

Abstract
Most of the stereocenters of polyketide natural products are established during assembly line biosynthesis. The body of knowledge for how stereocenters are set is now large enough to begin constructing physical models of key reactions. Interactions between stereocenter-forming enzymes and polyketide intermediates are examined here at atomic resolution, drawing from the most current structural and functional information of ketosynthases (KSs), ketoreductases (KRs), dehydratases (DHs), enoylreductases (ERs), and related enzymes. While many details remain to be experimentally determined, our understanding of the chemical and physical mechanisms utilized by the chirality-molding enzymes of modular PKSs is rapidly advancing.
I. Introduction
As a class of natural products, complex polyketides are characterized by their rich stereochemistries and potent bioactivities. These properties are linked since their diverse shapes and chemistries enable them to bind most biological targets. In synthesizing complex polyketides, Nature shows off her synthetic capabilities moreso than in any other pathway. Chemists dream of harnessing the stereocontrol observed in these reactions; however, understanding how these reactions naturally occur has been challenging enough.
The modular polyketide synthases (PKSs) that construct complex polyketides are the largest enzymes known to man.1, 2 They contain tens to hundreds of domains and very little is known about their higher-order architecture.3 Our understanding of the logic of polyketide synthesis has advanced considerably through the past twenty-five years, and in most cases the substituents and stereochemistries of a polyketide can be well predicted from the order of the enzymes in its synthase.4, 5 What the community has lacked for far too long are physical descriptions of the actual stereocontrolled reactions.
Recent crystal structures of the enzymatic domains of modular PKSs have now enabled visualization of many of the active sites in which stereocenters are set as well as physical descriptions of how polyketide intermediates undergo stereochemical transformation. Aside from the acyltransferases (ATs) that transfer extender units from malonyl-Coenzyme A (CoA) derivatives to acyl carrier protein (ACP) domains, each of the other enzymes within PKS modules can catalyze stereocontrolled reactions. Here we investigate how 1) “inversion of configuration” occurs through the condensation of an extender unit with a growing polyketide chain in the ketosynthase (KS) active site, 2) up to two chiral centers are set through the reduction of a β-ketoacyl intermediate in the ketoreductase (KR) active site, 3) trans- and cis-double bonds are generated from the dehydration of β-hydroxyacyl substrates in the dehydratase (DH) active site, and 4) the orientation of an α-substituent is set during the reduction of an α-substituted, α,β-trans-double bond in the enoylreductase (ER) active site. Both cis-AT PKSs, which contain embedded ATs, and trans-AT PKSs, which rely on separately encoded ATs, are considered since all of the enzymes, with the exception of ER, are structurally and functionally equivalent.2, 6 Other less common enzymes within modular PKSs, such as methyltransferases (MTs), enoyl isomerases (EIs), and pyran synthases (PSs), can also set stereocenters but are not as well characterized.7–11
II. KS inversion of configuration
Most KSs from modular PKSs condense malonyl extender units with growing polyketides and do not have an opportunity to control stereocenters. When extender units larger than malonyl-CoA are condensed, the building blocks themselves are chiral. The most common of these α-substituted extender units is (2S)-methylmalonyl-CoA (its epimer is not known to be utilized by modular PKSs).12 When an α-substituted malonyl extender unit is condensed with a polyketide intermediate, the α-substituent appears in the product in the opposite stereochemical orientation. This “inversion of configuration” was first demonstrated in fatty acid biosynthesis.13 Although several high-resolution structures of condensation-competent KSs from modular PKSs have been reported, no physical model has been proposed for how the “inversion of configuration” reaction proceeds, most likely due to the mysterious nature of the condensation reaction itself (PDBs: 2HG4, 3Q0P, 4NA1, 4MZ0, 4OPE, 4OQJ, 4QYR, 4TKT, 4WKY, 4ZDN).14–18
KSs belong to the thiolase superfamily.19 In contrast to the condensation mechanism of KSs, the condensation mechanism of thiolases is well described on a structural level from crystal structures of several intermediates along the reaction pathway (e.g., PDB: 1DM3).20 Most biosynthetic thiolases deprotonate acetyl-CoA to generate an enolate intermediate that can attack an acyl group bound to the catalytic cysteine. KSs form enolates through the loss of the extender unit carboxylate.21 Presumably, the enolates of thiolases and KSs are positioned equivalently for the attack of the acyl-enzyme (Figure 2).
Figure 2.
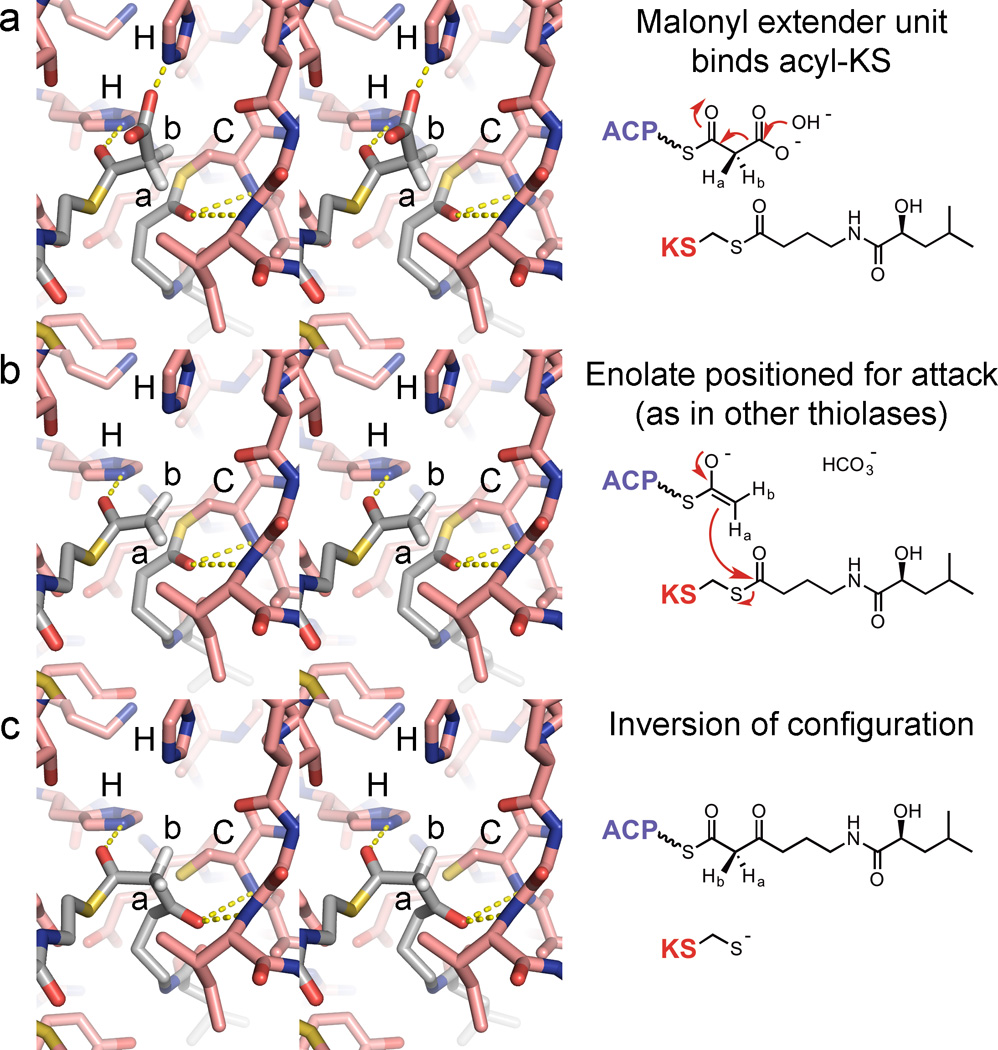
KS inversion of configuration. (a) A malonyl extender binds to an acylated KS (the illustrated KS was observed bound to a bacillaene intermediate; in the actual structure a serine substitutes for the catalytic cysteine; PDB: 4NA2). Attack of a hydroxide ion may release bicarbonate. (b) The enolate is positioned as observed in a crystal structure of a related thiolase incubated with acetyl-CoA (PDB: 1DM3). (c) After carbon-carbon bond formation, the stereochemical orientations of the Ha and Hb substituents are inverted. The methyl substituent of a methylmalonyl extender unit would replace Hb.
Recently, a crystal structure of a polyketide-bound KS was obtained, albeit with a serine substituting for the catalytic cysteine.17 The structure is of the KS from the second module of the bacillaene PKS covalently bound to its natural substrate (PksKS2; 2.30 Å-resolution; PDB: 4NA2). In the second half-reaction catalyzed by PksKS2, the thioester intermediate is attacked by the enolate generated in the first half-reaction through the loss of the malonyl carboxylate. Since this enolate is the same as that generated by biosynthetic thiolases that utilize acetyl-CoA, by utilizing the position of the enolate observed in the thiolase crystal structure 1DM3 the PksKS2 reaction can be modeled (Figure 2).20 A malonyl extender unit would bind to PksKS2 such that loss of the carboxylate generates an acetyl enolate as observed in 1DM3 (Figures 2a and 2b). This prepares the carbon-carbon bond-forming step that results in an “inversion of configuration” (Figure 2c). A reaction between a methylmalonyl extender unit and a polyketide-bound KS would proceed with a methyl group in place of the hydrogen labeled Hb in Figure 2.
III. KR stereoselectivity and stereospecificity
KRs are stereochemical workhorses, setting most of the stereocenters within polyketide scaffolds. Accordingly, they are the most studied of the enzymes embedded within modular PKSs and a relative abundance of structural and functional data exist for the many KR types.22 The stereocontrol exerted by KRs is exquisite, as they can be both stereoselective and stereospecific. KRs can be stereoselective in generating either an l-β-hydroxyl group (A-type KRs) or a d-β-hydroxyl group (B-type KRs) (the d/l system is preferred over the R/S system for polyketide intermediates since the assignment of R or S depends on the substituents present). They can also be stereospecific in reducing a substrate with either a d-α-substituent (A1- or B1-type KRs) or an l-α-substituent (A2- or B2-type KRs). Additionally, KRs can catalyze the epimerization of an α-substituent of an α-substituted-β-ketoacyl substrate preceding reduction (A2- or B2-type KRs) or in the absence of a subsequent reduction (C2-type).
The current body of structural knowledge can help inform us about the physical mechanisms underlying KR stereocontrol. In most biosynthetic pathways (e.g., fatty acids, polyketides, polyhydroxyalkanoates), ketoreductions are performed by short-chain dehydrogenase/reductase (SDR) enzymes on β-ketoacyl substrates to generate d-β-hydroxyacyl products.23 PKS KRs are SDR enzymes, and B-type KRs perform equivalent stereoselective reductions. Besides similar nicotinamide coenzyme binding sites and catalytic residues, these enzymes share other features such as a glutamine three residues before the catalytic tyrosine and an aspartate on a loop adjacent to the active site (in cis-AT PKS KRs this aspartate is the second aspartate of the “Leu-Asp-Asp motif”, and the loop is referred to as “Loop DE”).
The first determined KR crystal structure was of the B2-type KR (EryKR1, from the first module of the erythromycin PKS; 1.81 Å-resolution; PDB: 2FR0), which performs a stereoselective and stereospecific reduction reaction.24 Although some analogies could be made from structural and functional studies of the bacterial fatty acid synthase (FAS) ketoreductase FabG (PDB: 1Q7B)25, determining how EryKR1 exerts stereocontrol was, and continues to be, challenging. While a B-type KR that reduces an α-unsubstituted intermediate would have made for a more direct comparison, only recently has an informative structure of such an enzyme been determined, from the sixth module of the macrolactin PKS (MlnKR6, with NADP+ bound; 1.75-Å resolution; PDB: 5D2E).
The comparison of MlnKR6 with functionally equivalent enzymes has been made even more enlightening through recent crystal structures of SDR enzyme complexes. A high-molecular weight FabG (HMwFabG) has been observed complexed with hexanoyl-CoA and NADP+ (PDB: 3V1U)26, and an acetoacetyl-CoA reductase has been observed complexed with acetoacetyl-CoA (PDBs: 4N5M)27. The pantetheinyl arms of the substrates are equivalently bound - a glutamine forms a hydrogen bond with the thioester carbonyl and an aspartate forms a hydrogen bond with the NH of the amide closest to the thioester (Figure 3a). Together, the aspartate, the backbone NH of the residue preceding it, and the asparagine preceding the catalytic tyrosine coordinate a water molecule critical for the hydrogen bond network. This water molecule is equivalently coordinated in the crystal structure of MlnKR6. The residues of Loop DE possess high B-factors in each of the previous structures of B-type KRs, thus an equivalent hydrogen bond network had not been observed.24, 28–30
Figure 3.
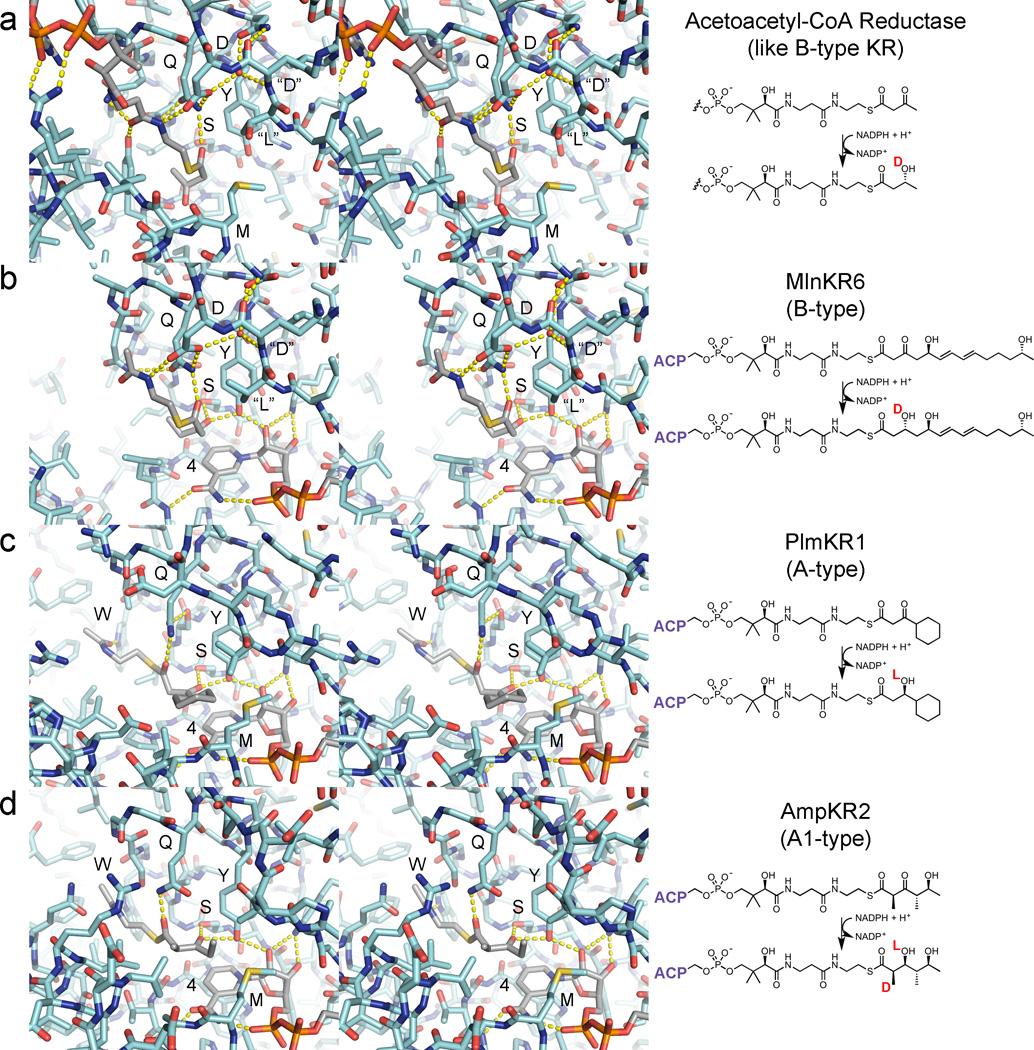
KRs stereoselectivity and stereospecificity. (a) An acetoacetyl-CoA reductase from a polyhydroxyalkanoate pathway was observed bound to acetoacetyl-CoA (PDB: 4N5M). Contacts with the acylated phosphopantetheinyl moiety include hydrogen bonds between the thioester and a glutamine NH2 as well as between the NH closest to the thioester and an aspartate. This hydrogen bond network is largely mediated through the interaction of loop residues (equivalent to the “Leu-Asp-Asp motif” in B-type KRs from cis-AT PKSs) with a well-ordered water molecule. (b) A crystal structure of the B-type KR MlnKR6 bound to NADP+ shows an equivalent interaction between the Leu-Asp-Asp motif and a well-ordered water molecule (PDB: 5D2E). A mimic of the natural substrate was modeled based on the acetoacetyl-CoA reductase complex. The β-keto group is able to form a hydrogen bond with the catalytic tyrosine such that reduction by NADPH will stereospecifically yield a β-hydroxyl group with a d-orientation. (c) A substrate mimic can readily be modeled in the crystal structure of the A-type KR PlmKR1 bound to NADP+ (PDB: 3HXY). Through positioning the β-keto group such that a β-hydroxyl group with an l-orientation will be generated through the reduction reaction, the cyclohexyl moiety becomes positioned to make hydrophobic interactions, the thioester carbonyl forms a hydrogen bond with the glutamine NH2 and the amide carbonyl closest the thioester forms a hydrogen bond with the indole NH of the signature A-type tryptophan. (d) A substrate mimic can also readily be modeled in the active site of the A1-type KR AmpKR2 bound to NADP+ (PDB: 3MJS). The α-methyl group may need to possess a d-orientation for stereospecific reduction to occur, since in the l-orientation it would sterically prevent the approach of the NADPH hydride to the β-keto carbon. The reactive NADPH hydrogen on the nicotinamide C4 carbon is labeled “4”.
The hexanoyl-CoA and acetoacetyl-CoA ligands complexed with the HMwFabG and acetoacetyl-CoA reductase indicate how pantetheinyl-bound β-ketoacyl substrates enter KR active sites and thus how B-type KRs enforce stereoselective reduction. From the positions of these molecules, a portion of the MlnKR6 substrate can be modeled in the MlnKR6 active site (Figure 3b). With its thioester bound equivalently to the thioesters of the hexanoyl-CoA and acetoacetyl-CoA ligands, the β-keto group is in position to form hydrogen bonds with the catalytic tyrosine and serine such that hydride transfer from NADPH will generate a d-β-hydroxy product.
Unlike B-type KRs, A-type KRs do not possess many functional equivalents from other pathways, thus little structural data is available to help explain their stereoselectivity. While several A-type KR structures have been solved (PDBs: 3MJS, 4L4X, 4IMP, 4HXY)31–34, perhaps the most insight into A-type stereoselectivity comes from the structure of the KR from the first module of the phoslactomycin PKS (PlmKR1; 1.68-Å resolution; PDB 4HXY)34. Its substrate has relatively few degrees of freedom and when its β-keto group is positioned between the catalytic tyrosine and serine such that reduction by NADPH will yield a β-hydroxyl group with an l-orientation, its cyclohexyl moiety contacts hydrophobic residues, its thioester carbonyl forms a hydrogen bond with the glutamine NH2, and its amide carbonyl forms a hydrogen bond with the indole NH of a tryptophan conserved in A-type KRs from cis-AT PKSs (Figure 3c).
The crystal structure of the KR from the second module of the amphotericin PKS bound to NADP+ (AmpKR2; 1.40-Å resolution, PDB: 3MJS) provides a starting point for understanding stereospecificity.31 The substrate mimic d-α-methyl-β-ketovaleroyl-S-NAC fits into the AmpKR2 active site equivalent to how the PlmKR1 active site is bound by its substrate, with the β-keto group between the catalytic tyrosine and serine, the thioester carbonyl forming a hydrogen bond with the glutamine NH2, and the amide carbonyl forming a hydrogen bond with the indole NH of the A-type tryptophan. This places the d-α-methyl substituent out of the way for the hydride transfer from NADPH. If an l-α-methyl-β-keto substrate were to bind, the methyl group would sterically prevent the approach of the substrate to NADPH. Molecular dynamics studies support this hypothesis.35 Experimentally, AmpKR2 almost exclusively prefers d-α-methyl-β-ketovaleroyl-S-NAC to l-α-methyl-β-ketovaleroyl-S-NAC.31, 32 When the glutamine is replaced with a histidine, commonly present in A2-type KRs, the mutant reduces the two enantiomers equally. When the position of the conserved tryptophan is also altered by replacing a glycine with a threonine, commonly present in many A2-type KRs, the double mutant almost exclusively prefers l-α-methyl-β-ketovaleroyl-S-NAC to d-α-methyl-β-ketovaleroyl-S-NAC. Curiously, the double mutant is four times more catalytically efficient than unmutated AmpKR2. The data may indicate that the A2-type ketoreduction reaction proceeds through a low-energy barrier and is discouraged by other KR types.
The aforementioned acetoacetyl-CoA reductase was also solved without acetoacetyl-CoA (PDBs: 4N5L, 4N5N).27 In the presence of acetoacetyl-CoA, the helix equivalent to the KR “lid helix” shifts 4.6 Å to close over the substrate. As observed for several A-type KRs, a methionine on the loop N-terminal to the “lid helix” is in position to latch over the nicotinamide coenzyme and make contact with the acyl portion of the substrate (Figure 3). When this methionine was replaced with alanine in AmpKR2, the resulting mutant was catalytically incompetent.31 From these data it is apparent that during the reduction reaction, KRs and related enzymes clamp down on both the nicotinamide coenzyme and the substrate undergoing reduction. A crystal structure of such a ternary complex is likely to reveal more of the detailed interactions that exist between KRs and their substrates to enable stereocontrolled reduction.
Epimerization of d-α-methyl-β-keto substrates is catalyzed in modular PKSs by A2-, B2-, and C2-type KRs, as well as epimerizing DHs.22, 28, 36–39 This phenomenon was first noticed from isotope-labeling studies of erythromycin in which hydrogens geminal to d-methyl groups were shown to be derived from propionate precursors and hydrogens geminal to l-methyl groups were shown to be derived from water.40 Thus the A1-type KRs of the erythromycin synthase (EryKR2, EryKR5, and EryKR6) perform reduction before epimerization can occur, while the B2-type (EryKR1) and C2-type (EryKR3) KRs catalyze epimerization before other reactions can ensue. That C2-type KRs, such as the KR in the third module of the erythromycin PKS, or epimerizing DHs, such as the DH in fourth module of the FK506 PKS, have been retained through evolution is telling of the relatively slow rate of uncatalyzed epimerization. How epimerization is catalyzed by KRs is unknown but may be as simple as positioning the thioester and β-carbonyl groups in the same plane to enable water to abstract the α-proton and generate an enolate that can accept a proton from its other side. The structure of the C2-type KR from the third module of the pikromycin PKS (PikKR3; 1.88-Å resolution, PDB: 3QP9) reveals that although its NADPH-binding motif is corrupted, the tyrosine, serine, glutamine, and tryptophan are still positioned as in A-type KRs to bind substrate.38
IV. DH double bond formation
The carbons of double bonds within polyketide scaffolds usually represent stereocenters installed by DHs during assembly line biosynthesis. These double-hotdog fold enzymes may install either cis- or trans-double bonds, although trans-double bonds are more frequently generated, especially by cis-AT PKSs.41, 2
Double bonds are formed through the syn-coplanar elimination of the β-hydroxy group and the l-α-proton.42 For elimination to occur, a DH needs to bind a substrate in a conformation in which the substituents of its α- and β-carbons are eclipsed. The substrate must also be bound such that the β-hydroxy group can be protonated by the catalytic aspartic acid and the l-α-proton can be abstracted by the catalytic histidine (Figure 4a). Through positioning a substrate mimic in the active site of a DH, it is apparent that it also can form a hydrogen bond with the NH of a conserved glycine through its thioester carbonyl and a conserved tyrosine through its hydroxyl group hydrogen. Bound in this fashion, the elimination reaction will generate a trans-double bond from a substrate possessing a d-β-hydroxy group and a cis-double bond from a substrate possessing an l-β-hydroxy group (Figure 4b). Thus, a cis-double bond can only be installed by a DH that operates on the product of an A-type KR. Whether a DH requires a differently shaped cavity to dehydrate l-β-hydroxy intermediates is unknown. Several structures of DHs from cis-AT PKSs have been determined, including one from the curacin PKS implicated in catalyzing the formation of a cis-double bond, and each possesses a remarkably similar active site.41, 43, 44 Holo-ACP substrates may dock to DHs as observed in a dehydratase/acyl carrier protein complex from a bacterial FAS.45
Figure 4.
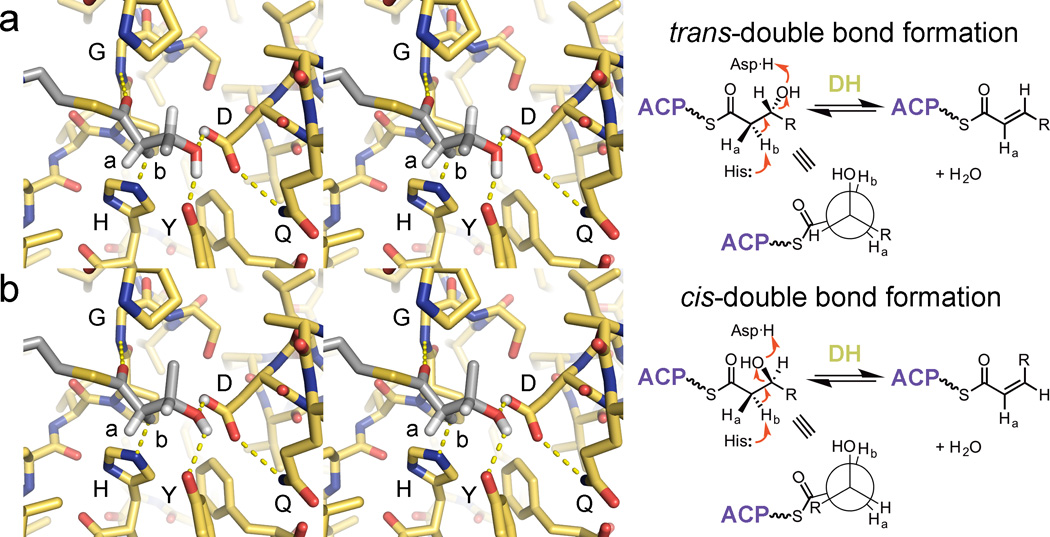
DH double bond formation. (a) Dehydration of a substrate to yield a trans-double bond is thought to occur through the syn-coplanar elimination of its d-β-hydroxyl group and l-α-proton (Hb). For this to occur, DH must bind its substrate so that its α- and β-substituents eclipse one another. All reported DH structures show the same orientation of the catalytic histidine and aspartic acid as well as the glycine NH; the tyrosine is also highly conserved (PDB: 3EL6). (b) The syn-coplanar dehydration reaction to generate a cis-double bonded product from an l-β-hydroxyacyl substrate is most likely catalytically equivalent to the syn-coplanar dehydration reaction that generates a trans-double bonded product from a d-β-hydroxyacyl substrate. DHs that generate cis-double bonds must accommodate for the different chain geometry.
V. ER stereoselectivity
A PKS module has a final opportunity to set a stereocenter if a trans-α,β-unsaturated intermediate possessing an α-subsituent is presented to an ER. The medium chain dehydrogenase/reductase (MDR) ERs from cis-AT PKSs are discussed here since much less is known about the TIM-barrel ERs of trans-AT PKSs, although a structure of a trans-ER from the difficidin PKS provides a basis for understanding its embedded relatives (DifA-ER; 2.30 Å-resolution; PDB: 4CW5).46
Only one crystal structure of a cis-AT PKS ER has been reported: the ER from the second module of the spinosyn PKS complexed with NADP+ (SpnER2; 3.0-Å resolution; PDB: 3SLK).29 SpnER2 does not set a stereocenter, but it does possess the tyrosine that has been demonstrated to deliver a proton to the enolate intermediate in l-type ERs.47, 48 The SpnER2 structure provides a framework to understand ER stereoselectivity, especially through a comparison with the ternary complex of a related MDR enzyme. Etr1p, a crotonyl-CoA reductase from the mitochondria of Candida tropicalis, was solved in complex with NADP+ and crotonyl-CoA (PDB: 4WAS) (Figure 5a).49 By placing a substrate mimic within the SpnER2 active site as crotonyl-CoA is positioned within the Etr1p active site, the relation of the enolate intermediate and the general acid that protonates it is made more apparent (Figure 5b). In l-type ERs, the tyrosine protonates the enolate intermediate to generate an l-α-substituent, whereas in d-type ERs the lysine is thought to protonate it from the opposite side to generate a d-α-substituent. Enolates may be generated by Etr1p through ene chemistry; whether ERs, and even KRs, also perform catalysis through such a mechanism remains an intriguing possibility.49, 50
Figure 5.
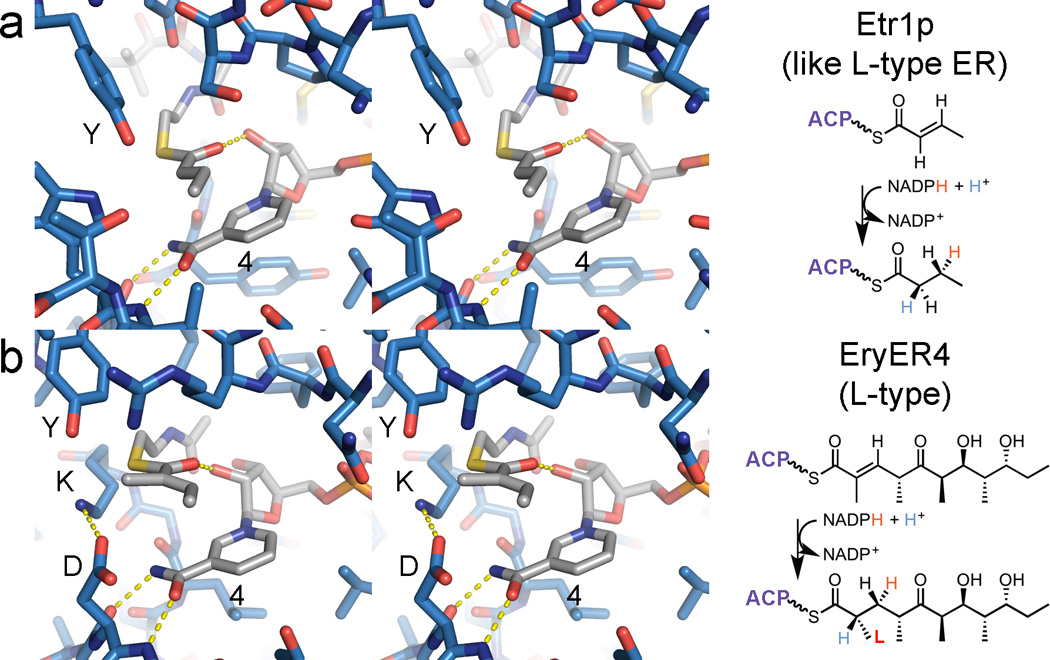
ER stereoselectivity. (a) The crystal structure of Etr1p bound to NADP+ and crotonyl-CoA elucidates how such MDR enzymes mediate reduction (PDB: 4WAS). (b) The structure of SpnER2 bound to NADP+ can help determine how PKS ERs perform a similar reaction (PDB: 3SLK). In the reduction performed by the l-type ER, EryER4, a tyrosine equivalent to the catalytic tyrosine of SpnER2 and Etr1p can add a proton to the enolate intermediate to stereoselectively generate an α-methyl substituent with an l-orientation. d-type ERs do not possess this tyrosine; the lysine, on the other face of the enolate intermediate, is hypothesized to donate a proton to generate a d-α-methyl substituent. The reactive NADPH hydrogen on the nicotinamide C4 carbon is labeled “4”.
VI. Conclusion
How PKS assembly line enzymes control stereochemistry is beginning to make both chemical and physical sense, especially owing to many recent crystal structures; however, much data remains to be collected. For example, while a binding mode presented here seeks to account for the stereoselectivity and stereospecificity of the A1-type KR AmpKR2, it needs to be tested experimentally and computationally through modifications of both substrate and enzyme. A crystal structure in which the AmpKR2 clamps over both its nicotinamide coenzyme and its ACP-tethered substrate should also be sought, although it may not be trivial to obtain. Since stereocontrol is intimately linked with reaction mechanism, many fundamental mechanistic details need to be worked out for KS, KR, DH, and ER. A topic that perhaps should have been discussed is whether the KS downstream of a module can select for a processed intermediate. A processing enzyme often cannot drive a reaction to completion by itself and may require a KS to choose between epimers or between hydrated and dehydrated intermediates. This will require more pioneering efforts in understanding the specificities of KSs.51–53 PKS enzymes have relatives in other pathways, and, as shown here, much can be learned from structural and functional studies of them. Hopefully, this compilation of stereocontrolled reactions will aid in designing experiments that further refine our understanding of how polyketide assembly line enzymes set stereocenters.
Supplementary Material
{kind=link}
Figure 1.

Modular PKS enzymes and the stereocenters they set. (a) Crystal structures of a representative KS, KR, DH, and ER domain (PDB: 4NA2, 3SLK, 3EL6) as well as an NMR structure of a representative holo-ACP (PDB: 2LIW) are shown above the primary structure of a full cis-AT PKS module (~2500 aa). Note the 18-Å phosphopantetheinyl arm of ACP. The methyltransferase (MT) domain is largely uncharacterized and will not be discussed here. (b) Extender units, selected by ATs, are condensed with polyketide intermediates by KSs. The elongation reaction and subsequent processing reactions are exquisitely stereocontrolled (stereochemical orientations are indicated). Any one of the displayed ketide units can by added by a PKS module containing the appropriate enzymes. All molecules are bound to an ACP. Enzyme types are defined in the text.
REFERENCES
- 1.Keatinge-Clay AT. The structures of type I polyketide synthases. Nat. Prod. Rep. 2012;29:1050. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 2.Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2010;27:996. doi: 10.1039/b816430b. [DOI] [PubMed] [Google Scholar]
- 3.Straight PD, Fischbach MA, Walsh CT, Rudner DZ, Kolter R. A singular enzymatic megacomplex from Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2007;104:305. doi: 10.1073/pnas.0609073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwan DH, Schulz F. The stereochemistry of complex polyketide biosynthesis by modular polyketide synthases. Molecules. 2011;16:6092. doi: 10.3390/molecules16076092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Breitling R, Takano E, Medema MH. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piel J. Accompanying review. Nat. Prod. Rep. 2015 [Google Scholar]
- 7.Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, Keasling JD. Divergent mechanistic routes for the formation of gem-dimethyl groups in the biosynthesis of complex polyketides. Angew. Chem. Int. Ed. Engl. 2015;54:2370. doi: 10.1002/anie.201410124. [DOI] [PubMed] [Google Scholar]
- 8.Kusebauch B, Busch B, Scherlach K, Roth M, Hertweck C. Functionally distinct modules operate two consecutive alpha,beta-->beta,gamma double-bond shifts in the rhizoxin polyketide assembly line. Angew. Chem. Int. Ed. Engl. 2010;49:1460. doi: 10.1002/anie.200905467. [DOI] [PubMed] [Google Scholar]
- 9.Gay DC, Spear PJ, Keatinge-Clay AT. A double-hotdog with a new trick: structure and mechanism of the trans-acyltransferase polyketide synthase enoyl-isomerase. ACS Chem. Biol. 2014;9:2374. doi: 10.1021/cb500459b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pöplau P, Frank S, Morinaka BI, Piel J. An enzymatic domain for the formation of cyclic ethers in complex polyketides. Angew. Chem. Int. Ed. Engl. 2013;52:13215. doi: 10.1002/anie.201307406. [DOI] [PubMed] [Google Scholar]
- 11.Luhavaya H, Dias MV, Williams SR, Hong H, de Oliveira LG, Leadlay PF. Enzymology of pyran ring A formation in salinomycin biosynthesis. Angew. Chem. Int. Ed. Engl. 2015;54:13622. doi: 10.1002/anie.201507090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan YA, Podevels AM, Kevany BM, Thomas MG. Biosynthesis of polyketide synthase extender units. Nat. Prod. Rep. 2009;26:90. doi: 10.1039/b801658p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sedgwick B, Cornforth JW. The biosynthesis of long-chain fatty acids. Stereochemical differentiation in the enzymic incorporation of chiral acetates. Eur. J. Biochem. 1977;75:465. doi: 10.1111/j.1432-1033.1977.tb11549.x. [DOI] [PubMed] [Google Scholar]
- 14.Tang Y, Kim C-Y, Mathews II, Cane DE, Khosla C. The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11124. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Y, Chen AY, Kim C-Y, Cane DE, Khosla C. Structural and mechanistic analysis of protein interactions in module 3 of the 6-deoxyerythronolide B synthase. Chem. Biol. 2007;14:931. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whicher JR, Smaga SS, Hansen DA, Brown WC, Gerwick WH, Sherman DH, Smith JL. Cyanobacterial polyketide synthase docking domains: a tool for engineering natural product biosynthesis. Chem. Biol. 2013;20:1340. doi: 10.1016/j.chembiol.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gay DC, Gay G, Axelrod AJ, Jenner M, Kohlhaas C, Kampa A, Oldham NJ, Piel J, Keatinge-Clay AT. A close look at a ketosynthase from a trans-acyltransferase modular polyketide synthase. Structure. 2014;22:444. doi: 10.1016/j.str.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohman JR, Ma M, Osipiuk J, Nocek B, Kim Y, Chang C, Cuff M, Mack J, Bigelow L, Li H, Endres M, Babnigg G, Joachimiak A, Phillips GN, Jr, Shen B. Structural and evolutionary relationships of "AT-less" type I polyketide synthase ketosynthases. Proc. Natl. Acad. Sci. USA. 2015;112:12693. doi: 10.1073/pnas.1515460112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haapalainen AM, Meriläinen G, Wierenga RK. The thiolase superfamily: condensing enzymes with diverse reaction specificities. Trends Biochem. Sci. 2006;31:64. doi: 10.1016/j.tibs.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Modis Y, Wierenga RK. Crystallographic analysis of the reaction pathway of Zoogloea ramigera biosynthetic thiolase. J. Mol. Biol. 2000;297:1171. doi: 10.1006/jmbi.2000.3638. [DOI] [PubMed] [Google Scholar]
- 21.Witkowski A, Joshi AK, Smith S. Mechanism of the beta-ketoacyl synthase reaction catalyzed by the animal fatty acid synthase. Biochemistry. 2002;41:10877. doi: 10.1021/bi0259047. [DOI] [PubMed] [Google Scholar]
- 22.Zheng J, Keatinge-Clay AT. The status of type I polyketide synthase ketoreductases. Med. Chem. Commun. 2013;4:34. [Google Scholar]
- 23.Kavanagh KL, Jörnvall H, Persson B, Oppermann U. Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 2008;65:3895. doi: 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keatinge-Clay AT, Stroud RM. The structure of a ketoreductase determines the organization of the beta-carbon processing enzymes of modular polyketide synthases. Structure. 2006;14:737. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Price AC, Zhang YM, Rock CO, White SW. Cofactor-induced conformational rearrangements establish a catalytically competent active site and a proton relay conduit in FabG. Structure. 2004;12:417. doi: 10.1016/j.str.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Dutta D, Bhattacharyya S, Roychowdhury A, Biswas R, Das AK. Crystal structure of hexanoyl-CoA bound to β-ketoacyl reductase FabG4 of Mycobacterium tuberculosis. Biochem. J. 2013;450:127. doi: 10.1042/BJ20121107. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Chang JH, Kim EJ, Kim KJ. Crystal structure of (R)-3-hydroxybutyryl-CoA dehydrogenase PhaB from R alstonia eutropha. Biochem. Biophys. Res. Commun. 2014;443:783. doi: 10.1016/j.bbrc.2013.10.150. [DOI] [PubMed] [Google Scholar]
- 28.Keatinge-Clay AT. A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem. Biol. 2007;14:898. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Zheng J, Gay DC, Demeler B, Keatinge-Clay AT. Divergent evolution of multimodular polyketide synthases revealed through the architecture of a ketoreductase-enoylreductase didomain. Nat. Chem. Biol. 2012;8:615. doi: 10.1038/nchembio.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piasecki SK, Zheng J, Axelrod AJ, Detelich M, Keatinge-Clay AT. Structural and functional studies of a trans-acyltransferase polyketide synthase ketoreductase the performs both alpha- and beta-ketoreduction. Proteins. 2014;82:2067. doi: 10.1002/prot.24561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng J, Taylor CA, Piasecki SK, Keatinge-Clay AT. Structural and functional analysis of A-type ketoreductases from the amphotericin modular polyketide synthase. Structure. 2010;18:913. doi: 10.1016/j.str.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 32.Zheng J, Piasecki SK, Keatinge-Clay AT. Structural studies of an A2-type modular polyketide synthase ketoreductase reveal features controlling alpha-substituent stereochemistry. ACS Chem. Biol. 2013;8:1964. doi: 10.1021/cb400161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng J, Fage CD, Demeler B, Hoffman DW, Keatinge-Clay AT. The missing linker: a dimerization motif located within polyketide synthase modules. ACS Chem. Biol. 2013;8:1263. doi: 10.1021/cb400047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonnett SA, Whicher JR, Papireddy K, Florova G, Smith JL, Reynolds KA. Structural and stereochemical analysis of a modular polyketide synthase ketoreductase domain required for the generation of a cis-alkene. Chem. Biol. 2013;20:772. doi: 10.1016/j.chembiol.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mugnai ML, Shi Y, Keatinge-Clay AT, Elber R. Molecular dynamics studies of modular polyketide synthase ketoreductase stereospecificity. Biochemistry. 2015;54:2346. doi: 10.1021/bi501401g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valenzano CR, Lawson RJ, Chen AY, Khosla C, Cane DE. The biochemical basis for stereochemical control in polyketide biosynthesis. J. Am. Chem. Soc. 2009;131:18501. doi: 10.1021/ja908296m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng J, Keatinge-Clay AT. Structural and functional analysis of C2-type ketoreductases from modular polyketide synthases. J. Mol. Biol. 2011;410:105. doi: 10.1016/j.jmb.2011.04.065. [DOI] [PubMed] [Google Scholar]
- 38.Garg A, Khosla C, Cane DE. Coupled methyl group epimerization and reduction by polyketide synthase ketoreductase domains, Ketoreductase-catalyzed equilibrium isotope exchange. J. Am. Chem. Soc. 2013;135:16324. doi: 10.1021/ja408944s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garg A, Xie X, Keatinge-Clay A, Khosla C, Cane DE. Elucidation of the cryptic epimerase activity of redox-inactive ketoreductase domains from modular polyketide synthases by tandem equilibrium isotope exchange. J. Am. Chem. Soc. 2014;136:10190. doi: 10.1021/ja5056998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cane DE, Liang TC, Taylor PB, Chang C, Yang CC. Macrolide biosynthesis. 3. Stereochemistry of the chain-elongation steps of erythromycin biosynthesis. J. Am. Chem. Soc. 1986;108:4957. [Google Scholar]
- 41.Keatinge-Clay AT. Crystal structure of the erythromycin polyketide synthase dehydratase. J. Mol. Biol. 2008;384:941. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valenzano CR, You Y-O, Garg A, Keatinge-Clay A, Khosla C, Cane DE. Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. J. Am. Chem. Soc. 2010;132:14697. doi: 10.1021/ja107344h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Crystal structures of dehydratase domains from the curacin polyketide biosynthetic pathway. Structure. 2010;18:94. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gay DC, You Y-O, Keatinge-Clay AT, Cane DE. Structural and stereospecificity of the dehydratase domain from the terminal module of the rifamycin polyketide synthase. Biochemistry. 2013;52:8916. doi: 10.1021/bi400988t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen C, Haushalter RW, Lee DJ, Markwick PR, Bruegger J, Caldara-Festin G, Finzel K, Jackson DR, Ishikawa F, O'Dowd B, McCammon JA, Opella SJ, Tsai S-C, Burkart MD. Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature. 2014;505:427–431. doi: 10.1038/nature12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bukhari HS, Jakob RP, Maier T. Evolutionary origins of the multienzyme architecture of giant fungal fatty acid synthase. Structure. 2014;22:1775. doi: 10.1016/j.str.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 47.Kwan DH, Sun Y, Schulz F, Hong H, Popovic B, Sim-Stark JC, Haydock SF, Leadlay PF. Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem. Biol. 2008;15:1231. doi: 10.1016/j.chembiol.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 48.Kwan DH, Leadlay PF. Mutagenesis of a modular polyketide synthase enoylreductase domain reveals insights into catalysis and stereospecificity. ACS Chem. Biol. 2010;5:829. doi: 10.1021/cb100175a. [DOI] [PubMed] [Google Scholar]
- 49.Rosenthal RG, Vögeli B, Quade N, Capitani G, Kiefer P, Vorholt JA, Ebert MO, Erb TJ. The use of ene adducts to study and engineer enoyl-thioester reductases. Nat. Chem. Biol. 2015;11:398. doi: 10.1038/nchembio.1794. [DOI] [PubMed] [Google Scholar]
- 50.Rosenthal RG, Ebert MO, Kiefer P, Peter DM, Vorholt JA, Erb TJ. Direct evidence for a covalent ene adduct intermediate in NAD(P)H-dependent enzymes. Nat. Chem. Biol. 2014;10:50. doi: 10.1038/nchembio.1385. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen T, Ishida K, Jenke-Kodama H, Dittmann E, Gurgui C, Hochmuth T, Taudien S, Platzer M, Hertweck C, Piel J. Exploiting the mosaic structure of trans-acyltransferase polyketide synthases for natural product discovery and pathway dissection. Nat Biotechnol. 2008;26:225. doi: 10.1038/nbt1379. [DOI] [PubMed] [Google Scholar]
- 52.Jenner M, Frank S, Kampa A, Kohlhaas C, Pöplau P, Briggs GS, Piel J, Oldham NJ. Substrate specificity in ketosynthase domains from trans-AT polyketide synthases. Angew. Chem. Int. Ed. Engl. 2013;52:1143. doi: 10.1002/anie.201207690. [DOI] [PubMed] [Google Scholar]
- 53.Jenner M, Afonso JP, Bailey HR, Frank S, Kampa A, Piel J, Oldham NJ. Acyl-chain elongation drives ketosynthase substrate selectivity in trans-acyltransferase polyketide synthases. Angew. Chem. Int. Ed. Engl. 2015;54:1817. doi: 10.1002/anie.201410219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.