suggesting a framework focused on the inducing stimuli (Martinez and Gordon, 2014; Murray et al., 2014). This diagram is based upon these papers and adapted from Martinez and Gordon (2014). Under the classic M1 state, exposure to LPS and/or IFN‐γ stimulates TLR4 or IFN‐γ receptors 1 and 2, respectively, leading to activation of transcription factors NF‐κΒ and STAT1 and increased expression of CD86 and MHC‐II. The increase in iNOS produces a burst of ROS and reactive nitrogen species (RNS) and the release of pro‐inflammatory cytokines, such as IL‐1α, IL‐1β, TNF‐α, IL‐6, IL‐12, IL‐23, the chemokines CCL2 and CCL20, and the receptor CCR2, macrophage receptor with collagenous structure (MARCO) and COX2.
M2 states have been proposed following various stimulatory factors. For example, upon stimulation with IL‐4/IL‐13, binding of the IL‐4 receptor (IL‐4R) initiates activation of STAT6, shifting the cells towards an anti‐inflammatory phenotype with an increase in Arg‐1, expression of CD206 and mannose receptor (MR), and release of anti‐inflammatory factors (IL‐4, IL‐6, IL‐10, IL‐13, IL‐1RA, FIZZ1, and PPARγ. Exposure of the cells to IL‐10 activates STAT6 via stimulation of IL‐10 receptors 1 and 2. This serves to shift the cells to a primary immunosuppressive state with an expression of CD206 and the release of IL‐10, TGF‐β, FIZZ1 and PPARγ.
A transcriptomic analysis of macrophages following discrete stimulation show activation pathways outside the standard M1/M2 polarization paradigm (Hume and Freeman, 2014) recommending the use of a combination of markers rather than isolated canonical markers of any specific activation state. It is becoming clear that any effort to determine whether or not macrophages exist within distinct activation or polarized states cannot rely on only one or two ‘markers’, but rather will require the examination of several markers including the mitochondrial bioenergetics of the cell. Further proposals for advancing our understanding of macrophage states and the heterogeneity of cellular responses include recommendations for the use of nomenclature linked to the activation standards rather than an M1/M2 classification (Murray et al., 2014). To translate these recommendation to microglia, efforts towards characterization would include assessments of morphological phenotype, discrimination between resident and infiltrating macrophages, metabolism, and where possible, functional features of the cells. Gaining a better understanding of the link between mitochondrial function and inflammation will support any future efforts to develop therapeutic approaches to support the normal and well‐regulated function of these dynamic cells.
Conflict of interest
None.
Supporting information
Figure S1 Representative schematic of mitochondrial function analysis using the Seahorse Bioscience extracellular flux analyzer (XF24) (Seahorse Bioscience, Billerica, MA, USA).
Table S1 Quantitative real‐time PCR primers sequences.
Acknowledgements
This work was supported by the Division National Toxicology Program, National Institute of Environmental Health Sciences (nos. 1Z01ES101623 and ES021164).
Orihuela, R. , McPherson, C. A. , and Harry, G. J. (2016) Microglial M1/M2 polarization and metabolic states. British Journal of Pharmacology, 173: 649–665. doi: 10.1111/bph.13139.
References
- Albina JE, Caldwell MD, Henry WL Jr, Mills CD (1989). Regulation of macrophages functions by L‐arginine. J Exp Med 169: 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B (1999). Microglia derived from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117: 145–152. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba‐Amer Y, van Rooijen N, Plonquet A et al (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into anti‐inflammatory macrophages to support myogenesis. J Exp Med 204: 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join‐Lambert O, Kayal S et al (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317: 666–670. [DOI] [PubMed] [Google Scholar]
- Barclay AN, Wright GJ, Brooke G, Brown MH (2002). CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol 23: 285–290. [DOI] [PubMed] [Google Scholar]
- Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G (2013). Macrophage polarisation: an immunohistochemical approach for identifying M1and M2 macrophages. PLoS ONE 8: e80908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M, Desnues B, Mege JL (2008a). Macrophage polarization in bacterial infections. J Immunol 181: 3733–3739. [DOI] [PubMed] [Google Scholar]
- Benoit M, Ghigo E, Capo C, Raoult D, Mege JL (2008b). The uptake of apoptotic cells drives Coxiella burnetii replication and macrophage polarization: a model for Q fever endocarditis. PLoS Path 5: e1000066. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bernhart E, Kollroser M, Rechberger G, Rechberger G, Reicher H, Heinemann A et al (2010). Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 10: 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Lopez‐Collazo E (2009). Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immun 30: 475–487. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Mantovani A (2012). Orchestration of metabolism by macrophages. Cell Metab 15: 432–437. [DOI] [PubMed] [Google Scholar]
- Blandini F (2013). Neural and immune mechanisms in the pathogenesis of Parkinson's disease. J Neuroimmune Pharmacol 8: 189–201. [DOI] [PubMed] [Google Scholar]
- Boche D, Perry VH, Nicoll JA (2013). Review: activation patterns of microglia and their identification in the human brain. Neuropath Appl Neurobiol 39: 3–18. [DOI] [PubMed] [Google Scholar]
- Bolaños JP, Garcia‐Nogales P, Almeida A (2004). Provoking neuroprotection by peroxynitrite. Curr Pharm Design 10: 867–877. [DOI] [PubMed] [Google Scholar]
- Bordt EA, Polster BM (2014). NADPH oxidase‐ and mitochondria‐derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radical Biol Med 76c: 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR et al (2002). Stimulation of toll‐like receptor 4 expression in human mononuclear phagocytes by interferon‐gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 99: 3427–3431. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH (1990). Isolation of nitric oxide synthetase, a calmodulin‐requiring enzyme. Proc Natl Acad Sci USA 87: 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briers TW, Desmaretz C, Vanmechelen E (1994). Generation and characterization of mouse microglial cell lines. J Neuroimmunol 52: 153–164. [DOI] [PubMed] [Google Scholar]
- Bruce‐Keller AJ (1999). Microglial‐neuronal interactions in synaptic damage and recovery. J Neurosci Res 58: 191–201. [DOI] [PubMed] [Google Scholar]
- Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G (2000). Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro‐ and anti‐inflammatory stimuli. J Leukoc Biol 67: 97–103. [PubMed] [Google Scholar]
- Butovsky O, Koronyo‐Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H (2006). Glatiramer acetate fights against Alzheimer's disease by inducing dendritic‐like microglia expressing insulin‐like growth factor 1. Proc Natl Acad Sci U S A 103: 11784–11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Fei L, Chang TT, DeLeo JA (2007). Induction of interleukin‐1beta by interldukin‐4 in lipopolysaccharide‐treated mixed glial cultures: microglial‐dependent effects. J Neurochem 102: 408–419. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9: 917–924. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC (2006). CNS immune privilege: hiding in plain sight. Immuno Rev 213: 48–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Lin JT, Cheng YF, Kuo CY, Huang CF, Kao SH et al (2014). Amelioration of LPS‐induced inflammation response in microglia by AMPK activation. BioMed Red Int'l 2014: 692061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G (2010). Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenais B, Morjani H, Drapier JC (2002). Impact of endogenous nitric oxide on microglial cell energy metabolism and labile iron pool. J Neurochem 81: 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O'Banion MK (2014a). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11: 98. doi: 10.1186/1742‐2094‐11‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O'Banion MK (2014b). Are ‘resting’ microglia more ‘m2'? Front Immunol 5: 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J et al (2013). Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro . Brain Behav Immun 32: 70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton C, Wilcock DM (2010). Assessing activation states in microglia. CNS Neurol Disorders Drug Targets 9: 174–191. [DOI] [PubMed] [Google Scholar]
- Colton CA (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 4: 399–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad AT, Dittel BN (2011). Taming of macrophage and microglial cell activation by microRNA‐124. Cell Res 21: 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane MJ, Daley JM, vanHoutte O, Brancato SK, Henry WL Jr, Albina JE (2014). The monocyte to macrophage transition in the murine sterile wound. PLoS ONE 9: e86660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti A, Benner C, Kerman BE, Gosselin D, Lagier‐Tourenne C, Zuccato C et al (2014). Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage‐determining factors. Nat Neurosci 17: 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashty M (2013). A quick look at biochemistry: carbohydrate metabolism. Clin Biochem 46: 1339–1352. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005). ATP mediates rapid microglial response to local brain injury in vivo . Nature Neurosci 8: 752–758. [DOI] [PubMed] [Google Scholar]
- Deng B, Wehling‐Henricks M, Villalta SA, Wang Y, Tidball JG (2012). IL‐10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol 189: 3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durafourt BA1, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC et al (2012). Comparison of polarization properties of human adult microglia and blood‐derived macrophages. Glia 60: 717–727. [DOI] [PubMed] [Google Scholar]
- El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao‐Tamayo M et al (2008). Toll‐like receptor‐induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9: 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrichmann G, Reick C, Saft C, Linker RA (2013). The role of the immune system in Huntington's disease. Clin Dev Immunol 2013: 541259. doi: 10.1155/2013/541259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MC, Couch Y, Sibson N, Turner MR (2013). Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol Cell Neurosci 53: 34–41. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE et al (2012). Commitment to glycolysis sustains survival of NO‐producing inflammatory dendritic cells. Blood 120: 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH (2014). TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol 15: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Henry CJ, Huang Y, Dugan A, Godbout JP (2012). Lipopolysaccharide‐induced interleukin (IL)‐4 receptor‐alpha expression and corresponding sensitivity to the M2 promoting effects of IL‐4 are impaired in microglia of aged mice. Brain Behav Immun 26: 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, Ludolph AC et al (2010). Effects of mitochondrial dysfunction on the immunological properties of microglia. J Neuroinflamm 7: 45. doi: 10.1186/1742‐2094‐7‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR et al (2006). A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311: 83–87. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF (2006). mRNA up‐regulation of MHC II and pivotal pro‐inflammatory genes in normal brain aging. Neurobiol Aging 27: 717–722. [DOI] [PubMed] [Google Scholar]
- Frank MG, Hershman SA, Weber MD, Watkins LR, Maier SF (2013). Chronic exposure to exogenous glucocorticoids primes microglia to pro‐inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 40: 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilich RW, Woodbury ME, Ikezu T (2013). Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE 8: e79416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai T, Folz RJ, Landmesser U, Harrison DG (2002). Extracellular superoxide dismutase and cardiovascular disease. Cardiovas Res 55: 239–249. [DOI] [PubMed] [Google Scholar]
- Funk JA, Gohlke J, Kraft AD, McPherson CA, Collins JB, Harry GJ (2011). Voluntary exercise protects hippocampal neurons from trimethyltin injury: possible role of interleukin‐6 to modulate tumor necrosis factor receptor‐mediated neurotoxicity. Brain Behav Immun 25: 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk N, Wieghofer P, Grimm S, Schaefer R, Buhring HJ, Gasser T (2013). Characterization of peripheral hematopoietic stem cells and monocytes in Parkinson's disease. Mov Disord 28: 392–395. [DOI] [PubMed] [Google Scholar]
- Galvan‐Peña S, O'Neill LA (2014). Metabolic reprograming in macrophage polarization. Front Immunol 5: 420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S et al (2012). Gene‐ expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13: 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW (1995). Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20: 269–287. [DOI] [PubMed] [Google Scholar]
- Ghigo E, Capo C, Raoult D, Mege JL (2001). Interleukin‐10 stimulates Coxiella burnetii replication in human monocytes through tumor necrosis factor down‐modulation: role in microbicidal defect of Q fever. Infect Immun 69: 2345–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigo E, Honstettre A, Capo C, Gorvel JP, Raoult D, Mege JL (2004). Link between impaired maturation of phagosomes and defective Coxiella burnetii killing in patients with chronic Q fever. J Infect Dis 190: 1767–1772. [DOI] [PubMed] [Google Scholar]
- Gimeno‐Bayon J, Lopez‐Lopez A, Rodriguez MJ, Mahy N (2014). Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci 92: 723–731. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Jung S (2014). Monocytes and macrophages: development pathways and tissue homeostasis. Nat Rev Immunol 14: 392–404. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleissner CA, Shaked I, Little KM, Ley K (2010). CXC chemokine ligand 4 induces a unique transcriptome in monocyte‐derived macrophages. J Immunol 184: 4810–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Nicola D, Schetters ST, Perry VH (2014). Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 62: 1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Prinz M (2013). Role of microglia in CNS autoimmunity. Clin Dev Immunol 2013: 208093. doi: 10.1155/2013/208093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S (2003). Alternative activation of macrophages. Nat Rev Immunol 3: 23–35. [DOI] [PubMed] [Google Scholar]
- Gordon S, Martinez FO (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32: 593–604. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR (2005). Monocyte and macrophage heterogeneity. Nat Rev Immunol 5: 953–964. [DOI] [PubMed] [Google Scholar]
- Graeber MB (2010). Changing face of microglia. Science 330: 783–788. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature Neurosci 10: 1387–1394. [DOI] [PubMed] [Google Scholar]
- Hardie DG (2007). AMP‐activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8: 774–785. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Kraft AD (2012). Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology 33: 191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haschemi A1, Kosma P, Gille L, Evans CR, Burant CF, Starkl P et al (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 15: 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M et al (2013). Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38: 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Zhao W, Appel SH (2009). Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol 4: 389–398. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Want LC, Means TK et al (2013). The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM et al (2000). Down‐regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290: 1768–1771. [DOI] [PubMed] [Google Scholar]
- Hooten KG, Beers DR, Zhao W, Appel SH (2015). Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics 12: 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume DA, Freeman TC (2014). Transcriptomic analysis of mononuclear phagocyte differentiation and activation. Immunol Rev 262: 74–84. [DOI] [PubMed] [Google Scholar]
- Innamorato NG, Lastres‐Becker I, Cuadrado A (2009). Role of microglial redox balance in modulation of neuroinflammation. Curr Opin Neurol 22: 308–314. [DOI] [PubMed] [Google Scholar]
- Italiani P, Mazza EM, Lucchesi D, Cifola I, Gemelli C, Grande A et al (2014). Transcriptomic profiling of the development of the inflammatory response in human monocytes in vitro . PLoS ONE 9: e87680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Schwartz M (2012). Non‐identical twins – microglia and monocyte‐derived macrophages in acute injury and autoimmune inflammation. Front Immunol 3: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannarkat GT, Boss JM, Tansey MG (2013). The role of innate and adaptive immunity in Parkinson's disease. J Parkinson Dis 3: 493–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch U‐K, Noda M, Verkhratsky A (2011). Physiology of microglia. Physiol Rev 91: 461–553. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M (2013). Factors regulating microglia activation. Front Cell Neurosci 7: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29: 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA et al (2013). Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 8: e78045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S et al (2012). Bacille Calmette‐Guerin induces NOD2‐dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA 109: 17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kletzien RF, Harris PK, Foellmi LA (1994). Glucose‐6‐phosphate dehydrogenase: a ‘housekeeping’ enzyme subject to tissue‐specific regulation by hormones, nutrients, and oxidant stress. FASEB J 8: 174–181. [DOI] [PubMed] [Google Scholar]
- Klimaszewska‐Łata J, Gul‐Hinc S, Bielarczyk H, Ronowska A, Zyśk M, Grużewska K et al (2014). Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J Neurochem 133: 284–297. [DOI] [PubMed] [Google Scholar]
- Knowles RG, Moncada S (1994). Nitric oxide synthases in mammals. Biochem J 298 (Pt 2): 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ et al (2010). Toll‐like receptor‐induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115: 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda E, Ho V, Ruschmann J, Antignano F, Hamilton M, Rauh MJ et al (2009). SHIP represses the generation of IL‐3‐induced M2 macrophages by inhibiting IL‐4 production from basophils. J Immunol 183: 3652–3660. [DOI] [PubMed] [Google Scholar]
- Kurowska‐Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S et al (2009). IL‐33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 183: 6469–6477. [DOI] [PubMed] [Google Scholar]
- Kurtz J, Franz K (2003). Innate defence: evidence for memory in invertebrate immunity. Nature 425: 37–38. [DOI] [PubMed] [Google Scholar]
- Labbe K, Saleh M (2008). Cell death in the host response to infection. Cell Death Differ 15: 1339–1349. [DOI] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ (2002). Shaping gene expression in activated and resting primary macrophages by IL‐10. J Immunol 169: 2253–2263. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Natoli G (2011). Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11: 750–761. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neurosci 39: 151–170. [DOI] [PubMed] [Google Scholar]
- Lisi L, Stigliano E, Lauriola L, Navarra P, Dello Russo C (2014). Proinflammatory‐activated glioma cells induce a switch in microglial polarization and activation status, from a predominant M2b phenotype to a mixture of M1 and M2a/B polarized cells. ASN Neuro 6: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HC, Zheng MH, Du YL, Wang L, Kuang F, Qin HY et al (2012). N9 microglial cells polarized by LPS and IL4 show differential responses to secondary environmental stimuli. Cell Immunol 278: 84–90. [DOI] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG (2007). Characterization and modeling of monocyte‐derived macrophages after spinal cord injury. J Neurochem 102: 1083–1094. [DOI] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA (2007). CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL‐4. J Neurosci 27: 8309–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SA, Kettenmann H (1998). Oligodendrocytes and microglia are selectively vulnerable to combined hypoxia and hypoglycemia injury in vitro . J Cereb Blood Flow Metabol 18: 521–530. [DOI] [PubMed] [Google Scholar]
- MacMicking J, Xie QW, Nathan C (1997). Nitric oxide and macrophage function. Annu Rev Immunol 15: 323–350. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25: 677–686. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000 Prime Rep 6: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Sica A, Mantovani A, Locati M (2008). Macrophage activation and polarization. Front Biosci 13: 453–461. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S (2009). Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27: 451–483. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J (2009). The inflammasomes: guardians of the body. Ann Rev Immunol 27: 229–265. [DOI] [PubMed] [Google Scholar]
- McPherson CA, Merrick BA, Harry GJ (2014). In vivo molecular markers for pro‐inflammatory cytokine M1 stage and resident microglia in trimethyltin‐induced hippocampal injury. Neurotox Res 25: 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Schneider DS, Soares MP (2012). Disease tolerance as a defense strategy. Science 335: 936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghari S, Bechah Y, Capo C, Lepidi H, Raoult D, Murray PJ et al (2008). Persistent Coxiella burnetii infection in mice overexpressing IL‐10: an efficient model for chronic Q fever pathogenesis. PLoS Pathog 4: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P (2009). Characterization of the microglial phenotype under specific pro‐inflammatory and anti‐inflammatory conditions: effects of oligomeric and fibrillar amyloid‐beta. J Neuroimmunol 210: 3–12. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP et al (2011). Distinct and non‐redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci 31: 11159–11171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD (2012). M1and M2 macrophages: oracles of health and disease. Crit Rev Immunol 32: 463–488. [DOI] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM (2000). M‐1/M‐2 macrophages and the Th1/Th2 paradigm. J Immunol 164: 6166–6173.10843666 [Google Scholar]
- Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R (2001). Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropath 101: 249–255. [DOI] [PubMed] [Google Scholar]
- Moehle MS, West AB (2014). M1 and M2 immune activation in Parkinson's disease: foe and ally? Neurosci S0306‐4522 00975‐0. doi: 10.1016/j.neuroscience.2014.11.018. [DOI] [PMC free article] [PubMed]
- Morris SM (2009). Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol 157: 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DW, Bates TE (2001). Activation of murine microglial cell lines by lipopolysaccharide and interferon‐gamma causes NO‐mediated decreases in mitochondrial and cellular function. Eur J Neurosci 13: 529–538. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP (2008). Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott RT, Ait‐Ghezala G, Town T, Mori T, Vendrame M, Zeng J et al (2004). Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 46: 369–379. [DOI] [PubMed] [Google Scholar]
- Murray PJ (2006). Understanding and exploiting the endogenous interleukin‐10/STAT3‐mediated anti‐inflammatory response. Curr Opin Pharmacol 6: 379–386. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA (2011). Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S et al (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonas KJ, Nair MG, Prieto‐Lafuente L, Paape D, Allen JE (2009). Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol 182: 3084–3094. [DOI] [PubMed] [Google Scholar]
- Nagamoto‐Combs K, Morecraft RJ, Darling WG, Combs CK (2010). Long‐term gliosis and molecular changes in the cervical spinal cord of the rhesus monkey after traumatic brain injury. J Neurotrauma 27: 565–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL et al (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204: 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan CF, Prendergast TJ, Wiebe ME, Stanley ER, Platzer E, Remold HG et al (1984). Activation of human macrophages. Comparison of other cytokines with interferon‐gamma. J Exp Med 160: 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG (2013). Training innate immunity: the changing concept of immunological memory in innate host defence. Eur J Clin Invest 43: 881–884. [DOI] [PubMed] [Google Scholar]
- Netea MG, Quintin J, vander Meer JW (2011). Trained immunity: a memory for innate host defense. Cell Host Microbe 9: 355–361. [DOI] [PubMed] [Google Scholar]
- Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA (2014, 2014). Inflammasome‐independent regulation of IL‐1‐family cytokines. Annu Rev Immunol 33: 49–77. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132 (Pt 2): 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- Novak ML, Koh TJ (2013). Macrophage phenotypes during tissue repair. J Leukoc Biol 93: 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A (2011). Alternative macrophage activation and metabolism. Ann Rev Pathol 6: 275–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Biber K, Vinet J, Boddeke HW (2011). Microglia phenotype diversity. CNS Neurol Disord Drug Targets 10: 108–118. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Hardie DG (2013). Metabolism of inflammation limited by AMPK and pseudo‐starvation. Nature 493: 346–355. [DOI] [PubMed] [Google Scholar]
- Ortega‐Gómez A, Perretti M, Soehnlein O (2013). Resolution of inflammation: an integrated view. EMBO Mol Med 5: 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458. [DOI] [PubMed] [Google Scholar]
- Park J, Choi H, Min JS, Park SJ, Kim JH, Park HJ et al (2013). Mitochondrial dynamics modulate the expression of pro‐inflammatory mediators in microglial cells. J Neurochem 127: 221–232. [DOI] [PubMed] [Google Scholar]
- Parwaresch MR, Wacker HH (1984). Origin and kinetics of resident tissue macrophages. Parabiosis studies with radiolabelled leucocytes. Cell Tissue Kinet 17: 25–39. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Pearce EJ (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Whitney N, Wu Y, Tian C, Dou H, Zhou Y et al (2008). HIV‐1‐infected and/or immune‐activated macrophage‐secreted TNF‐alpha affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia 56: 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF Jr, Cheever AW et al (2006). The IL‐21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest 116: 2044–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce JT, Ramalingam TR, Mentink‐Kane MM, Wilson MS, ElKasmi KC, Smith AM et al (2009). Arginase‐1‐expressing macrophages suppress Th2 cytokine‐driven inflammation and fibrosis. PLoS Pathol 5: e1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen JS, Fuentes‐Duculan J, Suárez‐Fariñas M, Pierson KC, Pitts‐Kiefer A, Fan L et al (2011). Tumor‐associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J Invest Dermatol 131: 1322–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H (2007). Neurotransmitter receptors on microglia. Trends Neurosci 30: 527–535. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN (2007). CNS‐derived interleukin‐4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci 27: 10714–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D et al (2009). Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappa B. Proc Natl Acad Sci U S A 106: 14978–14983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintin J, Cheng SC, vander Meer JW, Netea MG (2014). Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol 29: 1–7. [DOI] [PubMed] [Google Scholar]
- Rajaram MV, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS (2010). Mycobacterium tuberculosis activates human macrophage peroxisome proliferator‐activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J Immunol 185: 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Brown MA (2012). Innate immunity in the central nervous system. J Clin Invest 122: 1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE (2010). The myeloid cells of the central nervous system parenchyma. Nature 468: 253–262. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH (2009). Microglial physiology: unique stimuli, specialized responses. Ann Rev Immunol 27: 119–145. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Dietrich WD, Keane RW (2014). Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cerebral Blood Flow Metab 34: 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ (2009). CCL2 and interleukin‐6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2‐type macrophage polarization. J Biol Chem 284: 34342–34354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín‐Sanz P et al (2010). Substrate fate inactivated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185: 605–614. [DOI] [PubMed] [Google Scholar]
- Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N et al (2009). A CREB‐C/EBP beta cascade induces M2 macrophage‐specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci USA 106: 17475–17480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ (2001). Cutting edge: STAT6‐dependent substrate depletion regulates nitric oxide production. J Immunol 166: 2173–2177. [DOI] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A (2008). Amyloid‐beta oligomers set fire to inflammasomes and induce Alzheimer's pathology. J Cell Mol Med 12 (6a): 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini F, Franze A, Iervolino A, Filosa S, Salzano S, Ursini MV (1999). Enhanced glutathione levels and oxido resistance mediated by increased glucose‐6‐phosphate dehydrogenase expression. J Biol Chem 274: 2750–2757. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Melchior B, Masek K, Puntambekar SS, Danielson PE, Lo DD et al (2009). Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo . J Neurochem 109 (Suppl. 1): 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane M, Mosquera‐Miguel A, Gonzalez T, Fraga M, Salas A, Costoya JA (2011). The mitochondrial genome is a ‘Genetic Sanctuary’ during the oncogenic process. PLoS ONE 6: e23327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Schwartz M (2013). CNS sterile injury: just another wound healing? Trends Mol Med 19: 135–143. [DOI] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J et al (2013). Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38: 555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X et al (2012). The NALP3 inflammasome is involved in neurotoxic prion peptide‐induced microglial activation. J Neuroinflammation 9: 73. doi: 10.1186/1742‐2094‐9‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A (2012). Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet‐Wadiche LS et al (2010). Microglia shape adult hippocampal neurogenesis through apoptosis‐coupled phagocytosis. Cell Stem Cell 7: 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn S‐H, Lee J‐H, Chung H‐S, Lee H‐E, Lee J‐M, Bae H (2012). The effects of Foxp3 on gene expression profiles in activated microglial cells. Mol Cell Toxicol 8: 139–148. [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 176: 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N et al (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178. [DOI] [PubMed] [Google Scholar]
- Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J (2005). Macrophages sequentially change their functional phenotypes in response to changes in microenvironmental influences. J Immunol 175: 342–349. [DOI] [PubMed] [Google Scholar]
- Strachan‐Whaley M, Rivest S, Yong VW (2014). Interactions between microglia and T cells in multiple sclerosis pathobiology. J Interferon Cytokine Res 34: 615–622. [DOI] [PubMed] [Google Scholar]
- Sunnemark D, Eltayeb S, Nilsson M, Wallstrom E, Lassmann H, Olsson T et al (2005). CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein‐induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflam 2: 17. doi: 10.1186/1742‐2094‐2‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Mori T, Wu Y, Saxe M, Crawford F et al (2000). CD45 opposes beta‐amyloid peptide‐induced microglial activation via inhibition of p44/42 mitogen‐activated protein kinase. J Neurosci 20: 7587–7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Le W (2015). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Tang Y, Li T, Li J, Yang J, Liu H, Zhang XJ et al (2014). Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson's disease. Cell Death Differ 21: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, Majewska AK (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8: e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia PJ, Mena NP, Núñez MT (2014). The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol 5: 38. doi: 10.3389/fphar.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum MM, Ikezu T (2012). The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz) 60: 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR et al (2006). Oxidative metabolism and PGC‐1beta attenuate macrophage‐mediated inflammation. Cell Metabol 4: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch JR, McMurray MA, Nelson ZW, Gottschling DE (2009). Mitochondrial dysfunction leads to nuclear genome instability via an iron‐sulfur cluster defect. Cell 137: 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilalta A, Brown GC (2014). Deoxyglucose prevents neurodegenerationin culture by eliminating microglia. J Neuroinflammation 11: 58. doi: 10.1186/1742‐2094‐11‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG (2009). Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 18: 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovoz Y, Bogdan C, Paik J, Xie QW, Nathan C (1993). Mechanisms of suppression of macrophages nitric oxide release by transforming growth factor beta. J Exp Med 178: 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, vanderValk P et al (2013). Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation 10: 35. doi: 10.1186/1742‐2094‐10‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Emery JF, Sun X, Giffard RG (2013). Inflammatory response of microglial BV‐2 cells includes a glycolytic shift and is modulated by mitochondrial glucose‐regulated protein 75/mortalin. FEBS Lett 587: 756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29: 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG et al (2014). Rapid inflammasome activationinmicroglia contributes to brain disease in HIV/AIDS. Retrovirology 11: 35. doi: 10.1186/1742‐4690‐11‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhao Y, Sun M, Liu S, Li B, Zhang L et al (2014). 2‐Deoxy‐d‐glucose attenuates sevoflurane‐induced neuroinflammation through nuclear factor‐kappa B pathway in vitro . Toxicol Vitro 28: 1183–1189. [DOI] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123: 309–314. [DOI] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument‐Bromage H, Tempst P et al (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH et al (2011). Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118: e16–e31. [DOI] [PubMed] [Google Scholar]
- Wright GJ, Cherwinski H, Foster‐Cuevas M, Brooke G, Puklavec MJ, Bigler M et al (2003). Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol 171: 3034–3046. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R et al (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211: 1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M et al (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Appel SH (2013). Immune‐mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol 8: 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler‐Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN et al (2010). Nomenclature of monocytes and dendritic cells in blood. Blood 116: e74–e80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Representative schematic of mitochondrial function analysis using the Seahorse Bioscience extracellular flux analyzer (XF24) (Seahorse Bioscience, Billerica, MA, USA).
Table S1 Quantitative real‐time PCR primers sequences.