

Abstract
Relapsed and refractory hematologic malignancies have a very poor prognosis. Chimeric antigen receptor (CAR) T cells are emerging as a powerful therapy in this setting. Early clinical trials of genetically modified T cells for the treatment of non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL) have shown high complete response rates in patients with few therapeutic options. Exploration is ongoing for other hematologic malignancies including multiple myeloma (MM), acute myeloid leukemia (AML) and Hodgkin lymphoma (HL). At the same time, the design and production of CAR T cells is being advanced so that this therapy can be more widely utilized. Cytokine release syndrome (CRS) and neurotoxicity are common, but they are treatable and fully reversible. This review will review currently available data as well as future developments and challenges in the field.
Introduction
Genetic modification of T cells to express chimeric antigen receptors (CAR) offers a novel approach to treating hematologic malignancies. Like monoclonal antibodies, CAR T cells are targeted therapies directed against a cell-surface antigen on malignant cells. CAR T cells are potentially more potent than monoclonal antibodies and can establish long-lived immunity against the target antigen after a single infusion. As a result, compared with monoclonal antibodies, the criteria for target selection are more stringent due to severe toxicity that can develop if the target is expressed on non-cancerous cells. 1,2 Over the past decade, immunotherapy with CAR T cells has evolved from bench to bedside with promising early clinical results. In early-phase clinical trials at several centers, CAR T cells have induced impressive responses in chemotherapy-refractory chronic lymphocytic leukemia (CLL) and relapsed acute lymphoblastic leukemia (ALL). There are now a variety of CAR target antigens under investigation for multiple hematologic malignancies (Table 1). In this review we will discuss CAR T cell design, production and clinical use. We will review the early-phase clinical data on CAR T cells for non-Hodgkin lymphoma (NHL), CLL and ALL and touch upon the emerging use of CAR T cells in other hematologic malignancies. Additionally we will discuss the toxicities encountered and their management strategies.
Table 1. CAR Target Antigens Currently Under Investigation.
| CAR Target Antigen | Diseases | Expression of Normal Tissues | Study Phase | Toxicity Concerns | Other Targeted Therapies |
|---|---|---|---|---|---|
| CD19 | B-Cell NHLCLLALLMMHL | Early pro-B to mature B cell | Phase II ongoing | B-Cell Aplasia | Blinatumomab |
| CD20 | B-Cell NHL | B cells from Pre-B cell to B-cell lymphoblast | Phase I completed | B-Cell Aplasia | RituximabOfatumumabObinutuzumabIbrutumomab tiuxetan |
| CD22 | B-Cell NHLALL | Mature B cells | Phase I ongoing | Epratuzumab | |
| CD23 | CLL | Mature B cells | Preclinical data | ||
| CD30 | HLB-Cell NHL | Activated B and T cells, HRS cells | Phase I/II ongoing | Impaired T cell expansion | Brentuximab vedotin |
| CD33 | AML | Immature myeloid cells | Phase I/II ongoing | Myeloablation | Gemtuzumab ozogamicin |
| CD38 | MM | Plasma cells, T and B lymphocytes, NK cells, myeloid progenitor cells | Preclinical Data | Myelotoxicity | Daratumumab |
| CD70 | NHL | Activated Lymphocytes | Preclinical Data | Impaired cellular immune response | |
| CD123 | AMLHL | Hematopoietic progenitor cells | Phase I ongoing | Myeloablation | |
| CD133 | AMLALL | Hematopoietic progenitor cells | Phase I ongoing | Myeloablation, off-target effects (also expressed on endothelial progenitor cells, neuronal and glial stem cells) | |
| CD138 | MM | All plasma cells, epithelium | Phase I/II ongoing | Mucositis | |
| Kappa Light Chain | MMCLLB-Cell NHL | Plasma Cells, B Lymphocytes | Phase I ongoing | ||
| LeY | AMLMMMDS | Epithelium, early myeloid progenitor cells, multiple different malignancies | Phase I ongoing | Neutropenia, thrombocytopenia | |
| NKG2D | MMAMLMDS | NK cells, T cells | Phase I ongoing | Hypothermic, weight loss, pneumonitis in preclinical models | |
| CS1 | MM | Plasma Cells, T cells, NK cells, Dendritic cells | Preclinical Data | Immune deficiency | Elotuzumab |
| BCMA | MM | Plasma Cells, Mature B cells | Phase I ongoing | GSK287916 | |
| CD44v6 | AMLMM | Activated T cells, Monocytes, keratinocytes | Preclinical Data | Epithelial toxicity | |
| ROR1 | CLL | Pre-B cells, Lymphatic and epithelial malignancies | Preclinical Data | Off-target effects (expression on adipocytes, lung, pancreas) |
NHL: Non-Hodgkin Lymphoma; CLL: Chronic Lymphocytic Leukemia; ALL: Acute Lymphoblastic Leukemia; MM: Multiple Myeloma; HL: Hodgkin Lymphoma; AML: Acute Myeloid Leukemia; MDS: Myelodysplastic Syndrome; HRS: Hodgkin Reed-Sternberg; NK: Natural Killer
Chimeric Antigen Receptor Design
CARs are engineered receptors that artificially confer specificity of T lymphocytes for native cell-surface antigens. 3 CARs consist of an extracellular antigen binding domain, a hinge region, a trans-membrane domain, and an intracellular signaling domain (Figure 1). The antigen-binding domain typically comprises a single chain variable fragment (scFv), derived from a monoclonal antibody. Unlike the native T cell receptor, CARs recognize their targets independently of antigen processing by the target cell and independently of the major histocompatibility complex (MHC). 4-6
Figure 1.

Chimeric Antigen Receptor (CAR) Design. CARs consist of an extracellular antigen-binding domain, a hinge region, a trans-membrane domain and an intracellular signaling domain. First generation CARs include only CD3ζ as their signaling domain. Second generation CARs include a co-stimulatory domain derived from CD28 or 4-1BB. Third generation CARs include two co-stimulatory domains.
Binding of the antigen to the extracellular domain leads to T cell activation via the intracellular signaling domains. Early CAR designs contained the CD3ζ chain as the only intracellular signaling domain and are referred to as first-generation CARs. Preclinical and clinical studies have shown that the addition of co-stimulatory signaling domains to the intracellular component improves T cell proliferation and persistence. 7,8 Second-generation CARs incorporate a single co-stimulatory domain (e.g. CD28 or 4-1BB), whereas third-generation CARs incorporate two co-stimulatory domains (e.g. both CD28 and 4-1BB). The optimum choice and configuration of co-stimulatory signaling domains is uncertain and under investigation.
Production and Clinical Use of Chimeric Antigen Receptor T cells
Manufacturing of CAR T cells requires collection of autologous T cells via leukopheresis followed by ex vivo genetic modification to express the CAR on the T cell surface and expansion to generate a clinically effective cell dose (Figure 2). Several approaches to gene transfer and ex vivo expansion have been developed.
Figure 2.
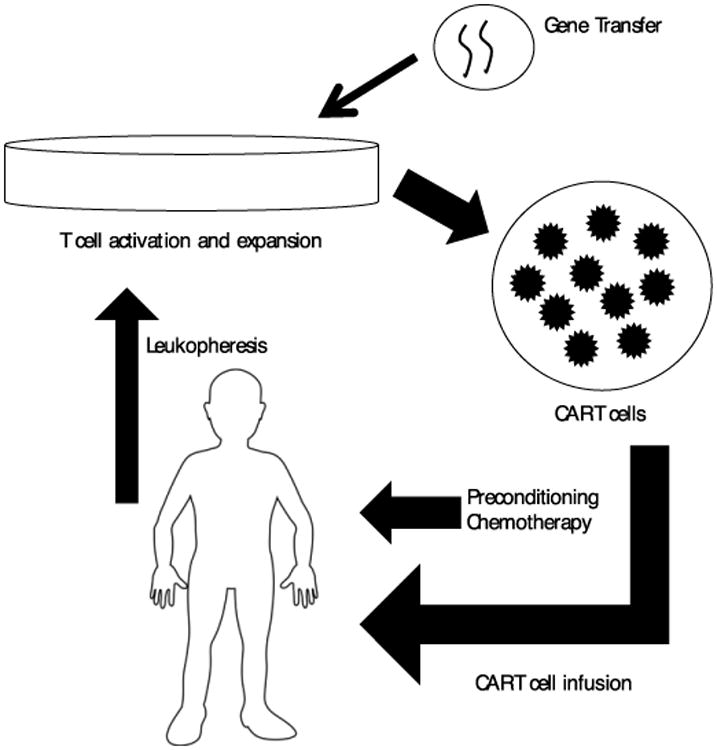
CAR T cell production and use. Peripheral blood mononuclear cells (PBMCs) are collected from the patient via apheresis and stimulated PMBCs are exposed to the viral or non-viral vector. T cells are stimulated using anti-CD3/anti-CD28 monoclonal antibody-coated magnetic beads with or without additional exogenous cytokines. Patients then receive lymphodepleting chemotherapy followed by infusion of CAR T cells.
Gene Transfer Techniques
The first gene transfer system used for CAR T cells employed a gamma retroviral vector. Gamma retroviruses integrate into genomic DNA leading to permanent and heritable CAR expression. CARs manufactured in this fashion are safe, relatively easy to produce and can efficiently and permanently transduce T cells. HIV-based lentiviral vectors are also able to efficiently and permanently transduce T cells. Lentiviral vectors enable higher and more stable CAR expression compared with gamma retroviruses. 9 They have a theoretical safety advantage; due to their preferred sites of integration into the genome they are considered less genotoxic than gamma retroviral vectors. 10 However, they are more costly to produce.
Potential disadvantages to viral vectors include cost, expertise required for production, and regulatory requirements. Transposon systems and electroporation of mRNA constructs have both been used as alternatives to viral vectors. Transposons (such as Sleeping Beauty), like retroviral vectors, can stably integrate into the genome, but they may require long duration of T cell culture to produce adequate cell doses. 11-13 Electroporation of mRNA constructs is inexpensive and less technically complex. It is considered safer than viral alternatives because there is no genome integration. However, mRNA is unstable, resulting in CAR expression that is only transient and not heritable. This may be useful in circumstances where only transient CAR activity is desirable, such as when the target antigen is also found in normal tissues. Though there is no potential for establishment of long-term immunity, the anti-tumor effect could be prolonged via serial CAR T cell infusions. In preclinical models, anti-CD19 CARs produced by mRNA electroporation showed comparable efficacy to those produced by lentiviral transduction, though clinical results, as discussed below suggest that long-lived in vivo CAR T cell persistence, which is not possible with mRNA transfection, is associated with superior outcomes. 14-16
T-cell Expansion Ex Vivo
After introduction of the vector into the T cells, the cells must then be expanded ex vivo. The T cells are most often stimulated using anti-CD3/anti-CD28 monoclonal antibody-coated magnetic beads. Additional exogenous cytokines (such as IL-2, IL-7, IL-12 or IL-15) can be added to enhance T cell expansion and proliferation. 17-20 The optimal time for ex vivo expansion is unknown and studies have used a culture period ranging from 1 to 6 weeks. Most groups achieve several hundred to several thousand-fold T cell expansion within this short period of time. 18-28
Adoptive Transfer
Both murine models and clinical trials have showed that in vivo proliferation and persistence of adoptively transferred T cells are augmented when patients receive some form of lymphodepleting therapy, either radiation or chemotherapy, a few days prior to T cell infusion. 24,29-31 Lymphodepletion prior to infusion of CAR T cells is thought to create homeostatic space for T cell expansion and deplete other cells that would compete for available cytokines. 32,33 The optimal T cell dose and number of infusions is unknown, and the level of T cell engraftment and expansion does not always correlate with the dose of T cells given. Once CAR T cells are infused back into the host, they rapidly redistribute throughout the body. 34-36 CAR T cells then undergo accumulation at the sites of target recognition. It is unclear how long CAR T cells must persist within the host to continue immunosurveillance and prevent relapse.
Chimeric Antigen Receptor T cells in Hematologic Malignancies
An ideal target for CAR T cells is an antigen detected on tumor cells but not on normal tissues, therefore limiting the “on target, off tumor” toxicity. In this regard, both CD19 and CD20 are good CAR targets for most B cell malignancies, including B-cell ALL, CLL and B-cell NHL, as the only expected antigen-specific toxicity is B cell aplasia and subsequent hypoglobulinemia, which is manageable with immunoglobulin replacement therapy.
Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia
The earliest clinical work with CAR T cells in hematologic malignancies was in patients with B-cell NHL. Although initial studies were small, they helped inform further research. At City of Hope, first-generation anti-CD20 and anti-CD19 CAR T cells were tested in patients with diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL). Although the therapy was well tolerated, T cell persistence was limited. At 1 week post-infusion, detectable levels of transferred T cells were found only after 3 of 15 infusions, and poor persistence of CAR T cells was observed. 21
T cell persistence improved with the addition of a co-stimulatory domain. The National Cancer Institute (NCI) used a second-generation anti-CD19 CAR with a CD28 co-stimulatory domain in a patient with advanced FL. The patient received lymphodepleting chemotherapy followed by CAR T cell infusion and intravenous IL-6 every 8 hours for 8 doses. The patient had a partial response (PR) that lasted 32 weeks. B cell aplasia was present in the peripheral blood from approximately 9 weeks post-T cell infusion until at least 39 weeks. The anti-CD19 CAR T cells persisted in the peripheral blood until 27 weeks post-T cell infusion. 29
Subsequently, our institution, the University of Pennsylvania (Penn) reported results from treatment of three patients with advanced, chemotherapy resistant CLL using an anti-CD19 CAR with a CD137 (4-1BB) co-stimulatory domain (CTL019, previously referred to as CART19) administered following lymphodepleting chemotherapy. Notably, CTL019 cells persisted in the blood of all patients for at least 6 months. Of the three patients treated, there were 2 CRs and 1 PR lasting greater than 8 months after CART19 infusion. 26,37
The importance of lymphodepleting chemotherapy for in vivo persistence of CAR T cells was highlighted in a study at Memorial Sloan Kettering Cancer Center (MSKCC) in which ten patients with refractory CLL and relapsed ALL were treated with an anti-CD19 CAR T cell with a CD28 co-stimulatory domain (19-28z). The seven patients who received 19-28z immediately following lymphodepleting chemotherapy had significantly more durable persistence of 19-28z cells in the peripheral blood and bone marrow compared to the three patients who did not receive lymphodepleting chemotherapy. 24
Over the last year, results have been reported from multiple centers evaluating second-generation anti-CD19 CAR T cells in B-cell NHL and CLL. Kochendorfer et al. 38 at the NCI treated 15 patients with NHL with an anti-CD19 CAR with a CD28 co-stimulatory domain. Of the 7 evaluable patients with DLBCL, 4 obtained CR, 2 obtained PR and 1 had stable disease (SD) after infusion of CAR T cells. This was the first report of anti-tumor efficacy of an anti-CD19 CAR T cell in aggressive lymphoma. At Penn, Schuster et al. 39 recently reported interim results from 19 NHL patients treated with CTL019. The overall response rate (ORR) for the entire cohort was 68%; by histology, 6 of 13 DLBCL patients, 7 of 7 FL patients and 1 of 2 mantle cell lymphoma patients exhibited at least PRs. It is too early to make conclusions about the long-term durability of the responses observed in NHL. A recently published update of the initial CTL019 study in CLL is encouraging regarding response durability, however, as the two initially reported CRs are ongoing more than 4 years post-treatment, and two additional subjects from this cohort of 14 also attained durable MRD-negative CRs. 40
There are many ongoing trials of anti-CD19 CAR T cells in patients with lymphoma. Alternative clinical settings are being evaluated including the administration of CAR T cells after autologous stem cell transplantation (NCT01840566) and as a bridge to allogeneic transplant (NCT02431988). 41,42 Third-generation constructs are being studied (NCT02132624). Targets other than CD19 are also being investigated including CD30 (NCT02259556; NCT01316146; NCT02274584) and CD22 (NCT02315612).
Acute Lymphoblastic Leukemia
Perhaps the most exciting results from the early experience with CAR T cell therapy have been in B-cell ALL. The prognosis of ALL in the relapsed or refractory setting is dismal, with remission rates with standard chemotherapy of about 30%. 43 The MSKCC group was the first to show antitumor efficacy in a small group of patients with ALL. Five patients with relapsed ALL underwent lymphodepleting chemotherapy, were treated with 19-28z, and eligible patients then underwent allogeneic stem cell transplantation. All 5 patients achieved minimal residual disease (MRD) negative status after T cell infusion, enabling them to undergo transplantation if they were otherwise eligible. 44 Investigators at Penn and the Children's Hospital of Philadelphia published a report of 30 adults and children with relapsed or refractory ALL who received CTL019. CR was achieved in 27 of the 30 patients at 6 months. Patients had persistence of CAR T cells and B cell aplasia for up to 2 years. Remissions of 2 to 24 months were seen in 19 of the 27 responding patients. Event free survival (EFS) and overall survival (OS) at 6 months were 67% and 78% respectively. 45,46 Similarly encouraging results have been reported at other centers. Davila et al. 47 at MSKCC treated 16 patients with 19-28z and had a CR rate of 88%. The 19-28z cells persisted for 1 to 3 months and 7 patients were able to proceed to allogeneic stem cell transplant. All of these patients were in CR at the time of publication. Lee et al 48 at the NCI reported 21 children and young adults who received a second-generation anti-CD19 CAR T cells with a CD28 co-stimulatory domain. The CR rate was 70% and the longest persistence of CAR T cell was 68 days. OS at a median follow-up of 10 months was 51.6% and leukemia-free survival in the 12 patients who achieved MRD-negative CR was 78.8%. Ongoing studies are evaluating larger patient cohorts.
Additional Targets in Hematologic Malignancies
Acute Myeloid Leukemia
A challenge of adapting CAR T cell therapy for use myeloid malignancies is the identification of a target that is present on tumor cells but absent from normal myeloid cells and their precursors. Two AML targets have been identified that would be expected to spare normal myelopoiesis: The Lewis Y antigen (LeY) and NKG2D. The LeY antigen is an oligosaccharide that is structurally related to the Lewis blood group antigens but not expressed on erythrocytes and has limited expression on other normal tissues. This antigen is however expressed in high number in many malignancies including AML. 49 A phase I study of 4 patients with high-risk AML using a second-generation anti-LeY CAR T cell with a CD28 co-stimulatory domain showed that the therapy was well tolerated and effective. Three of the 4 patients had responses, but all eventually had progressive disease (PD) despite persistence of CAR T cells. 35 NKG2D is an activating cell surface receptor expressed on natural killer (NK) cells and a variety of other immune cells; its ligand is overexpressed by many tumor cells. Preclinical models have demonstrated the antitumor efficacy of a NKG2D CAR construct in a variety of settings. 50-55 However, the expression on multiple different immune cells can lead to severe off-target toxicity in preclinical models. 56 An ongoing clinical trial is evaluating the safety of a CAR T cell directed against NKG2D ligands in patients with AML, myelodysplasic syndrome (MDS) and multiple myeloma (MM) (NCT02203825).
Preclinical studies have investigated CD33 or CD123 as targets. While both are expressed on AML blasts and normal hematopoietic stem/progenitor cells, CD123 is expressed to a much lesser degree on normal tissues. Preclinical models have showed that both antigens are effective targets for CAR T cells in reduction of tumor burden, but, as expected, there is less killing of normal progenitor cells by anti-CD123 CAR T cells. 57,58 An ongoing trial in China (NCT01864902) reported results of one patient treated with anti-CD33 CAR T cell therapy with a 4-1BB co-stimulatory domain. Although the patient had a marked decrease in blast percentage at 2 weeks, this was followed by a gradual increase and disease progression. This occurred despite the persistence of CAR T cells at high levels in the bone marrow and peripheral blood for at least 2 months. Additionally, the patient experienced toxicities including cytokine release syndrome and pancytopenia. 59 A study of an anti-CD123 CAR T cell at City of Hope is not yet open for recruitment (NCT02159495). Myelotoxicity of CARs against these targets might be manageable if they employ transient expression systems such as mRNA transfection and are used as a bridge to allogeneic stem cell transplantation. 57,58
Multiple Myeloma
Multiple targets have been investigated in preclinical and clinical settings for CAR T cells in multiple myeloma (MM). 60 CARs targeting CS1, CD38, CD138, B Cell Maturation Antigen (BCMA), CD44v6, LeY, kappa light chain, and NKG2D have demonstrated preclinical anti-MM activity. The clinical studies mentioned above testing LeY- and NKG2D-targeted CARs in myeloid malignancies are also enrolling patients with MM. Additional clinical studies of CARs targeting CS1, BCMA, CD138, and kappa light chain are ongoing but have not yet published results.
BCMA and CS1 are the targets with the most substantiated rationale in MM. These antigens are expressed on MM plasma cells in nearly all patients with little expression on non-malignant cells. BCMA expression is restricted to late-stage B cells and plasma cells. 61 CS1 is expressed on plasma cells and, at low levels, on NK cells and subsets of T cells and activated monocytes and dendritic cells. 62 CARs against both BCMA and CS1 have anti-myeloma activity in vitro and in murine models. 63,64 Phase-1 clinical trials of CARs targeting BCMA are open at the NIH and Penn (NCT02546167, NCT02215967); phase-1 studies of CARs targeting CS1 are expected to open soon at multiple sites.
We have recently reported a case in which CTL019 administered after autologous stem cell transplantation (ASCT) led to durable complete response despite this patient's very poor response to prior ASCT. 65 Though CD19 is not expressed on the dominant population of MM plasma cells in most patients, it can be found on minor subsets of the MM clone, and several lines of evidence suggest that these CD19+ subsets might be uniquely drug-resistant and clonogenic. 66 Further study is required to determine which patients might benefit from this approach and to better understand the mechanism of action of CTL019 in this predominantly CD19-negative neoplasm.
Hodgkin Lymphoma
An attractive target in Hodgkin lymphoma (HL) is CD30, which is expressed on Hodgkin Reed-Sternberg (HRS) cells. However, CD30 is also present on some activated T cells and so CAR T cells with this target could theoretically induce fratricide, limiting the utility of the therapy. There are preclinical data demonstrating that CD30-directed CAR T cells can target CD30+ Hodgkin cell lines and there are a number of ongoing clinical trials evaluating this target (NCT02259556; NCT01316146; NCT02274584). 67,68 However, the HRS cells make up only a small portion of the tumor, which is characterized largely by the immunosuppressive microenvironment. Therefore the ideal target would be expressed on both populations of cells. A group at our institution identified CD123 as a potential target because it is expressed on both HRS cells and immune cells of the microenvironment, and they have presented preclinical data showing the efficacy of a CD123 CAR T cell construct against for disseminated HL. 69
Toxicity of Chimeric Antigen Receptor T Cells
B-Cell Aplasia
Toxicities from CAR T cells can result from the effects of the target antigen on non-tumor cells, the so called “on target, off tumor” effect, or truly off-target effects in which the CAR cross-reacts to an unintended target antigen. B cell aplasia is seen in patients treated with CD19 CAR T cells. 23,29,38,45,46 The duration of B cell aplasia is variable depending on the construct and co-stimulatory molecules used. It is associated with hypogammaglobulinemia that responds to intravenous immunoglobulin replacement therapy. Other targets would be expected to have different “on target, off tumor” effects, and these are described in more detail above. Fortunately, off-target toxicity of CARs for hematologic malignances has not been identified, but our group recently reported off-target fatal cardiotoxicity of autologous T cells redirected towards the cancer-testis antigen MAGE-A3 using a lentivirally transduced affinity-enhanced T cell receptor. 70,71
Cytokine Release Syndrome
In some patients, CAR T cells induce an inflammatory reaction called the cytokine release syndrome (CRS). Clinical manifestations can range from isolated fever to hemodynamic collapse with multisystem organ failure. This syndrome can resemble macrophage activation syndrome with hepatosplenomegaly, hyperferritinemia, and hypofibrinogemia. 45,46 CRS generally develops within 1-2 weeks of CAR T cell infusion. 23,38,44,46 The onset of clinical symptoms correlates with the peak in vivo expansion of CAR T cells and the levels of inflammatory cytokines, particularly IFN-γ, IL-10, and IL-6. 37,38,44-46,48 Patients with greater disease burden at the time of CAR T cell infusion tend to have a more severe syndrome. 44,46 Occurrence of CRS may not be a prerequisite to achieve antitumor control. 44
Though in mild cases the syndrome resolves uneventfully without intervention, severe CRS patients often require intensive supportive care including the administration of vasopressors and respiratory support. 47 Reversal of severe CRS requires the use of either corticosteroids or the IL-6 receptor-blocking antibody, tocilizumab. While corticosteroids can in some cases quickly reverse symptoms, they can also decrease the expansion of CAR T cells. Suppression of CAR T cells presumably has a negative effect on tumor control. For example, in one study, all 3 patients who were treated with steroids experienced disease recurrence despite achieving CR after initial CAR T cell therapy. Conversely, tocilizumab can improve clinical symptoms and is not thought to impair expansion or function of T cells. 47 Thus, tocilizumab is typically the first choice for management of CRS that requires intervention, except in severe cases where combination of tocilizumab and corticosteroids might be considered.
Neurotoxicity
Patients who receive CAR T cells can develop reversible neurotoxicity independently of CRS. While global encephalopathy is the most common toxicity, other symptoms have been reported (seizures, aphasia, hallucinations). Most commonly, the symptoms are brief and self-limited. They resolve over several days without intervention and patients do not appear to have any long-term sequelae. In fact, the administration of tocilizumab or corticosteroids does not appear to alter outcome. 38,46 Imaging (CT or MRI) and lumbar puncture have not identified an etiology for these symptoms. 38,46-48 Neurotoxicity does not appear to be associated with the presence of CAR T cells in the cerebrospinal fluid. 47 Similar toxicity has been described with the anti-CD19 biospecific T cell engager blinatumomab. 72 This toxicity needs to be further characterized going forward.
Conclusions
The optimal CAR T cell design and application continues to be actively explored for many hematologic malignancies. In small studies at several centers, CD19 seems to be a safe and effective target for CAR T cells in early-phase clinical trials in ALL, CLL, NHL and more recently MM. There are several promising targets for CAR therapy in MM, notably BCMA and CS1 that are currently in phase-1 clinical trials. Identification of safe targets for myeloid neoplasms is more challenging, but transient or reversible CAR expression techniques may enable use of CARs against myeloid antigens such as CD123 or CD33, particularly in patients with available donors for allogeneic transplantation.
Target identification is only one consideration in clinical development for CAR T cells. As indicated by the very different response rates with anti-CD19 CARs between CLL and ALL, disease-specific factors may affect CAR function. Choices regarding CAR design and manufacturing processes also affect efficacy and toxicity profiles and may need to be tailored to host- and disease-specific immune parameters. Similarly, the optimal position of CAR T cell therapy in relation to existing therapies will need to be determined. Determining, for each disease, the optimal targets, CAR designs, and points of integration with existing therapies will require continued innovation and collaboration between laboratory-based and clinical investigators.
References
- 1.Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J Clin Oncol. 2006;24(13):e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 2.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional expression of chimeric receptor genes in human T cells. J Immunol Methods. 2001;248(1-2):67–76. doi: 10.1016/s0022-1759(00)00343-4. [DOI] [PubMed] [Google Scholar]
- 4.Hombach A, Heuser C, Gerken M, et al. T cell activation by recombinant FcepsilonRI gamma-chain immune receptors: An extracellular spacer domain impairs antigen-dependent T cell activation but not antigen recognition. Gene Ther. 2000;7(12):1067–1075. doi: 10.1038/sj.gt.3301195. [DOI] [PubMed] [Google Scholar]
- 5.Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 6.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 9.Morgan RA, Kakarla S. Genetic modification of T cells. Cancer J. 2014;20(2):145–150. doi: 10.1097/PPO.0000000000000033. [DOI] [PubMed] [Google Scholar]
- 10.Modlich U, Navarro S, Zychlinski D, et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17(11):1919–1928. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol Ther. 2010;18(4):674–683. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh H, Manuri PR, Olivares S, et al. Redirecting specificity of T-cell populations for CD19 using the sleeping beauty system. Cancer Res. 2008;68(8):2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiti SN, Huls H, Singh H, et al. Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother. 2013;36(2):112–123. doi: 10.1097/CJI.0b013e3182811ce9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrett DM, Zhao Y, Liu X, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22(12):1575–1586. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birkholz K, Hombach A, Krug C, et al. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009;16(5):596–604. doi: 10.1038/gt.2008.189. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Zheng Z, Cohen CJ, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13(1):151–159. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinrichs CS, Spolski R, Paulos CM, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111(11):5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollyman D, Stefanski J, Przybylowski M, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32(2):169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maus MV, Thomas AK, Leonard DG, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol. 2002;20(2):143–148. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 21.Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter DL, Levine BL, Bunin N, et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006;107(4):1325–1331. doi: 10.1182/blood-2005-08-3373. [DOI] [PubMed] [Google Scholar]
- 23.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh H, Figliola MJ, Dawson MJ, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71(10):3516–3527. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen MC, Clarke P, Tan G, et al. Human T lymphocyte genetic modification with naked DNA. Mol Ther. 2000;1(1):49–55. doi: 10.1006/mthe.1999.0012. [DOI] [PubMed] [Google Scholar]
- 28.Ye Q, Loisiou M, Levine BL, et al. Engineered artificial antigen presenting cells facilitate direct and efficient expansion of tumor infiltrating lymphocytes. J Transl Med. 2011;9:131–5876. 9–131. doi: 10.1186/1479-5876-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116(19):3875–3886. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174(5):2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parente-Pereira AC, Burnet J, Ellison D, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol. 2011;31(4):710–718. doi: 10.1007/s10875-011-9532-8. [DOI] [PubMed] [Google Scholar]
- 37.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuster SJ, Svoboda J, Nasta SD, et al. Oral presentations: Phase II trial of chimeric antigen receptor modified T cells directed against CD19 in relapsed/refractory diffuse large B cell, follicular, and mantle cell lymphomas. Hematol Oncol. 2015;33:175–176. [Google Scholar]
- 40.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauter CS, Riviere I, Bernal Y, et al. Phase I trial of 19-28z chimeric antigen receptor modified T cells (19-28z CAR-T) post-high dose therapy and autologous stem cell transplant (HDT-ASCT) for relapsed and refractory (rel/ref) aggressive B-cell non-hodgkin lymphoma (B-NHL) ASCO Meeting Abstracts. 2015;33(15):8515. [Google Scholar]
- 42.Sauter CS, Riviere I, Bernal YJ, et al. Interim safety analysis: A phase I trial of high dose therapy and autologous stem cell transplantation followed by infusion of chimeric antigen receptor modified T-cells (19-28z CAR-T) directed against CD19+ B-cells for relapsed and refractory aggressive B cell non-hodgkin lymphoma (B-NHL) Blood. 2014;124(21):677–677. [Google Scholar]
- 43.Fielding AK, Richards SM, Chopra R, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109(3):944–950. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 44.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuriev E, Farrugia W, Scott AM, Ramsland PA. Three-dimensional structures of carbohydrate determinants of lewis system antigens: Implications for effective antibody targeting of cancer. Immunol Cell Biol. 2005;83(6):709–717. doi: 10.1111/j.1440-1711.2005.01374.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106(5):1544–1551. doi: 10.1182/blood-2004-11-4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66(11):5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 52.Zhang T, Barber A, Sentman CL. Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Cancer Res. 2007;67(22):11029–11036. doi: 10.1158/0008-5472.CAN-07-2251. [DOI] [PubMed] [Google Scholar]
- 53.Barber A, Zhang T, DeMars LR, Conejo-Garcia J, Roby KF, Sentman CL. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67(10):5003–5008. doi: 10.1158/0008-5472.CAN-06-4047. [DOI] [PubMed] [Google Scholar]
- 54.Barber A, Meehan KR, Sentman CL. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. Gene Ther. 2011;18(5):509–516. doi: 10.1038/gt.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barber A, Zhang T, Megli CJ, Wu J, Meehan KR, Sentman CL. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Exp Hematol. 2008;36(10):1318–1328. doi: 10.1016/j.exphem.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.VanSeggelen H, Hammill JA, Dvorkin-Gheva A, et al. T cells engineered with chimeric antigen receptors targeting NKG2D ligands display lethal toxicity in mice. Mol Ther. 2015 doi: 10.1038/mt.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kenderian SS, Ruella M, Shestova O, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647. doi: 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang QS, Wang Y, Lv HY, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garfall AL, Fraietta JA, Maus MV. Immunotherapy with chimeric antigen receptors for multiple myeloma. Discov Med. 2014;17(91):37–46. [PubMed] [Google Scholar]
- 61.Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsi ED, Steinle R, Balasa B, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775–2784. doi: 10.1158/1078-0432.CCR-07-4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chu J, He S, Deng Y, et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin Cancer Res. 2014;20(15):3989–4000. doi: 10.1158/1078-0432.CCR-13-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garfall AL, Maus MV, Hwang WT, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373(11):1040–1047. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hajek R, Okubote SA, Svachova H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br J Haematol. 2013;163(5):551–564. doi: 10.1111/bjh.12563. [DOI] [PubMed] [Google Scholar]
- 67.Hombach A, Heuser C, Sircar R, et al. Characterization of a chimeric T-cell receptor with specificity for the hodgkin's lymphoma-associated CD30 antigen. J Immunother. 1999;22(6):473–480. doi: 10.1097/00002371-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Savoldo B, Rooney CM, Di Stasi A, et al. Epstein barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of hodgkin disease. Blood. 2007;110(7):2620–2630. doi: 10.1182/blood-2006-11-059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruella M, Kenderian SS, Shestova O, et al. Novel chimeric antigen receptor T cells for the treatment of hodgkin lymphoma. Blood. 2014;124(21):806–806. [Google Scholar]
- 70.Cameron BJ, Gerry AB, Dukes J, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Topp MS, Gokbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]