

Abstract
Covering: 2003 to July 2015 for polyketide synthase starter units and 2012 to September 2015 for polyketide synthase extender units
This highlight provides an overview of recent advances in understanding the diversity of polyketide synthase (PKS) substrate building blocks. Substrates functioning as starter units and extender units contribute significantly to the chemical complexity and structural diversity exhibited by this class of natural products. This article complements and extends upon the current comprehensive reviews that have been published on these two topics (Moore and Hertweck, Nat. Prod. Rep., 2002, 19, 70; Chan et. al., Nat. Prod. Rep., 2009, 1, 90; Wilson and Moore, Nat. Prod. Rep., 2012, 29, 72).
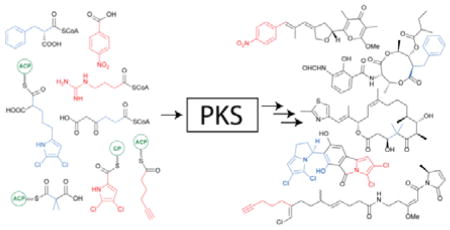
Graphical Abstract
Polyketides comprise a highly diverse class of natural products, with many important biological and pharmacological activities. Substrates functioning as starter units and extender units during their assembly significantly contribute to the chemical complexity and structural diversity exhibited by this class of natural products. This highlight provides an overview of the recent advances in understanding the diversity of these polyketide synthase (PKS) building blocks.
Introduction
Polyketides comprise a large class of natural products exhibiting a high degree of structural diversity, encompassing molecules such as macrolides, aromatics and polyenes. The structural diversity exhibited by polyketides is exemplified by the wide range of important biological and pharmacological activities they possess, including antimicrobial, antiproliferative and immunosuppressant. Polyketides are derived from successive decarboxylative Claisen condensation reactions between relatively simple malonate precursors and acylthioesters, in a manner analogous to that observed in fatty acid biosynthesis, resulting in a two carbon extension to the growing polyketide chain (Figure 1).1
Figure 1.

Mechanism of the decarboxylative Claisen condensation catalyzed by a KS domain during polyketide chain extension.
Polyketides are assembled by enzymes known as polyketide synthases (PKSs), which are classified into three distinct groups (type I, type II and type III PKSs) depending upon their structure and biochemistry. The classification of PKSs and indeed many aspects of polyketide biosynthesis have been the subject of several comprehensive reviews over the years, consequently a brief description of each shall be provided in this highlight.1–4 In summary, type I PKSs are large multifunctional, multimodular enzymes, with the catalytic domains responsible for polyketide assembly and β-keto group processing encoded within a polypeptide. The archetypal example of a type I PKS is the 6-deoxyerythronolide B synthase which is responsible for the biosynthesis of the aglycone core of the antibiotic erythromycin.1,2 Within type II PKSs, however, the catalytic domains required for polyketide chain assembly are encoded within discrete proteins that are used in an iterative manner generating intermediates in which the β-keto groups remain intact. Subsequent cyclizations yield the aromatic molecules typically associated with type II PKSs, such as for example actinorhodin.1,3 The third and simplest class is the type III PKSs. These homodimeric ketosynthases (KSs) differ from both type I and type II PKSs as they utilize free coenzyme A (CoA)-linked thioester substrates in an acyl carrier protein (ACP)-independent fashion, including such examples as 1,3,6,8-tetrahydroxynapthalene synthase.1,4
The structural diversity of polyketides can be attributed to a variety of factors, including the polyketide chain length, variations in the oxidation state and the stereochemistry of the β-keto groups, the mechanism of offloading and subsequent modifications to the polyketide carbon skeleton introduced by post-PKS tailoring enzymes.5,6 However, in recent years both polyketide starter and extender units have been illuminated as playing a significant role in adding to the structural diversity and chemical complexity observed within polyketide natural products.7–9 Their incorporation during thiotemplate-driven chain assembly represents an efficient means to add unusual moieties such as p-nitrobenzenes, alkynes, branched-alkyl chains and halogenated pyrroles, into polyketides.
Polyketide starter units
PKSs are known to incorporate a wide variety of starter unit carboxylates.7 With an ever-increasing number of starter units being identified and novel mechanisms for their incorporation being elucidated, they represent a significant route for the introduction of structural diversity into polyketide scaffolds.
New strategies for the biosynthesis of known starter units
The ease with which genome, and indeed metagenomic, sequencing data sets can be obtained has vastly improved in recent years, aiding both the discovery of new secondary metabolites and biosynthetic investigations into known compounds. The ability to link known secondary metabolites to their biosynthetic gene clusters and subsequent biosynthetic investigations has proved to be an invaluable tool, resulting in the identification of unprecedented biosynthetic routes towards commonly utilized PKS substrates. Such an example is the provision of the propionyl-ACP (1) starter unit incorporated into the lomaiviticins (2), biosynthesised by a type II PKS.10
In the absence of a priming ketosynthase (KS) domain, Balskus and co-workers identified a bifunctional acyltransferase/decarboxylase (AT/DC), lom62, within the biosynthetic gene cluster10,11 that was required for the formation of this starter unit. In vitro analysis demonstrated the ability of recombinant Lom62 to selectively load methylmalonyl-CoA onto the standalone ACP, Lom63, and subsequently decarboxylate this methylmalonyl-ACP intermediate to yield 1 (Figure 2).10 Intriguingly AT/DCs are typically associated with type I PKS systems. Not only does this represent a new mechanism in the biosynthesis of 1, it represents the first instance of an AT/DC being utilized by a type II PKS.10
Figure 2.

Biosynthesis of propionyl-ACP (1) via an unprecedented mechanism involving Lom62, a bifunctional AT/DC, for incorporation as a starter unit into lomaiviticin A (2).
Biosynthesis of new PKS starter units
In recent years, a wealth of unusual PKS starter units have been identified following both the discovery of new PKS-derived natural products and from investigations into previously isolated and characterized polyketides. An example of the latter includes the identification of the PKS starter unit p-nitrobenzoic acid (pNBA), which introduces a nitro aryl moiety into polyketide carbon scaffolds. Incorporated into aureothin (3), a polyketide first isolated from Streptomyces thioluteus in the 1960’s, pNBA was first elucidated as its starter unit approximately 40 years later following isotope labelling studies in which d4-pNBA was selectively incorporated into 3.12,13 Hertweck and co-workers also reported the genetic basis for pNBA formation alongside these chemical studies, revealing that a highly unusual p-aminobenzoate N-oxygenase, AurF, was responsible for the biosynthesis of pNBA. Deletion of aurF in vivo abolished the production of 3, which could subsequently be restored following chemical complementation with pNBA.13
In vitro characterization of AurF succeeded this initial work, with Zhao and co-workers demonstrating that AurF is a non-heme di-iron monooxygenase. Catalyzing sequential oxidation reactions, AurF catalyzes the addition of oxygen to p-aminobenzoic acid (pABA) yielding a hydroxyamine intermediate, p-hydroxylaminobenzoate (pHABA). Subsequent oxidation of this, via a nitroso intermediate, which was isolated and characterized, yields the pNBA starter unit (Figure 3A).14
Figure 3.

Biosynthesis of newly identified PKS starter units. (A) Biosynthesis of p-nitrobenzoic acid (pNBA), the starter unit for aureothin (3) biosynthesis. (B) Biosynthesis of the L-proline derived starter units pyrrolyl-CP (5) and 4,5-dichloropyrrolyl-CP (8). (C) Biosynthesis of the L-arginine derived starter unit 4-guanidinobutyryl-CoA (12). (D) Biosynthesis of 5-hexynoyl-ACP (16) for (I) incorporation into jamaicamide (17) and (II) utilization of its biosynthetic pathway for the bioengineering of an antimycin analogue (19). In this figure and all others in this article, we highlight the unusual starter unit and extender unit derived atoms in red and blue, respectively.
Many PKS pathways utilize modified amino acids as starter units to prime their thiotemplate assembly lines. Pyrrolyl-S-carrier protein (pyrrolyl-CP, 5), derived from L-proline, was first hypothesised as a starter unit in undecylprodiginine (4) biosynthesis following analysis of the biosynthetic gene cluster from Streptomyces coelicolor A3(2).15 Confirmation ensued with the in vitro characterization of SC3F7.11, a L-prolyl-AMP ligase, SC3F7.09, a CP, and RedW, a L-prolyl-S-CP dehydogenase.16 The selective adenylation of L-proline by SC3F7.11 allowed for subsequent loading of the L-prolyl-AMP intermediate onto the stand alone CP SC3F7.09, yielding L-prolyl-S-CP (6). Upon incubation with RedW, 6 was converted into 5, in a FAD-dependent manner (Figure 3B).16
Pyoluteorin (7), isolated from Pseudomonas fluorescens Pf-5, was also hypothesised to incorporate 5 as a starter unit; consequently characterization of PltF (SC3F7.11), PltL (SC3F7.09), and PltE, (RedW), was completed concomitantly with the above in vitro analysis.16 However characterization of a subsequent halogenation event illuminated 4,5-dichloropyrrolyl-CP (8) as the true starter unit for pyoluteorin biosynthesis (Figure 3B). The FADH2-dependent halogenase, PltA, was found to catalyze an unprecedented dichlorination of the pyrrole moiety at positions 4 and 5 following attachment to the CP, PltL.17 The carrier protein was observed to be essential for substrate recognition as the free pyrrole-2-carboxylate was not accepted as a substrate by recombinant PltA. 8 has since been postulated as the starter unit in pyrrolomycin (9), marinopyrrole (10) and chlorizidine A (11) biosynthesis, following identification of the genes encoding enzymes homologous to those in the pyoluteorin biosynthetic pathway (Figure 3B).18–20
L-Arginine was discovered to be the precursor of 4-guanidinobutyryl-CoA (12), the starter unit which primes the PKS assembly lines of the polyene polyketides azalomycin F3a (13) and clethramycins. The biosynthetic gene clusters of these secondary metabolites were identified in Streptomyces violaceusniger DSM 4137 following genome sequencing. Four enzymes which are duplicated in the S. violaceusniger genome – an arginine monooxygenase (AM, STRVN_6565 and STRVN_2699), a CoA ligase (STRVN_7500 and STRVN_2700), an AT (STRVN_7501 and STRVN_2704) and an amino hydrolase (AH, STRVN_6564 and STRVN_7510) – were found to be responsible for this transformation via the in vitro reconstitution of this pathway by Leadlay and co-workers.
Firstly, both flavin-dependent AMs were shown to catalyze the specific decarboxylation of L-arginine to 4-guanidinobutyramide (14). No conversion was observed when the AMs were challenged with different amino acid precursors including tryptophan, lysine, and glutamine. The activity of the AHs was confirmed via HPLC-MS analysis, with the conversion of 14 into 4-guanidinobutyric acid (15) observed. Finally the CoA ligases were found to transform 14 into the thioester 12. Interestingly there is extensive crosstalk between these two pathways, as determined genetically. Upon deletion of either AH, both 13 and the clethramycins were produced. However deleting both genes encoding the AHs resulted in the abolishment in production of 13 and the clethramycins.21 This ability of the azalomycin and clethramycin pathways to complement each other extends to the AT domains responsible for the loading of 12 onto the ACPs within the loading module of the PKS.21
The most intriguing of the recently characterized PKS starter units is 5-hexynoyl-ACP (16), the starter unit utilized during jamaicamide (17) biosynthesis.22 The incorporation of 16 into 17 results in the installation of a terminal alkyne moiety, a highly sought after functionality in natural products due to its chemical biology tagging properties via “Click” chemistry.23 Uncovered by Zhang and co-workers, the biosynthesis of 16 is mediated by a fatty-acyl-CoA ligase, a membrane-bound fatty acid desaturase and an ACP (JamA, JamB and JamC, respectively). Both in vivo and in vitro analysis revealed that JamA catalyzes the activation of 5-hexenoic acid by ATP and subsequent loading onto JamC, to furnish a 5-hexenoic-ACP (18) intermediate. Following this, JamB desaturates the terminal alkene of 18 to yield the alkyne 16, which is used to prime the PKS (Figure 3D(i)).24 In vitro investigations suggested that JamB does not exhibit much substrate tolerance. When challenged with alkenoyl-JamC thioesters of varying length (5 to 8 carbons) conversion to the corresponding alkynoyl-JamC was only observed for 16. Such substrate specificity was also observed with regards to the terminal alkene moiety, as no activity was detected when the position of unsaturation was altered in the starting hexenoic acid.24 The genes responsible for the provision of 16 are arranged in a single operon, forming a convenient cassette which can be utilized as a biosynthetic “tool” in the engineering of PKS pathways. Indeed, introduction of the jamA,B,C cassette into a heterologous Escherichia coli host in which a portion of the antimycin PKS/non-ribosomal peptide synthetase (NRPS) was coexpressed, resulted in the in situ generation of 16. Following subsequent extension, presumably via primary metabolic fatty acid synthase (FAS), and conversion into the malonate derivative via AntE (see below), 5-hexynoylmalonyl-CoA was incorporated into an antimycin analogue (19) as an extender unit (Figure 3D(ii)).24 This result paves the way to utilizing this trio of genes to generate “taggable” polyketides without the need for the feeding of alkyne precursors.
Novel incorporation mechanisms for PKS starter units
Alongside the wealth of PKS starter units that have been uncovered in recent years, advances have also been made with regards to their priming onto the PKS assembly line. These fall outside of the loading of PKS systems via a canonical AT domain within the loading module or via a KSQ domain, which decarboxylates malonate precursors that have been loaded onto the ACP within the initiating module by the loading AT domain.7
The potent antitumor compound FR901464 (20), isolated from Pseudomonas sp. No 2663, has been found to be the product of an NRPS/trans-AT PKS hybrid. Elucidation of the biosynthetic pathway of 20 unveiled a remarkable loading module for this canonical assembly line, which is comprised of 4 domains: a dehydratase-like domain (DH*), a ketoreductase-like domain (KR*), a domain with high sequence homology to the bifunctional glyceryl transferase/phosphatase (GAT) FkbH and an ACP.25 Previous reports of FkbH-like enzymes have illuminated their pivotal role in making glyceryl units available for PKS biosynthesis from D-1,3-bisphosphoglycerate (1,3-BPG).26–28 Whilst this domain architecture of the 20 PKS loading module had previously been observed in bryA which encodes the first 4 modules of the putative bryostatin PKS, both genetic and biochemical characterization had remained elusive29–31
Following the identification of the DH*-KR*-GAT-ACP loading module in the 20 PKS, Tan and co-workers set out to biochemically characterize the chain initiation mechanism in the assembly of 20. Following cloning and expression of individual GAT and ACP domains, they were able to demonstrate the activation of 1,3-BPG by GAT with simultaneous removal of the phospho group. The resulting glyceryl intermediate was then transferred to the ACP, allowing the DH* and KR* domains act upon this glyceryl intermediate, with the DH* catalyzing an initial dehydration to yield an ACP-linked pyruvate intermediate. Subsequent keto-reduction at the α-position by KR* generates the L-lactyl-ACP starter unit (21), thus priming the assembly line with this modified glycerate moiety.25,32
Another alternative mechanism for the loading of starter units to initiate chain elongation is via a GCN5-related N-acetyltransferase loading (GNATL) domain. GNATL has been observed in association with several PKS initiation modules, including the onnamide and theopederin PKSs, both identified from an uncultivated symbiont of the marine sponge Theonella swinhoei,33 and the curacin (22) PKS, from the marine cyanobacterium Lyngbya majuscula.34 Loading modules containing GNATL have been found to prime PKSs with an acetyl moiety, and are typically coupled with an ACPL. In some cases, including the loading module of the 22 PKS, an N-terminal adapter domain (AR) is present. In vitro and structural studies of the 22 PKS loading module by Sherman and coworkers illuminated GNATL as a bifunctional enzyme, with the ability to firstly catalyze the decarboxylation of malonyl-CoA into acetyl-CoA and subsequently catalyze the transfer of acetyl-CoA onto the ACP of this module, thus priming the PKS for 22 assembly.
To determine the mechanism with which the priming occurs, in vitro assays utilizing the full AR-GNATL-ACP and smaller GNATL-ACP didomain in both their apo and holo forms were performed. Whilst loading of the acetyl group was observed when holo forms of the curacin enzymes were challenged with malonyl-CoA, no covalent adduct was observed when utilizing the apo forms, thus confirming that the decarboxylated acetyl-CoA intermediate is directly transferred to the ACP. Comparisons between the efficiency of PKS priming between the full AR-GNATL-ACPL loading module and the truncated GNATL-ACPL didomain suggest that AR facilitates acyl transfer of the acetyl-CoA intermediate onto the ACP.34
Polyketide extender units
Whilst not as structurally diverse as starter units, PKS extender units are ever-increasingly being recognised for their ability to introduce chemical complexity into polyketides due to the identification of a number of highly unusual pathway-specific extender units, including chloroethylmalonyl-CoA (23), allylmalonyl-CoA (24) and hexylmalonyl-CoA (25) (Figure 5).35–37 Selected for by AT domains within the PKS (either by cis-acting or trans-acting ATs), these malonate derivatives can be linked to either CoA or a standalone ACP via a thioester linkage.9 The biosynthesis of atypical PKS extender units is generally encoded within the biosynthetic gene cluster of the secondary metabolite into which they are incorporated. Central to their biosynthesis are crotonyl-CoA carboxylase/reductases (CCRs), which catalyze the NADPH-dependent carboxylation of α,β-unsaturated acylthioesters in a highly stereoselective fashion.8,38,39 Recently, the intricate nature of this mechanism was illuminated by Erb and coworkers with the identification of a covalent ene adduct between NADPH and the acyl-CoA thioester during the catalytic cycle. Firstly, the pro-(4R) hydrogen on NADPH is transferred to the re face of C3 with concomitant addition of the substrate at C2 to NADPH yielding a covalent ene adduct intermediate. Elimination of the enolate allows the carboxylation to proceed via the addition of CO2 in an anti-fashion to C2 yielding the malonate derivatives (Figure 5).40
Figure 5.

Proposed catalytic mechanism of the reductive carboxylation catalyzed by CCRs, with the regiosepecificity and the stereoselectivity of the reaction indicated. The structures of previously characterized atypical PKS extender units, biosynthesised by CCRs, are also highlighted.
With the pivotal role of CCRs in the biosynthesis of atypical extender units well established,8 their presence within a biosynthetic gene cluster is highly indicative that the encoded polyketide is structurally distinct. Thus in the genome mining era, CCRs have become useful markers for the potential prioritization of polyketide biosynthetic gene clusters.
Biosynthesis of new PKS extender units
Several unusual CoA-linked extender units have been unveiled in recent years including benzylmalonyl-CoA and 4-oxoadipyl-CoA, incorporated into splenocins (26) isolated from Streptomyces sp. CNQ431,41 and pamamycins (27) isolated from S. alboniger,42,43 respectively. Benzylmalonyl-CoA represents the first example of a characterized aromatic PKS extender unit. Derived from phenylalanine, deamination via an ammonia lyase, EnccP, yields cinnamic acid, which then undergoes conversion into the CoA derivative by SpnF, a CoA ligase. A final reductive carboxylation by the CCR, SpnE, yields benzylmalonyl-CoA (Figure 6A). This relatively simple biosynthetic pathway effectively transforms amino acids into CoA-linked extender units. As a consequence, this knowledge has contributed to the development of an efficient biosynthetic tool kit in the engineering of PKS pathways.41
Figure 6.

Biosynthesis of newly identified PKS extender units. (A) Biosynthesis of benzylmalonyl-CoA incorporated into the splenocins. (B) Biosynthesis of 3-oxoadipyl-CoA for incorporation into the pamamycins. (C) Biosynthesis of dichloropyrrolepropyl-ACP for incorporation into chlorizidine A (11, see blue highlight in Figure 3B). (D) Two characterized routes for the incorporation of gem-dimethyl groups present in yersinibactin and epothilone. The route exclusively utilized by EpoM8 (the epothilone PKS) proceeds via dimethylmalonyl-ACP (top pathway).
The provision of the extender unit 4-oxoadipyl-CoA (28), and its methylated derivative 5-methyl-4-oxoadipyl-CoA, provides a mechanism for the incorporation of succinate into the pamamycin series of polyketides. Genetic and biochemical characterization of PamA, a highly unusual KS domain encoded within the biosynthetic gene cluster for 27, demonstrated its importance in the provision of 3-oxoadipyl-CoA, an intermediate towards 28. Recombinant PamA was shown to facilitate the direct condensation of malonyl-CoA with succinyl-CoA to furnish 3-oxoadipyl-CoA (Figure 6B). Upon incubation of PamA with methylmalonyl-CoA and succinyl-CoA, 2-methyl-3-oxoadipyl-CoA was also produced. Subsequent rotation of the 3-oxoadipyl-CoA derivatives by the highly atypical AT domain PamB yields 28 and its methylated derivatives, which are the unusual extender units incorporated into the pamamycins.44,45
Whilst CoA-linked extender units are more prevalent, ACP-linked extender units typically introduce a wider variety of functionality into the carbon skeletons of polyketides. Examples include hydroxyl and amino groups via (2R)-hydroxymalonyl-ACP and (2S)-aminomalonyl-ACP, respectively.28 This trend is exemplified by the dichloropyrrolypropylmalonyl-ACP (29) extender unit incorporated into 11, a halogenated marine alkaloid from Streptomyces sp. CNH-287.46 Moore and co-workers described how 11 is derived from the starter unit 8. Extension of this with malonyl-CoA is mediated by a dedicated FabF, Clz11, which acts in parallel with primary metabolic FAS enzymes resulting in a fully saturated acyl chain. Oxidation by the enoyl-ACP-reductase, Clz3, yields the α,β-unsaturated thioester which is then reductively carboxylated by Clz4, the CCR, giving rise to the highly unusual extender unit 29 (Figure 6C).20
The ACP-linked family of extender units has been expanded further through the identification of dimethylmalonyl-ACP (30), which is responsible for the installation of geminal dimethyl groups into epothilone (31) and yersiniabactin (32). Keasling and co-workers sought to characterize the in vitro function of EpoM8, module 8 within the PKS responsible for the biosynthesis of 31. Contrary to an original hypothesis, the SAM-dependent methylation catalyzed by the methyltransferase (MT) domain of EpoM8, was found to precede the decarboxylative condensation reaction catalyzed by the KS-domain, thus resulting in the identification of this novel extender unit, 30 (Figure 6D).47 Unlike EpoM8, however, the 32 PKS was far less selective. In vitro analysis demonstrated the ability of the 32 PKS to utilize both methylmalonyl-ACP and 30, with the methylation event able to take place before or after the decarboxylative Claisen condensation catalyzed by the KS (Figure 6D).47
Expanding our knowledge of CCR-dependent extender unit biosynthetic pathways; structural and biochemical studies
With an ever-increasing interest in combinatorial biosynthesis and bioengineering of polyketide pathways, the biosynthesis and incorporation of PKS extender units provides a convenient means for altering their basic carbon skeletons. As CCRs form the basis of the paradigm for atypical PKS extender unit biosynthesis, it is essential that we have a greater understanding of both the structural and mechanistic basis of substrate selectivity, so that they can be exploited in the engineering of polyketides. The biochemical characterization and utilization of the antimycin (33—36) CCR, AntE, in combinatorial biosynthetic studies has significantly contributed towards our understanding of the substrate scope of this family of enzymes.
The total in vitro enzymatic synthesis of the antimycins provided the first biochemical characterization of AntE, which catalyzes the reductive carboxylation of (2E)-hexenoyl-CoA into butylmalonyl-CoA in an NADPH-dependent manner (Figure 7A). Non-native substrates were tested in vitro demonstrating that AntE displays a relaxed substrate specificity, as both crotonyl-CoA and (2E)-octenoyl-CoA were reductively carboxylated to yield (2S)-ethylmalonyl-CoA and (2S)-hexylmalonyl-CoA, respectively.48 Subsequent biochemical investigations by Abe, Liu and co-workers illuminated AntE as the most promiscuous CCR characterized to date.49 When challenged with a series of 12 α,β-unsaturated acyl-CoA thioesters (37—48), varying in chain length, chain branching and halogenation, all substrates with the exception of (2E)-undecanoyl-CoA (48), were converted by AntE into their corresponding malonate derivatives. Of all the substrates tested, 48 represented the longest acyl chain length; it is therefore likely that this surpassed the linear length of the acyl chain tolerated by AntE.49 Alongside these extensive in vitro studies, in vivo feeding experiments unveiled that chloropentenoyl-CoA (49) and cyclohexanepropenoyl-CoA (50) were also accepted as substrates by AntE (Figure 7B).49
Figure 7.

Biochemical and structural characterization of CCRs involved in PKS extender unit biosynthesis. (A) Native reactions catalyzed by AntE, the pathway specific CCR, involved in antimycin biosynthesis. (B) Substrates used to probe the promiscuity of AntE – the substrates in black were tolerated in vitro by AntE, substrates in green were tolerated in vivo by AntE and the malonate derivative highlighted in red was not accepted as a substrate. (Ci) Native reaction catalyzed by CinF, for incorporation of hexanoylmalonyl-CoA into cinnabaramide A. (Cii) Active site of CinF, crystallized in complex with both NADH and 2-octenoyl-CoA; residues essential for substrate specificity of CinF are highlighted. The PDB accession number for CinF with ligands bound is 4A0S and the image was generated in Pymol.70 (Ciii) Sequence alignments of characterized CCRs, with the residues conferring substrate specificity highlighted in blue. SalG is responsible for the biosynthesis of chloroethylmalonyl-CoA (23) in salinosporamide A biosynthesis, RevT is responsible for the biosynthesis of alkylmalonyl-CoAs incorporated into the reveromycins, CCR is from Streptomyces collinus and catalyzes the reductive decarboxylation of crotonyl-CoA into methylmalonyl-CoA, AntE is the highly promiscuous CCR from the antimycin pathway, SpnE is involved in the biosynthesis of benzylmalonyl-CoA from the splenocin biosynthetic gene cluster, and Clz4 is responsible for the biosynthesis of ACP-linked dichloropyrrolepropyl extender unit incorporated into chlorizidine A.
Recently the cinnabaramide (51) hexylmalonyl-CoA synthase, CinF, from Streptomyces sp. JS360 was structurally characterized by Müller and co-workers (Figure 7C(i)). The crystal structure of CinF illustrated how the stereochemical outcome of the carboxylation reaction is tightly controlled within the active site of this tetrameric protein. Each monomer is comprised of two distinct domains, a cofactor binding domain and a catalytic domain, with the specificity of the enzyme defined by a large hydrophobic pocket within the catalytic domain. The acyl chain of 2-ocetenoyl-CoA is bound within this hydrophobic pocket, which orients C3 for optimal hydride transfer from NADPH to the re face of the α,β-unsaturated acyl-CoA. Furthermore in silico docking demonstrated the binding of CO2 via a glutamate and asparagine increases the susceptibility of CO2 to undergo a nucleophilic attack from the α,β-unsaturated bond of 2-ocetenoyl-CoA to yield (2S)-hexylmalonyl-CoA (Figure 7C(ii)).50
Two specific residues, Ala163 and Gly362, within the hydrophobic pocket are proposed to be responsible for the substrate specificity of CinF. Indeed mutating residues A163 and G362 for amino acids with significantly larger side chains (G362F, G362I and A163I) resulted in the inability of CinF to accept 2-octenoyl-CoA as a substrate (Figure 7C(ii)).50 Sequence alignments of CinF with characterized CCRs highlights the importance of these residues in determining substrate specificity. These sites within the bona fide CCR from Streptomyces collinus, which accepts crotonyl-CoA as its native substrate, are occupied by isoleucine and phenylalanine residues, respectively, considerably condensing the size of the active site hydrophobic pocket (Figure 7C(iii)). It is interesting to note that the sequence of Clz4 differs substantially from other characterized CCRs; however this CCR acts upon ACP-linked α,β-unsaturated acylthioesters, rather than a CoA-linked substrate (Figure 7C(iii)).
A brief in vitro characterization of CinF reported alongside the crystal structure, provided some evidence that the acyl chain of the substrate, rather than the CoA moiety, influences the rate at which the carboxylation reaction is catalyzed. Whilst CinF was able to accept crotonyl-CoA, 2-octenoyl-CoA and 2-octenoic acid N-acetylcysteamine (SNAC) as substrates, a higher affinity was displayed towards the 2-octenoyl thioesters when compared to crotonyl-CoA.50 This result can be easily interpreted in light of the crystal structure, as it is the acyl chain of the α,β-unsaturated acyl-CoA thioester that is located within the active site. The ribose and adenine moieties of the CoA are bound to the surface of the protein, away from the active site.50
Despite the importance of CCRs in the provision of atypical extender units, the structural characterization of this class of enzymes is limited. Only two CCR crystal structures have been reported to date, the aforementioned CinF representing the first and AntE representing the second.51 Whilst the core active site residues responsible for the binding of NADPH and CO2 are conserved between CinF and AntE, distinct differences in the cavities which accommodate the acyl-chains of the α,β-unsaturated acyl-CoA thioesters are observed. Within AntE, the active site pocket is lined by an alanine residue (Ala182), whereas in CinF, this position is occupied by leucine (Leu200 in CinF). This substitution significantly increases the size of the active site pocket within AntE, thus rationalizing the promiscuity exhibited by AntE at a molecular level according to Abe, Liu and co-workers.51
Engineering PKSs for the incorporation of unusual PKS extender units
The scope of PKS extender units that are incorporated into polyketides is varied and has substantially grown in size in recent years due to the discovery of the CCR superfamily. Ultimately, the nature of the extender molecule is governed and controlled by AT domains associated with the thiotemplate assembly lines. Known as “gatekeepers”, AT domains can be classified depending upon whether they are encoded within the PKS modules or if they are discrete proteins individually encoded within the biosynthetic gene cluster, referred to as cis-AT domains and trans-AT domains, respectively.
The manipulation of AT domains for the incorporation of non-natural PKS substrates has been at the forefront of PKS engineering for many years, with traditional methodologies focusing upon the swapping of AT domains or the generation of hybrid-AT domains, thus altering module specificity.52–54 The replacement of native AT domains however, frequently results in non-functional PKS assembly lines due to significant disruptions in protein-protein interactions, which are essential for large multifunctional enzyme complexes.55,56 Consequently, a more promising and attractive approach for the modification to AT domains is through the engineering of “native” AT domains via precise alterations to individual amino acid residues conferring specificity. Whilst allowing for the incorporation of non-natural PKS substrates, this minimally invasive strategy preserves critical protein-protein interactions yielding functional polyketide assembly lines. Such endeavours have been significantly aided by advances in the structural and mechanistic understanding of AT domains.55,57,58
A multifaceted approach was taken by Schulz and coworkers for the production of an erythromycin analogue, 2-propargylerythromycin (52), resulting from the incorporation of a 2-propargylmalonate (53) precursor.59 Utilizing a combination of protein engineering and computational modelling, a highly conserved valine residue at position 295 was identified in the AT of DEBS module 6 (AT6DEBS). This amino acid was found to be integral in defining the active site pocket into which the side chain of the malonate building blocks extend.59 The mutation of this residue to an alanine (Val295Ala) significantly lowered the steric hindrance within this hydrophobic pocket allowing for the incorporation of this synthetic precursor to yield 52 with “clickable” alkyne functionality. This was validated through an analogous experiment in which the Val295Lys mutation was tested, and as anticipated, the steric bulk of the side chain of this amino acid residue prevented the incorporation of 53, resulting in the production of wild-type erythromycin (Figure 8A).59
Figure 8.

(A) Structures of the engineered polyketide 2-propargylerythromycin, resulting from the selection of 2-propargylmalonate by AT5DEBS, and the pre-monensin series, derived from the feeding a variety of “non-native” precursors for generation of novel derivatives. (B) The two distinct routes established for the provision of fluoromalonyl-CoA (58) and the suite of fluorinated polyketide molecules generated from the minimal DEBS system.
Whilst no engineering was undertaken, molecular docking simulations were also applied to the monensin PKS. This played a pivotal role in establishing if there were any “non-native” substrates that could be recognized by the AT domain of module 5 (AT5mon), thus enabling informed decisions to be made with regards to precursor-directed biosynthesis of the monensins.60 Various malonyl-SNAC analogues, including propargyl-SNAC and propyl-, butryl- and allylmalonyl-SNAC, were fed to a premonensin producing strain, S. cinnamonensis A495, resulting in the selective incorporation of these precursors by AT5mon adding to the series of premonensin derivatives produced (54—57) by the strain (Figure 8A).60
The concept of pathway engineering was taken a step further by Chang and co-workers with the production of both fluorinated triketide lactones and fluorinated tetraketides (Figure 8B).61 Rather than depending upon the feeding of fluoromalonate analogues for their subsequent incorporation by a PKS assembly line, a biosynthetic route for the generation of fluoromalonyl-CoA (58) was established, thus enabling the generation of this precursor in situ. Two distinct routes for the provision of 58 were described, the first utilizing malonyl-CoA synthetase (MatB) to couple CoA to fluoromalonate. In the second, an acetate kinase - phosphotransacetylase (AckA-Pta) pair activated fluoroacetate and subsequent carboxylation, via the action of an ACCase, yielded 58 (Figure 8B).61 Previous work by Keatinge-Clay and co-workers established the high degree of substrate promiscuity exhibited by MatB,62 which was exploited in the provision of 58.61 Indeed, Keatinge-Clay and co-workers were able to generate several PKS extender units in vitro including methylmalonyl-CoA, methoxymalonyl-CoA and hydroxymalonyl-CoA.62
Chang and co-workers then probed 58 as a substrate for modules 2, 3 and 6 from the DEBS PKS assembly line. Utilization of the entire module not only demonstrated the recognition of 58 by the AT, it highlighted the acceptance of the fluorine extender unit by the KR and thioesterase domains, which resulted in fully processed triketide lactones and tetraketides (Figure 8B). Whilst 58 was naturally accepted as a substrate, attempts were made to increase the selectivity of the AT domains towards this “non-native” extender unit through engineering. Mutating the highly conserved serine residue within the AT domains (Ser2107 in AT6DEBS) to an alanine improved the selectivity towards 58. However, it was found that the incorporation of 58 was enhanced following complementation of DEBS modules in which the AT domain was inactivated with the trans-AT domain from the disorazole PKS.61 When combined with previous observations that a 4-fluorobutyrate moiety could be accepted as a starter unit by the DEBS PKS through precursor directed biosynthesis, this is highly suggestive that the entirety of the DEBS system is amenable to fluorinated substrates.63 With fluorine being a highly desirable entity,64 the ability to generate 58 in situ and its subsequent incorporation into a simple polyketide, represents a significant stride towards the site-specific engineering of fluorinated (polyketide) natural products.
Outlook
Over the past couple of decades a plethora of PKS substrates have been described, primarily from modular type I PKS systems, highlighting their important contributions to the vast structural diversity exhibited by the polyketide class of natural products. As a result, significant efforts have been made to understand the mechanisms for the provision of both PKS starter and extender unit substrates. Investigations into the generation of highly unusual PKS extender units has highlighted the prevalence of CCRs in these pathways and resulted in the definition of a new biosynthetic paradigm associated with polyketide biosynthesis.8 The biosynthesis of the antimycin series of polyketides has illuminated AntE as the poster child for this class of enzymes, due to the extreme levels of tolerance it exhibits towards many substrates.49,51 Indeed, AntE has been shown to complement various polyketide pathways for the biosynthesis of unusual PKS extender units, paving the way for the utilization of AntE as a synthetic biology “tool” for designer PKS extender unit generation.49
With the increased ability to manipulate the metabolic pathways associated with PKS substrate production, one of the remaining challenges involves being able to reliably and consistently incorporate diverse substrates at will into PKSs. Solving this bioengineering problem should prove to be a highly valuable endeavour in the development of new pharmaceuticals and biofuels.65,66 Recent progress showing that single point mutations within AT domains can alter the specificity of a module whilst maintaining protein integrity are an important step forward in realizing this goal. As a more complete picture is beginning to emerge in understanding the detailed choreography of PKS catalysis at the molecular level,67,68 we may finally be on the cusp of being able to fine tune PKSs into productive synthetic biology tools for the construction of ad hoc “designer” molecules.69
Supplementary Material
Figure 4.

mechanisms for the incorporation of PKS starter units. (A) Incorporation of a glycyl moiety into FR901464 (20) via the unusual GAT domain located within the loading module for this PKS assembly line. (B) Priming of the curacin A (22) PKS via an unusual GNATL domain located within the initiating module of the PKS.
Acknowledgments
We appreciate the generosity of the NIH through grants R01-AI047818 and R01-CA127622 for supporting our work on the biosynthesis and bioengineering of PKS substrates and products.
References
- 1.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 3.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 4.Austin MB, Noel JP. Nat Prod Rep. 2003;20:79–110. doi: 10.1039/b100917f. [DOI] [PubMed] [Google Scholar]
- 5.Hertweck C. Angew Chem, Int Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 6.Hertweck C. Trends Biochem Sci. 2015;40:189–199. doi: 10.1016/j.tibs.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Moore BS, Hertweck C. Nat Prod Rep. 2002;19:70–99. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- 8.Wilson MC, Moore BS. Nat Prod Rep. 2012;29:72–86. doi: 10.1039/c1np00082a. [DOI] [PubMed] [Google Scholar]
- 9.Chan YA, Podevels AM, Kevany BM, Thomas MG. Nat Prod Rep. 2009;26:90–114. doi: 10.1039/b801658p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waldman AJ, Balskus EP. Org Lett. 2014;16:640–643. doi: 10.1021/ol403714g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. ChemBioChem. 2013;14:955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirata Y, Nakata H, Yamada K, Okuhara K, Naito T. Tetrahedron. 1961;14:252–274. [Google Scholar]
- 13.He J, Hertweck C. J Am Chem Soc. 2004;126:3694–3695. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- 14.Choi YS, Zhang HJ, Brunzelle JS, Nair SK, Zhao HM. Proc Natl Acad Sci U S A. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerdeño AM, Bibb MJ, Challis GL. Chem Biol. 2001;8:817–829. doi: 10.1016/s1074-5521(01)00054-0. [DOI] [PubMed] [Google Scholar]
- 16.Thomas MG, Burkart MD, Walsh CT. Chem Biol. 2002;9:171–184. doi: 10.1016/s1074-5521(02)00100-x. [DOI] [PubMed] [Google Scholar]
- 17.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Parry RJ. Antimicrob Agents Chemother. 2007;51:946–957. doi: 10.1128/AAC.01214-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamanaka K, Ryan KS, Gulder TA, Hughes CC, Moore BS. J Am Chem Soc. 2012;134:12434–12437. doi: 10.1021/ja305670f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani SM, Moore BS. J Am Chem Soc. 2013;135:18032–18035. doi: 10.1021/ja409520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong H, Fill T, Leadlay PF. Angew Chem, Int Ed. 2013;52:13096–13099. doi: 10.1002/anie.201308136. [DOI] [PubMed] [Google Scholar]
- 22.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 23.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004–2012. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, Liu J, Zhang W. Nat Chem Biol. 2015;11:115–120. doi: 10.1038/nchembio.1718. [DOI] [PubMed] [Google Scholar]
- 25.Zhang F, He HY, Tang MC, Tang YM, Zhou Q, Tang GL. J Am Chem Soc. 2011;133:2452–2462. doi: 10.1021/ja105649g. [DOI] [PubMed] [Google Scholar]
- 26.Dorrestein PC, Van Lanen SG, Li W, Zhao C, Deng Z, Shen B, Kelleher NL. J Am Chem Soc. 2006;128:10386–10387. doi: 10.1021/ja0639362. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Hong H, Gillies F, Spencer JB, Leadlay PF. ChemBioChem. 2008;9:150–156. doi: 10.1002/cbic.200700492. [DOI] [PubMed] [Google Scholar]
- 28.Chan YA, Boyne MT, 2nd, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL, Thomas MG. Proc Natl Acad Sci U S A. 2006;103:14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hildebrand M, Waggoner LE, Liu H, Sudek S, Allen S, Anderson C, Sherman DH, Haygood M. Chem Biol. 2004;11:1543–1552. doi: 10.1016/j.chembiol.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Lopanik NB, Shields JA, Buchholz TJ, Rath CM, Hothersall J, Haygood MG, Hakansson K, Thomas CM, Sherman DH. Chem Biol. 2008;15:1175–1186. doi: 10.1016/j.chembiol.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trindade-Silva AE, Lim-Fong GE, Sharp KH, Haygood MG. Curr Opin Biotechnol. 2010;21:834–842. doi: 10.1016/j.copbio.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He HY, Yuan H, Tang MC, Tang GL. Angew Chem, Int Ed. 2014;53:11315–11319. doi: 10.1002/anie.201406602. [DOI] [PubMed] [Google Scholar]
- 33.Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S. Proc Natl Acad Sci U S A. 2004;101:16222–16227. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu LC, Geders TW, Wang B, Gerwick WH, Hakansson K, Smith JL, Sherman DH. Science. 2007;318:970–974. doi: 10.1126/science.1148790. [DOI] [PubMed] [Google Scholar]
- 35.Eustaquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Proc Natl Acad Sci U S A. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mo S, Kim DH, Lee JH, Park JW, Basnet DB, Ban YH, Yoo YJ, Chen SW, Park SR, Choi EA, Kim E, Jin YY, Lee SK, Park JY, Liu YA, Lee MO, Lee KS, Kim SJ, Kim D, Park BC, Lee SG, Kwon HJ, Suh JW, Moore BS, Lim SK, Yoon YJ. J Am Chem Soc. 2011;133:976–985. doi: 10.1021/ja108399b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoo HG, Kwon SY, Kim S, Karki S, Park ZY, Kwon HJ. Biosci, Biotechnol, Biochem. 2011;75:1191–1193. doi: 10.1271/bbb.110003. [DOI] [PubMed] [Google Scholar]
- 38.Erb TJ, Berg IA, Brecht V, Muller M, Fuchs G, Alber BE. Proc Natl Acad Sci U S A. 2007;104:10631–10636. doi: 10.1073/pnas.0702791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erb TJ, Brecht V, Fuchs G, Muller M, Alber BE. Proc Natl Acad Sci U S A. 2009;106:8871–8876. doi: 10.1073/pnas.0903939106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenthal RG, Ebert MO, Kiefer P, Peter DM, Vorholt JA, Erb TJ. Nat Chem Biol. 2014;10:50–U85. doi: 10.1038/nchembio.1385. [DOI] [PubMed] [Google Scholar]
- 41.Chang CC, Huang R, Yan Y, Ma HM, Dai Z, Zhang BY, Deng ZX, Liu W, Qu XD. J Am Chem Soc. 2015;137:4183–4190. doi: 10.1021/jacs.5b00728. [DOI] [PubMed] [Google Scholar]
- 42.McCann PA, Pogell BM. J Antibiot. 1979;32:673–678. doi: 10.7164/antibiotics.32.673. [DOI] [PubMed] [Google Scholar]
- 43.Kondo S, Yasui K, Katayama M, Marumo S, Kondo T, Hattori H. Tetrahedron Lett. 1987;28:5861–5864. [Google Scholar]
- 44.Rebets Y, Brotz E, Manderscheid N, Tokovenko B, Myronovskyi M, Metz P, Petzke L, Luzhetskyy A. Angew Chem, Int Ed. 2015;54:2280–2284. doi: 10.1002/anie.201408901. [DOI] [PubMed] [Google Scholar]
- 45.Rong J, Nelson ME, Kusche B, Priestley ND. J Nat Prod. 2010;73:2009–2012. doi: 10.1021/np100421v. [DOI] [PubMed] [Google Scholar]
- 46.Alvarez-Mico X, Jensen PR, Fenical W, Hughes CC. Org Lett. 2013;15:988–991. doi: 10.1021/ol303374e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, Keasling JD. Angew Chem, Int Ed. 2015;54:2370–2373. doi: 10.1002/anie.201410124. [DOI] [PubMed] [Google Scholar]
- 48.Sandy M, Rui Z, Gallagher J, Zhang W. ACS Chem Biol. 2012;7:1956–1961. doi: 10.1021/cb300416w. [DOI] [PubMed] [Google Scholar]
- 49.Yan Y, Chen J, Zhang L, Zheng Q, Han Y, Zhang H, Zhang D, Awakawa T, Abe I, Liu W. Angew Chem, Int Ed. 2013;52:12308–12312. doi: 10.1002/anie.201305569. [DOI] [PubMed] [Google Scholar]
- 50.Quade N, Huo L, Rachid S, Heinz DW, Muller R. Nat Chem Biol. 2012;8:117–124. doi: 10.1038/nchembio.734. [DOI] [PubMed] [Google Scholar]
- 51.Zhang L, Mori T, Zheng Q, Awakawa T, Yan Y, Liu W, Abe I. Angew Chem, Int Ed. 2015;54:13462–13465. doi: 10.1002/anie.201506899. [DOI] [PubMed] [Google Scholar]
- 52.Petkovic H, Lill RE, Sheridan RM, Wilkinson B, McCormick EL, McArthur HAI, Staunton J, Leadlay PF, Kendrew SG. J Antibiot. 2003;56:543–551. doi: 10.7164/antibiotics.56.543. [DOI] [PubMed] [Google Scholar]
- 53.Gregory MA, Kaja AL, Kendrew SG, Coates NJ, Warneck T, Nur-e-Alam M, Lill RE, Sheehan LS, Chudley L, Moss SJ, Sheridan RM, Quimpere M, Zhang M-Q, Martin CJ, Wilkinson B. Chem Sci. 2013;4:1046–1052. [Google Scholar]
- 54.Dunn BJ, Khosla C. J R Soc, Interface. 2013;10 doi: 10.1098/rsif.2013.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunn BJ, Cane DE, Khosla C. Biochemistry. 2013;52:1839–1841. doi: 10.1021/bi400185v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong FT, Chen AY, Cane DE, Khosla C. Biochemistry. 2010;49:95–102. doi: 10.1021/bi901826g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keatinge-Clay AT. Nat Prod Rep. 2012;29:1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 58.Park H, Kevany BM, Dyer DH, Thomas MG, Forest KT. PLoS One. 2014;9:e110965. doi: 10.1371/journal.pone.0110965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. ACS Chem Biol. 2013;8:443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- 60.Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, Kushnir S, Ismail S, Fansa EK, Wittinghofer A, Schulz F, Sanchez-Garcia E. ChemBioChem. 2014;15:1991–1997. doi: 10.1002/cbic.201402206. [DOI] [PubMed] [Google Scholar]
- 61.Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MCY. Science. 2013;341:1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hughes AJ, Keatinge-Clay A. Chem Biol. 2011;18:165–176. doi: 10.1016/j.chembiol.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 63.Goss RJ, Hong H. Chem Comm. 2005;31:3983–3985. doi: 10.1039/b506635b. [DOI] [PubMed] [Google Scholar]
- 64.Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 65.Khosla C, Keasling JD. Nat Rev Drug Discovery. 2003;2:1020–1028. doi: 10.1038/nrd1256. [DOI] [PubMed] [Google Scholar]
- 66.Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. Nature. 2012;488:320–328. doi: 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- 67.Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan ARH, Hakansson K, Sherman DH, Smith JL, Skiniotis G. Nature. 2014;510:512–517. doi: 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whicher JR, Dutta S, Hansen DA, Hale WA, Chemler JA, Dosey AM, Narayan ARH, Hakansson K, Sherman DH, Smith JL, Skiniotis G. Nature. 2014;510:560–564. doi: 10.1038/nature13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim E, Moore BS, Yoon YJ. Nat Chem Biol. 2015;11:649–659. doi: 10.1038/nchembio.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.