

ABSTRACT
Infection by alphaherpesviruses, including herpes simplex virus (HSV) and pseudorabies virus (PRV), typically begins at epithelial surfaces and continues into the peripheral nervous system (PNS). Inflammatory responses are induced at the infected peripheral site prior to invasion of the PNS. When the peripheral tissue is first infected, only the innervating axons are exposed to this inflammatory milieu, which includes the interferons (IFNs). The fundamental question is how do PNS cell bodies respond to these distant, potentially damaging events experienced by axons. Using compartmented cultures that physically separate neuron axons from cell bodies, we found that pretreating isolated axons with beta interferon (IFN-β) or gamma interferon (IFN-γ) significantly diminished the number of herpes simplex virus 1 (HSV-1) and PRV particles moving in axons toward the cell bodies in a receptor-dependent manner. Exposing axons to IFN-β induced STAT1 phosphorylation (p-STAT1) only in axons, while exposure of axons to IFN-γ induced p-STAT1 accumulation in distant cell body nuclei. Blocking transcription in cell bodies eliminated antiviral effects induced by IFN-γ, but not those induced by IFN-β. Proteomic analysis of IFN-β- or IFN-γ-treated axons identified several differentially regulated proteins. Therefore, unlike treatment with IFN-γ, IFN-β induces a noncanonical, local antiviral response in axons. The activation of a local IFN response in axons represents a new paradigm for cytokine control of neuroinvasion.
IMPORTANCE
Neurons are highly polarized cells with long axonal processes that connect to distant targets. PNS axons that innervate peripheral tissues are exposed to various situations that follow infection, inflammation, and damage of the tissue. After viral infection in the periphery, axons represent potential front-line barriers to PNS infection and damage. Indeed, most viral infections do not spread to the PNS, yet the mechanisms responsible are not well studied. We devised an experimental system to study how axons respond to inflammatory cytokines that would be produced by infected tissues. We found that axons respond differentially to type I and type II interferons. The response to type I interferon (IFN-β) is a rapid axon-only response. The response to type II interferon (IFN-γ) involves long-distance signaling to the PNS cell body. These responses to two interferons erect an efficient and rapid barrier to PNS infection.
INTRODUCTION
Most viruses infecting the nervous system are opportunistic pathogens, but the alphaherpesviruses (e.g., herpes simplex virus [HSV], varicella-zoster virus [VZV], and pseudorabies virus [PRV]) have evolved to enter the nervous system efficiently to establish life-long infections (1, 2). These viruses enter the axons of peripheral nervous system (PNS) neurons by direct fusion of the viral membrane with the axonal plasma membrane (3). Virus capsids with associated inner-tegument proteins are then transported to the neuronal cell bodies along microtubules. This process is mediated by dynein, a minus-end-directed microtubule motor (4). In immunocompetent natural hosts, alphaherpesvirus infections usually result in a quiescent infection in the PNS. Spread to the central nervous system (CNS) is rare, but when it happens, the consequences are severe. However, in nonnatural hosts, productive infection of the cell bodies of PNS neurons and spread to CNS neurons are frequently observed (5).
Alphaherpesvirus infection of epithelial cells leads to production of many inflammatory and antiviral cytokines, such as type I interferon (alpha interferon [IFN-α] and beta interferon [IFN-β]) (6). These cytokines induce both autocrine signaling to help clear the viral infection and paracrine signaling to alert other cells in the surrounding tissue of the infection (7). If the infection in epithelial cells progresses without effective control, more global innate and adaptive immune responses are activated. Cells that mediate these responses produce copious amounts of cytokines, including type II IFN (gamma interferon [IFN-γ]) (8). It is widely held that by binding to their cognate receptors, IFNs stimulate a cascade of phosphorylation events that ultimately result in phosphorylation and activation of the signal transducer and activator of transcription (STAT) proteins. The STAT proteins then translocate to the nucleus to induce the transcription of hundreds of IFN-stimulated genes (ISGs) (9). These gene products carry out a multitude of effector functions to combat virus infection.
Neurons have highly specialized intracellular signaling, transport, and gene expression patterns that maintain a highly polarized morphology (10). Because axons can be meters in length, the time required for intracellular signal transduction to the cell bodies can be too long for a rapid response. Accordingly, axons are capable of responding autonomously to environmental stimuli (11, 12). For example, local protein translation of mRNAs in axons is induced by nerve growth factors and axonal injury. The subsequent transport of locally synthesized signaling molecules to the nuclei mediates retrograde communication with the cell bodies (11, 13). Such local responses and long-distance communication in neurons play an essential role in the development and movement of growth cones as well as axon regeneration (14, 15). During viral infection of epithelial tissues, innervating axons are bathed in the proinflammatory and antiviral cytokines produced by the infected epithelial cells, but their cell bodies remain unexposed. Since PNS neurons do not divide, the usual apoptotic response of infected cells to inflammatory cytokines is not appropriate. The fundamental question is how do PNS axons respond to inflammatory cytokines? The coordination of axonal and cell body responses is critical, as it determines whether neurons produce appropriate responses to limit subsequent viral invasion of the nervous system.
In this study, we established a system to study how isolated PNS neuron axons respond to IFNs. We found that preexposure of axons to either IFN-β or IFN-γ significantly reduced retrograde transport of PRV and herpes simplex virus 1 (HSV-1) particles in axons. IFN-γ but not IFN-β reduced replication of PRV in the neuron cell bodies. Importantly, the antiviral effects of IFN-β in axons primarily depend on autonomous axonal mechanisms. In contrast, the IFN-γ response required nuclear transcription. Exposure of axons to IFN-β induced phosphorylation of STAT1 (p-STAT1) only in the axons, whereas axonal treatment with IFN-γ resulted in localization of p-STAT1 to the nucleus. Analysis of the axon proteome using mass spectrometry and quantitative spectral counting showed that IFN-β and IFN-γ induced unique changes in abundance in several axonal proteins. Overall, these results suggest that the axonal antiviral response to IFN-β and IFN-γ are different. The activation of local events in axons represents a new paradigm for cytokine control of neuroinvasion.
RESULTS
IFN pretreatment of axons reduces PRV retrograde infection.
To test whether exposure of axons to IFN has an effect on PRV infection, we cultured primary rodent superior cervical ganglion (SCG) neurons in vitro in trichambers (Fig. 1A) (16). This system allowed us to physically limit the site of IFN treatment and virus inoculation to the axons in the neurite (N) compartment. The distant and physically separated cell bodies in the soma (S) compartment remained unaffected, except by intracellular signaling through their axons. Similar to a prior report (17), we found no significant difference in PRV titer in the S compartment when axons were pretreated with IFN-β and infected with wild-type PRV-Becker (data not shown). However, the virus titer in the S compartment is dependent on the number of cell bodies that extend axons into the N compartment. To quantify infection in the cell bodies that extended axons to the N compartment, we infected axons with PRV-Becker expressing monomeric red fluorescent protein (mRFP)-tagged capsid protein VP26 (PRV-180), so that virus infection can be monitored by accumulation of the mRFP-VP26 fusion protein in the nucleus (Fig. 1B) (18). We then added DiO, a green fluorescent lipophilic dye, to the N compartment to label all the cell bodies that have extended axons to the N compartment (19). The extent of primary infection was calculated by the ratio of dual-colored cells (red and green) divided by the total number of green cell bodies. Pretreating axons with either IFN-β or IFN-γ reduced the fraction of primarily infected neurons to 66.07% ± 4.63% and 60.60% ± 12.82%, respectively, relative to untreated controls at 24 h postinfection (hpi) (Fig. 1C).
FIG 1 .

IFN pretreatment of axons reduces PRV retrograde infection. (A) The trichamber neuron culture system physically separates the neuron cell bodies and axons into soma (S), middle (M), and neurite (N) compartments. (B and C) IFN-β or IFN-γ was added to the N-compartment axons for 24 h before inoculation of PRV-180 and addition of DiO to the N compartment. Images of neuron cell bodies in the S compartment were taken at 8, 16, and 24 hpi, and representative sections at 24 hpi are shown in panel B (bars, 200 µm). (C) Quantification of the primarily infected cells. The results are normalized to the values for samples not treated with IFN (No Treatment) at 24 hpi. Data are shown as means ± standard errors of the means (SEMs) (error bars). Values that are statistically significantly different by two-way ANOVA are indicated as follows: *, P < 0.05; ***, P < 0.001 (n ≥ 3). (D and E) IFN-β or IFN-γ was added to the N compartment for 6 or 24 h before infection with PRV-180. Movement of fluorescent particles in the N compartment was recorded at ~1 frame per second (fps). (D) Stack images of representative movies. Each dot represents a stalled particle, and each line represents a moving particle (bars, 10 µm). (E) Quantification of the percentage of moving particles. (F) IFN-β or IFN-γ was added to the N-compartment axons for 24 h before inoculation of PRV-180 in the S compartment and addition of DiO to the N compartment. The percentages of primarily infected cells at 10 hpi were quantified and are shown in the graph. The results are normalized to the values for samples not treated with IFN. Values are shown as means plus SEMs (error bars). Values that are significantly different by Student’s t test (n = 3) are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01. Values that are not significantly different (ns) are also indicated.
Axonal IFN-β pretreatment reduces the number of moving PRV particles in axons, but not PRV replication in the cell bodies.
Next, we determined whether PRV particle transport in axons or replication in cell bodies is limited by axonal IFN pretreatment. We used live-cell fluorescence microscopy to examine the dynamics of fluorescent PRV particles immediately after infection (15 to 45 min postinfection) in IFN-treated axons (Fig. 1D). After 6 h of axonal IFN-β or IFN-γ treatment, the percentages of moving particles in axons were reduced to 74.35 ± 5.38 and 69.32 ± 4.24, respectively, relative to untreated controls (Fig. 1D and E; see Movie S1, top panel, in the supplemental material). After 24 h of IFN treatment, the percentages of moving particles were further reduced to 49.87 ± 4.13 and 37.30 ± 7.71, respectively (Fig. 1D and E; Movie S1, bottom panel).
To determine whether axonal IFN treatment affects PRV replication in the cell bodies, we exposed axons to IFN and then inoculated the cell bodies with PRV-180 (Fig. 1F). Compared to the control samples, there was no effect of IFN-β on PRV replication. However, after IFN-γ treatment of axons, the percentage of infected cells was reduced to 54.90 ± 6.51 (Fig. 1F). Together, these results indicate that axonal IFN-β treatment affects PRV retrograde transport in axons but not replication in the cell bodies, while axonal IFN-γ treatment restricts both.
The observed antiviral effect of IFN requires the IFN receptors.
To confirm that IFN-β signaling in axons is responsible for limiting PRV retrograde infection, we treated axons with an antibody that blocks the IFN-α/β receptor (IFNAR) prior to IFN-β treatment and PRV infection (20). This antibody completely blocked STAT1 phosphorylation in IFN-β-treated noncompartmentalized SCG neurons (Fig. 2A). Moreover, the IFNAR-blocking antibody rescued both the PRV retrograde transport defect and infection defect in IFN-β-treated samples to levels similar to those treated with the control antibody (Fig. 2B to D; see Movie S2 in the supplemental material). When the axons were incubated with no antibodies or with a nonspecific control antibody, axonal IFN-β treatment restricted both PRV retrograde transport and infection (Fig. 2B to D; Movie S2). To show that this finding was not due to off-target effects of the antibody, we repeated the assays in SCG neurons isolated from wild-type (WT) versus IFN-α/β receptor (IFNAR) and IFN-γ receptor (IFNGR) double knockout (KO) mice (21). Noncompartmentalized neurons from KO mice failed to phosphorylate STAT1 upon both IFN-β and IFN-γ treatment (Fig. 2E). Similarly, neurons from the KO mice showed no antiviral effect when their axons were treated with IFN-β (Fig. 2F to H; Movie S3). However, axonal IFN-γ treatment of KO neurons still reduced the percentage of moving PRV particles to 78.50 ± 5.78. This reduction was statistically significant, although less in comparison to 51.07 ± 0.93 in WT neurons (Fig. 2F and G; Movie S3). Together, these results indicate that IFNAR-mediated signaling is primarily responsible for the antiviral effects, while signaling through IFNGR may be only partially responsible.
FIG 2 .

The observed antiviral effect of IFN requires the IFN receptors. (A to D) Mouse SCG neurons were treated with a nonspecific control antibody (Ab) or IFNAR antibody or not treated with antibody prior to the addition of IFN-β for 24 h. (E to H) SCG neurons from WT or IFNAR and IFNGR double knockout mice were treated with IFN-β or IFN-γ for 24 h (or the indicated time) or not treated with IFN (No Treatment). (A and E) Immunoblotting of p-STAT1 in noncompartmentalized mouse SCG neurons. (B, C, F, and G) After axonal IFN treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. (B and F) Stack images of representative movies. Each dot represents a stalled particle, and each line represents a moving particle (bars, 10 µm). (C and G) Quantification of the percentage of moving particles. The results are normalized to the values for no-treatment samples. Data are shown as means plus SEMs. Statistical significance by Student’s t test (n ≥ 3) is indicated as follows: ns, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005. (D and H) After axonal IFN treatment, PRV-180 and DiO were added to the N compartment. The percentage of primarily infected cells was quantified. The results are normalized to the values for no-treatment samples at 24 hpi. Data are shown as means ± SEMs. Statistical significance by two-way ANOVA (n ≥ 3) is indicated as follows: ns, not significant; ***, P < 0.001; ****, P < 0.0001.
Axonal IFN pretreatment reduces the number of moving HSV-1 particles, but not LysoTracker-positive organelles in axons.
We next determined whether IFN treatment reduces global retrograde transport in axons or whether the transport deficiency is specific to alphaherpesvirus particles. We treated axons with IFN-β or IFN-γ for 24 h and then inoculated the N-compartment axons with HSV-1 OK-14, which expresses mRFP-VP26 red capsid protein (O. Kobiler and L. W. Enquist, unpublished data). Similar to the results with PRV, we found a reduction to 62.18% ± 1.85% and 54.94% ± 5.85% of moving HSV-1 particles in axons of IFN-β- and IFN-γ-treated samples, respectively (Fig. 3A and B; see Movie S4 in the supplemental material). To determine whether IFN treatment affects nonviral cargos, we used LysoTracker to label acidic organelles, such as lysosomes and late endosomes in axons. Axonal IFN treatment had no effect on either anterograde or retrograde transport of these acidic organelles (Fig. 3C and D; Movie S5). These results suggest that the transport restriction induced by axonal IFN treatment is specific to entering PRV and HSV-1 particles.
FIG 3 .
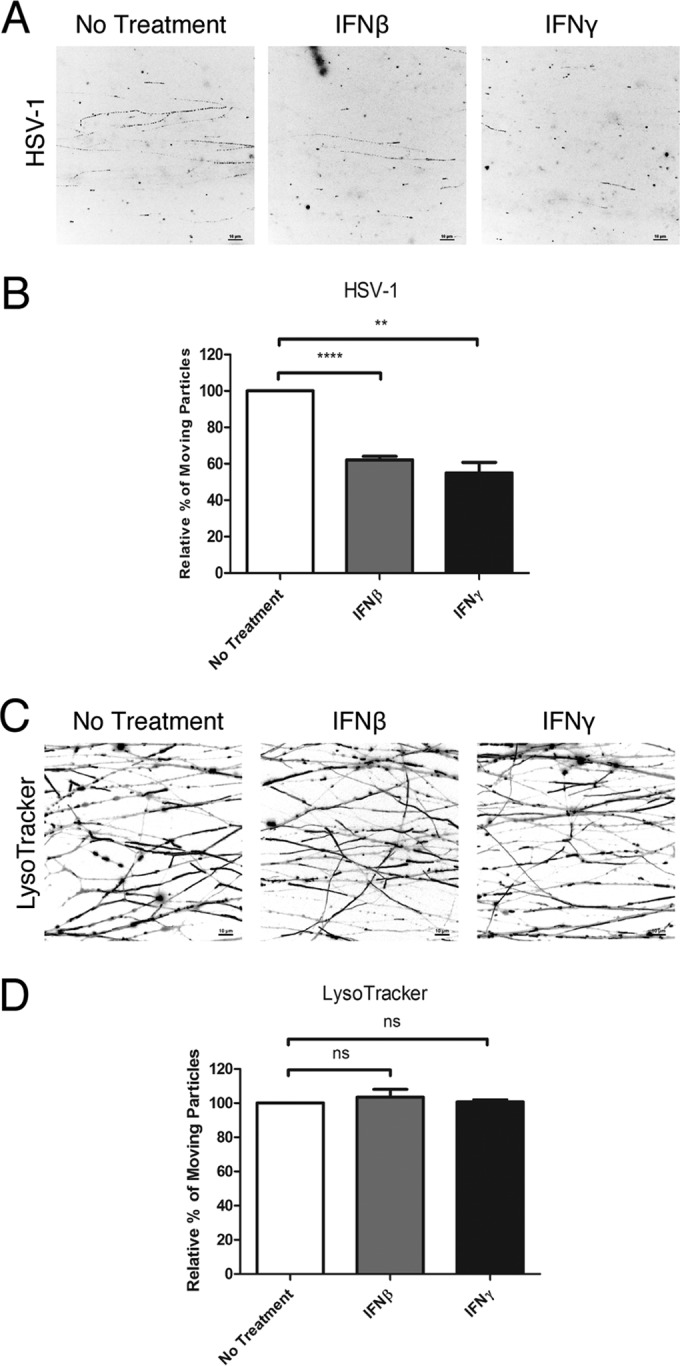
Axonal IFN pretreatment reduces the number of moving HSV-1 particles, but not LysoTracker-positive organelles in axons. (A and B) IFN-β or IFN-γ was added to the N compartments for 24 h before infection with HSV-1 OK-14. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. (A) Stack images of representative movies. Each dot represents a stalled particle, and each line represents a moving particle (bars, 10 µm). (B) Quantification of the percentage of moving particles. (C and D) IFN-β or IFN-γ was added to the N-compartment axons for 24 h before staining with LysoTracker. Movement of fluorescent particles in the N compartment was recorded at ~5 fps. (C) Stack images of representative movies. Each dot represents a stalled particle, and each line represents a moving particle (bars, 10 µm). (D) Quantification of the percentage of moving particles. The results are normalized to the values for no-treatment samples. Data are shown as means plus SEMs. Statistical significance by Student’s t test (n ≥ 3) is indicated as follows: ns, not significant; **, P < 0.005; ****, P < 0.0001.
Axonal IFN-β treatment induces local rapid phosphorylation of STAT1.
STAT1 is phosphorylated when IFN binds the cognate receptor. We measured STAT1 phosphorylation in neurons that were exposed to IFN only through their axons. We detected p-STAT1 only in the axons of neurons whose axons were exposed to IFN-β. In contrast, we detected p-STAT1 only in the neuron cell bodies after axonal IFN-γ treatment (Fig. 4A). The increased level of p-STAT1 in axons exposed to IFN-β was seen as early as 1 h after treatment and lasted for at least 24 h (Fig. 4B). By using immunofluorescence staining, we did not detect p-STAT1 in cell body nuclei after axonal IFN-β treatment, while nuclear accumulation of p-STAT1 was detected in ~2/3 of the neurons that extend axons to the N compartment after IFN-γ axonal treatment (Fig. 4C). As STAT1 nuclear translocation is required for expression of ISGs in canonical IFN signaling, these results suggest that, unlike IFN-γ, axonal IFN-β treatment will not induce nuclear transcription.
FIG 4 .

Axonal IFN-β treatment induces local antiviral responses in axons. (A) Immunoblotting of p-STAT1 in cell bodies in the S compartment and axons in the N compartment after axonal IFN-β or IFN-γ treatment for 24 h. (B) Immunoblotting of p-STAT1 in axons in the absence (−) or presence of IFN-β treatment in the N compartment for 1, 6, or 24 h. (C) Immunofluorescence staining of p-STAT1 in neuron cell bodies after IFN-β or IFN-γ treatment in the N compartment for 24 h. CellTracker Orange (CMRA) labeling in cell bodies indicates that they have axonal connection to the N compartment. The numbers of cells visualized were 78, 118, and 117 for no-treatment, IFN-β-treated, and IFN-γ-treated samples, respectively. Representative images are shown (bars, 20 µm). (D and E) q-PCR quantification of Mx1, IFIT1, or GBP2 expression level in rat SCG with S- or N-compartment treatment of IFN-β (D) or IFN-γ (E). Raw threshold cycle (CT) values were normalized to the values for internal control β-actin, and the ΔCT value was calculated by subtracting the normalized CT value of the experimental condition from that of no-treatment sample. Data are shown as means plus SEMs. Statistical significance by Student’s t test (n = 3) is indicated as follows: ns, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005. (F and G) Dimethyl sulfoxide (DMSO) (carrier) or actinomycin D was added to the S compartment, while simultaneously, axons in the N compartment were treated with IFN-β or IFN-γ or not treated with IFN. Twenty-four hours after treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~2 fps. (F) Stack images of representative movies. Each dot represents a stalled particle, and each line represents a moving particle (bars, 10 µm). (G) Quantification of the percentage of moving particles. The results are normalized to the values for DMSO-treated, non-IFN-treated samples. Data are shown as means plus SEMs. Statistical significance by Student’s t test (n = 3) is indicated as follows: ns, not significant; *, P < 0.05; **, P < 0.01.
IFN-β-mediated inhibition of PRV particle transport does not require transcription in the nucleus.
Treatment of cell bodies with either IFN-β or IFN-γ induced high levels of ISG expression (Fig. 4D and E). As predicted, we found no transcription of Mx1 or IFIT1 after axonal IFN-β exposure (Fig. 4D). However, while axonal IFN-γ treatment did not induce a significant change in GBP2 level, we detected a 1.63-fold ± 0.15-fold increase with IFIT1 level (Fig. 4E). This result likely reflects the possibility of a different subset of ISGs being induced and/or the low number of cell bodies that extend axons into the N compartment where IFN is applied. To test whether the effect of axonal IFN treatment is dependent on de novo transcription in the nucleus, we added the global transcription inhibitor actinomycin D (ActD). We applied ActD to the S-compartment cell bodies while adding IFN-β or IFN-γ to N-compartment axons. The addition of ActD slightly restored the IFN-β-mediated restriction of PRV retrograde axonal transport from 53.61% ± 5.36% to 64.52% ± 4.34% (Fig. 4F and G; see Movie S6 in the supplemental material). However, it fully restored the IFN-γ-mediated restriction of PRV retrograde axonal transport (Fig. 4F and G; Movie S6). These data confirm that the antiviral effect of axonal IFN-β treatment does not require nuclear transcription. The antiviral response is primarily produced locally in axons upon IFN-β treatment.
Axonal IFN treatment induces changes in the axonal proteome.
To our knowledge, there are no previous reports of autonomous axonal signaling of IFN-β through local STAT1 phosphorylation. As a first step in understanding this local IFN response, we determined the axonal proteome after IFN-β or IFN-γ treatment, using mass spectrometry-based proteomic methods (Fig. 5A). A total of five biological replicates were performed for the untreated sample, four replicates for IFN-β treatment, and three replicates for IFN-γ treatment. We identified 2,193 ± 45, 2,227 ± 60, and 2,288 ± 6 proteins, in untreated, IFN-β-treated, and IFN-γ-treated axons, respectively. Differential relative protein abundances between samples were assessed using label-free spectral counting. We identified 48 proteins in IFN-β-treated axons and 47 proteins in IFN-γ-treated axons that changed significantly in comparison to the untreated condition (P < 0.05) (Fig. 5A; see Table S1 in the supplemental material). Specifically, in IFN-β-treated samples, 38 proteins increased and 10 proteins decreased in abundance (Fig. 5B; Table S1). Similarly with IFN-γ, 38 proteins increased and 9 proteins decreased (Fig. S1A; Table S1). The majority of differentially abundant proteins were unique to either IFN-β or IFN-γ treatment, while only nine proteins were common to both conditions (Fig. 5A; Table S1). The most prominent dichotomy between the proteins affected by IFN treatment was the unique association of ATPase activities with IFN-β. These included two subunits of the plasma membrane calcium-transporting ATPase (PMCA) and three subunits of the sodium-potassium-transporting ATPase, all of which were downregulated upon IFN-β treatment. This finding is consistent with previous reports demonstrating that a cytokine mixture (including IFN-β) downregulated PMCA (22) and that IFN treatment results in an increase in intracellular calcium levels (23). It is also likely these ATPase activity-linked protein abundance changes are a consequence of and/or function to maintain IFN-mediated signaling processes.
FIG 5 .

Axonal IFN treatment induces changes in the axonal proteome. (A) N-compartment axons were treated with IFN-β or IFN-γ for 24 h or not treated with IFN (No Treatment) before collection and one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) separation. Samples were subsequently subjected to in-gel digestion with trypsin. The resulting peptides were analyzed by nanoliquid chromatography-electrospray ionization-tandem mass spectrometry (nLC-MS/MS) using an LTQ orbitrap XL spectrometer. The Venn diagram shows the number of proteins that have significant changes in abundance in axons after IFN-β or IFN-γ treatment. The numbers of biological replicates are 5, 4, and 3 for non-IFN-treated, IFN-β-treated, and IFN-γ-treated samples, respectively. (B) Tabulation of proteins with significant changes in abundance in axons after IFN-β treatment. Proteins were manually categorized based on their biological function or subcellular localization and color coded based on the changes in spectral counts following IFN-β treatment. A purple ring around a protein indicates that similar changes were also seen in IFN-γ-treated samples. Proteins that were detected only in IFN-β-treated sample and not in the non-IFN-treated sample are indicated with an asterisk.
The ratio of increased proteins to decreased proteins was almost 4:1 in either IFN-treated axons, which is remarkably different from the ratio of 1:1 reported for axonal proteomic changes induced by PRV infection in 1 h (24). These data suggest that IFN treatment may induce local protein synthesis as well as degradation or transport of axonal proteins. Proteins that increased after IFN-β treatment were assigned to multiple functional categories, including cytoskeleton regulation, translation regulation, signal transduction, cell growth regulation, secretion and exocytosis, metabolism regulation, and cell adhesion. One observation of particular interest was the consistent detection of increased levels of the eight proteins involved in regulating protein translation, five of which were also increased in IFN-γ-treated axons (Fig. 5B; see Fig. S1A and Table S1 in the supplemental material). These proteins include translation regulation factors (Eif2s1 and Tsfm), ribosomal component (Rps3a), enzymes involved in amino acid synthesis and modification (Psat1, Bcat2, and Pycr1) and aminoacyl-tRNA synthetases (Hars and Tars2). Using the methionine homologue l-azidohomoalanine (l-AHA) and click chemistry to label newly synthesized axonal proteins, we found that IFN-β treatment greatly increased local axonal protein synthesis. In contrast, IFN-γ treatment induced minimal increases in local synthesis (Fig. S1B). The increase in protein level after axonal IFN-γ treatment may be due to inhibition of protein degradation or increase in transport of effector proteins into the axons. Furthermore, we found that blocking protein translation in axons using emetine had no effect on IFN-β-induced axonal STAT1 phosphorylation (Fig. S1C). These results indicate that STAT1 phosphorylation occurs before IFN-β-induced local protein synthesis. The increase in axonal protein translation after IFN-β may contribute to the observed antiviral response. However, we were not able to determine whether new protein synthesis is required for IFN-induced antiviral effect in axons because efficient PRV transport is also dependent on new protein synthesis (25).
A common mechanism for IFN-β and IFN-γ reduction of PRV particle transport.
Treatment of axons with IFN-β or IFN-γ reduced both PRV and HSV-1 particle transport. As shown in the mass spectrometry data, cytoplasmic linker 2 (Clip2; also known as Clip115) was the only protein whose levels were consistently reduced in both IFN-β- and IFN-γ-treated axons (Fig. 5B; see Fig. S1A and Table S1 in the supplemental material). We confirmed this reduction in Clip2 level by Western blotting (Fig. S1D). Clip2 and its homologue Clip1 have roles in regulating microtubule dynamics (26). Since axonal treatment with either IFN-β or IFN-γ specifically restricts PRV and HSV-1 retrograde transport, the reduced Clip2 levels may contribute to this phenotype. To determine whether PRV retrograde transport in axons depends on the Clip proteins, we knocked down Clip1 or Clip2 expression in chambered SCG neurons using small interfering RNA (siRNA) magnectofection. This method effectively reduced the protein level of Clip1 or Clip2 in both cell bodies and axons in S and N compartments, respectively (Fig. 6A and B). The siRNA has no off-target cross-reaction between the two types of Clip proteins (Fig. S1E). We then inoculated axons in the N compartment with PRV-180 and monitored virus particle transport in the axons (Fig. 6C). The movement of PRV particles in axons was reduced to 78.03% ± 6.22% with Clip1 knockdown and to 72.16% ± 6.00% with Clip2 knockdown, relative to nontargeting siRNA controls (Fig. 6C and D; see Movie S7 in the supplemental material). These results suggest that Clip1 and Clip2 are required for efficient retrograde axonal transport of PRV particles. Consequently, reducing Clip2 by treatment of axons with either IFN-β or IFN-γ may be a common mechanism to reduce the number of viral particles that reach the PNS cell bodies.
FIG 6 .

Reducing the level of Clip1 or Clip2 restricts PRV particle transport. (A and B) Immunoblotting of Clip1 (A) and Clip2 (B) in cell bodies in the S compartment and axons in the N compartment after transfection with nontargeting (NT), Clip1 or Clip2 siRNA in the S compartment. Quantification of Clip1 (A) and Clip2 (B) bands is shown below each blot. Bands were normalized to internal control β-actin of each sample and to the NT siRNA sample. (C and D) Cell bodies were transfected with NT, Clip1, or Clip2 siRNA before PRV-180 inoculation (5 × 105 PFU) in the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~3 fps. (C) Stack images of representative movies. Each dot represents a stalled particle, and each line represents a moving particle (bars, 20 µm). (D) Quantification of the percentage of moving particles. The results are normalized to the values for NT siRNA samples. Data are shown as means plus SEMs. Values that are significantly different (P < 0.05) by Student’s t test (n = 4) are indicated by bars and an asterisk.
DISCUSSION
The nervous system has evolved finely tuned mechanisms to maintain neuronal homeostasis and to respond to environmental insults. The hierarchical organization of neurons (i.e., CNS neurons at the top of the pyramid with PNS axons at the bottom of the pyramid, as they are in contact with the peripheral tissues) and their highly differentiated state requires multistep and dynamic control of defenses. In this study, we found that exposure of axons to IFN-β and IFN-γ activated distinct mechanisms to specifically restrict alphaherpesvirus retrograde infection of PNS cell bodies, while axonal transport of cellular organelles was unaffected. These effects were mediated through IFN receptors, since both IFN receptor knockout mouse neurons and IFNAR-blocking antibody treatment blocked the antiviral effects. The IFN-β response is local and confined to axons: STAT1 is phosphorylated only in axons, and the antiviral effect (reduced retrograde transport) does not require transcription of ISGs in the nucleus. In contrast, axons treated with IFN-γ produce a cell-wide response: IFN-γ treatment of axons triggers phosphorylation and nuclear translocation of STAT1. The subsequent antiviral effects of IFN-γ not only inhibit retrograde transport of PRV particles in axons but also block virus replication in the cell bodies.
When axons were exposed to IFN-β, STAT1, the master regulator of the IFN response, was phosphorylated and retained in axons. p-STAT1 is a well-characterized transcription factor, and its role in neurons is essential for protection against HSV-1 infection (27). We suggest that STAT1 acts as a signaling hub to reduce the risk of alphaherpesvirus spread into the nervous system. The presence of p-STAT1 in axons may reduce capsid transport to cell bodies by inducing axonal protein synthesis and formation of retrograde injury signaling complexes that compete for the fast retrograde transport machinery mediated by dynein. STAT1 is reported to interact with many signaling molecules including STAT2 (28), STAT3 (29, 30), and mammalian target of rapamycin (mTOR) (31, 32). Among these proteins, STAT3 is translated and phosphorylated locally in axons and transported to cell bodies following axonal injury to regulate survival and regeneration of axons (13, 33–35). mTOR is characterized as the major regulator of protein synthesis. During exposure of axons to IFN, p-STAT1 may activate mTOR and mediate the local translation of STAT3 as well as other signaling molecules. We have shown that axonal injury retards PRV retrograde transport (25). As cellular proteins that mediate retrograde trafficking of any cargo in axons may be limited, we suggested that the reduction of virus particle transport after axonal injury reflects competition for the fast axonal transport complexes (24, 25). Here we suggest that similar axonal damage response mechanisms may result when axons are exposed to the cytokine milieu produced by infected epithelial cells.
Unlike the response observed when axons are exposed to IFN-β, axonal IFN-γ treatment led to translocation of p-STAT1 to the nucleus and reduction in both PRV particle transport in the axons and subsequent viral replication in the cell bodies. Nuclear transcription is also required for IFN-γ inhibition of PRV transport, and it is likely that induction of ISG expression is responsible for these antiviral effects. However, the levels of ISG transcripts, including GBP2 and IFIT1, were low, perhaps reflecting the fact that not all cell bodies are connected to the N compartment where axons were exposed to IFN-γ. A comprehensive study of axonal IFN-γ-induced transcription is required to determine which genes may be involved in this antiviral response. Axonal IFN-γ exposure may also activate local axonal translation, although this response was not as prominent as found with IFN-β treatment (see Fig. S1B in the supplemental material).
Previous studies have come to conflicting conclusions regarding whether axonal IFN exposure induces an antiviral response. Svennerholm et al. found no antiviral effect against HSV-1 infection when neurites of dorsal root ganglion neurons were pretreated with IFN-α and IFN-β (17). In contrast, a recent study demonstrated that axonal IFN-β signaling restricted HSV-1 infection of trigeminal ganglion neurons (27). However, the mechanism of antiviral response activation and the steps of viral replication that are affected were not determined. Our results show that exposure of axons to IFN-β or IFN-γ reduced the number of moving particles of both PRV and HSV-1 in axons. The mechanism is under study, but it may include inefficient uptake of viral particles as well as deficiency in retrograde transport. It is important to note that the transport of other axonal cargo, including acidic endosomes, was not affected by IFN treatment.
To understand the specific antiviral effects of IFN, we determined the axonal proteome after IFN treatment. Mass spectrometry-based analysis of axons before and after IFN-β and IFN-γ treatment revealed a subset of proteins with significant increases or decreases after IFN exposure. While some changes were specific to the type of IFN, others showed similar trends after IFN-β and IFN-γ treatment. One such protein, Clip2, is a brain-specific microtubule plus-end binding protein (+TIP) with high homology to Clip1 (also known as Clip170) (36–38). Clip1 regulates growth and dynamics of the microtubule cytoskeleton, interacts with dynein/dynactin complexes and LIS1, and initiates retrograde axonal transport of various cellular cargos (26, 39). Clip1 also affects postentry movement of HSV-1 in nonneuronal cells (40). Various studies have implicated Clip2 functions in microtubule-based transport. Clip2 competes with Clip1 and the dynactin large subunit, p150Glued, for microtubule plus-end binding (41). Clip2 also interacts with bicaudal D, dynein light chain LC8, and it may mediate binding of peroxisomes to microtubules (42–44). In our study, knocking down Clip2 reduced PRV particle transport by ~25%, which is less than the ~40 to 50% reduction by IFN treatment for 24 h. It is important to note that we could achieve only ~50% knockdown efficiency in axons. Also, Clip2 is only one of several other axonal proteins that are jointly regulated by both IFNs. Even though we demonstrated that knocking down Clip1 also reduced PRV transport, IFN treatment does not affect Clip1 levels. Clip1 is generally involved in initiating retrograde transport of various cellular cargos in axons. We demonstrated that axonal IFN treatment does not affect transport of other cellular cargos such as lysosomes/late endosomes. Therefore, by reducing axonal Clip2, but not Clip1, the IFN response may preferentially affect viral transport, without more general effects on transport of cellular cargos. Interestingly, axonal infection of PRV also rapidly reduces Clip2 levels (24). This result could reflect an axonal antiviral effect against infection, similar to IFN pretreatment, or depletion of Clip2 from axons via cotransport with virus particles to the cell bodies. In general, we believe Clip2 may play important roles in initiating alphaherpesvirus retrograde transport and in the axonal antiviral response upon PRV entry or exposure to IFN.
Our working model to account for our results is shown in Fig. 7. In the PNS, axons extend long distances from the cell bodies to innervate various peripheral tissues. The PNS axon termini in tissues are both primary sites of neuroinvasion and are exposed to inflammatory cytokines, such as IFN-β and IFN-γ, produced by the infected epithelial cells and surveilling immune cells. Such a differentiated cellular architecture requires finely tuned and dynamic response machinery in both the axon termini and cell bodies to enable timely, efficient, and well-controlled response to different peripheral signals. We suggest that the inflammatory milieu in damaged peripheral tissue induces a biphasic activation of immune defense against infectious agents, involving local autonomous action by axons at the site of infection and more-distant responses in the cell bodies. According to this hypothesis, in the early stages of infection, while virus replication is controlled by the intrinsic and innate defenses of the epithelial cells, exposure of nerve termini to IFN-β stimulates local responses in axons, independent of actions in the distant cell bodies. This local response serves to limit the transport of alphaherpesvirus particles that enter the axons. In this first phase, neuronal cell bodies are not engaged, which minimizes the risk of any unnecessary responses such as apoptosis. In the second phase, when primary intrinsic and innate defense against the peripheral virus infection is not effective and the infection progresses, further innate and adaptive immune cells (e.g., natural killer cells and T cells) enter the tissues. IFN-γ produced by these cells signals through axons and alarms neuronal cell bodies of a potential viral invasion from the periphery. In this phase of immune activation, a global response, involving canonical IFN signaling and induction of ISGs in the cell body, shuts down both virus particle transport in axons and replication in the cell bodies.
FIG 7 .
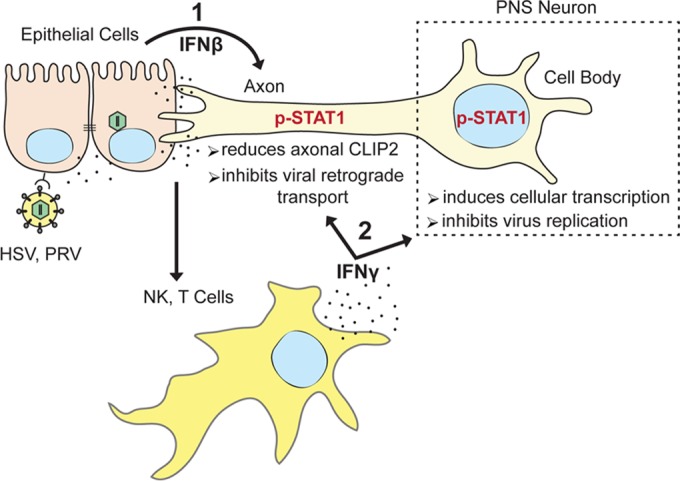
Biphasic immune activation against alphaherpesvirus spread from epithelial cells to the PNS neurons. Alphaherpesvirus infection of epithelial cells induces production and secretion of various cytokines, including IFN-β. Exposure of innervating nerve termini to IFN-β limits the transport of alphaherpesviruses. In the first phase (1), STAT1 is phosphorylated and retained in axons, which may cause the reduction of Clip2 proteins that limit the transport of virus capsids into the connected cell body. If the virus infection is not contained in the first phase and the infection progresses without effective control, innate and adaptive immune cells are activated (e.g., natural killer and T cells). These cells produce copious amounts of cytokines, including IFN-γ. In the second phase (2), exposure of axons to IFN-γ results in the nuclear translocation of p-STAT1, which may activate expression of numerous ISGs. In this phase, the transport of virus capsids is limited through reduction of Clip2, while the replication of virus in the cell bodies is also restricted.
Although we observed a consistent and statistically significant reduction of PRV particle retrograde transport, the antiviral effect of both types of IFN does not completely block the high-MOI (multiplicity of infection) virus infection. The effect is time dependent, with longer axonal exposure to IFNs producing a stronger effect. These findings pose two questions: why is the antiviral response not more robust, and what is its biological significance? The moderate response may reflect the prosurvival, antiapoptotic nature of neurons. Since the majority of neurons are fully differentiated nonreplicating cells, losing a population after each infection or reactivation may cause an ultimate unrecoverable loss of neurological function. However, the moderate antiviral response after axonal IFN exposure may be important, especially in vivo, where the number of virus particles to which axon termini are exposed may be considerably fewer than the number we artificially provide in vitro. Our recent studies showed that by reducing the number of viral particles moving in axons, the outcome of infection in cell bodies is dramatically affected. Fewer incoming particles lead to a quiescent infection, as opposed to the productive infection typically observed in vitro with high doses of inoculum (24). Therefore, the effect of peripheral cytokine exposure may facilitate the quiescent, nondamaging infection of the PNS, the hallmark of alphaherpesvirus neuroinvasion.
Our work has revealed a novel and noncanonical axonal response after IFN-β exposure and provided insight into the coordination of defenses that must occur at the interface between peripheral tissues and the nervous system. We identified specific proteins that determine the mode (p-STAT1 in axons versus nucleus) and extent (axonal or global) of this defensive response. Further work is needed to dissect the interaction map of the proteins and signaling pathways that determine the defensive reactions against different neurotropic virus infections in different tissues. These efforts will also contribute to understanding the molecular mechanisms of neuropathies and to the development of targeted therapies for different types and levels of neurological diseases.
MATERIALS AND METHODS
Cell lines and virus strains.
Porcine kidney (PK15) and African green monkey (Vero) epithelial cells were used to produce and determine the titers of PRV and HSV strains, respectively. Cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin. PRV-Becker is a wild-type laboratory strain (45). PRV-180 expresses mRFP-VP26 in a PRV-Becker background (18). HSV-1 OK14 was constructed by cotransfecting BamHI-digested pHSV1(17+)Lox-mRFPVP26 and purified HSV-1(17+) DNA (O. Kobiler and L. W. Enquist, unpublished data). The pHSV1(17+)Lox-mRFPVP26 was a kind gift from Katinka Döhner and Beate Sodeik (46, 47).
Neuronal cultures.
SCGs were isolated from day 17 Sprague-Dawley rat embryos (Hilltop Labs) and day 14.5 wild-type (Jackson Laboratory) or IFNAR and IFNGR KO (kind gift from Herbert W. Virgin) C57BL/6J mouse embryos. Neurons were cultured in trichambers (Tyler Research) as described previously (16) (see also Text S1 in the supplemental material). All animal work was performed in accordance with the Princeton Institutional Animal Care and Use Committee (protocols 1947-13 and 1851-14).
Viral infection and drug treatment in compartmented neuronal cultures.
For all experiments in trichambers, 1% methylcellulose in neuronal medium was added in the middle (M) compartment to prevent any possible leakage from either the soma (S) or neurite (N) compartment. Five hundred units/ml IFN-β or IFN-γ was added to the N compartment for the indicated time. Neuronal infections were done using 106 PFU of PRV or HSV-1 unless otherwise noted. DiO was added at 2.5 µg/ml (Life Technologies) to the N compartment at 1 hpi. CellTracker Orange (Life Technologies) was added at 10 µM to the N compartment. Low-endotoxin, azide-free (LEAF)-purified mouse IgG1, κ isotype control antibody, or anti-mouse IFNAR-1 antibody (BioLegend) was added at 10 µg/ml for 3 h prior to IFN-β. Actinomycin D (Sigma-Aldrich) was added at 0.5 µg/ml to the S compartment simultaneous to axonal IFN treatment. LysoTracker Red DND-99 (Life Technologies) staining was done at 50 nM for 1 h. Emetine (Sigma-Aldrich) was added at 100 nM to the N compartment 1 h prior to IFN-β for 6 h.
Microscopy.
All imaging was performed on a previously described Nikon Ti-Eclipse inverted epifluorescence microscope (48). Tiled images of the entire S compartment were captured using a Cool Snap ES2 camera (Photometrics) at ×4 magnification, and movies of fluorescent particles were taken using an Andor iXon Ultra EMCCD camera at a magnification of ×60. The movies of fluorescent particles were acquired within 15 to 45 min after infection or staining. During live-cell imaging, neuron cultures were kept in a humidified stage-top incubator at 37°C with 5% CO2. The percentage of infected cell bodies and moving fluorescent particles were calculated manually using NIS-Elements imaging software (Nikon). For fixed samples, images were captured using the Cool Snap ES2 camera at a magnification of 60×. All images and movies were assembled for publication using ImageJ. For comparative analysis, fluorescence intensity, exposure time, and other parameters were consistent for all conditions in the same experiment.
Western blot analysis.
Immunoblotting was performed using the following antibodies: anti-β-actin (1:5,000) (Sigma), anti-p-STAT1 (1:500), and anti-Clip1 (1:500) (Cell Signaling Technology), anti-STAT1 (1:500) (Santa Cruz), anti-Clip2 (1:500) (Abcam), and horseradish peroxidase-conjugated secondary mouse or rabbit antibodies (1:10,000) (KPL) (see Text S1 in the supplemental material).
Immunofluorescence staining.
Neurons were cultured in chambered optical plastic dishes (Ibidi). Staining was performed using anti-p-STAT1 antibody (1:100), Alexa Fluor 488-conjugated secondary antibody against rabbit (1:500) (Life Technologies) and 4′,6′-diamidino-2-phenylindole (DAPI) (1:1,000) (see Text S1 in the supplemental material).
Quantitative reverse transcription-PCR.
Total RNA was extracted from neuron cell bodies in the S compartment using RNeasy Plus minikit (Qiagen). cDNA synthesis was performed using SuperScript III first-strand synthesis kit (Life Technologies) with oligo(dT) primer. Quantitative reverse transcription-PCR (q-RT-PCR) was performed using the Eppendorf Realplex Mastercycler. Reaction mixture was prepared using Kapa Sybr Fast quantitative PCR (q-PCR) master mix. Samples were prepared in triplicate. Plotted values were calculated using the –ΔΔCT method normalized to β-actin samples and samples not treated with IFN (see Text S1 in the supplemental material).
Mass spectrometry-based proteomic sample preparation and data analysis.
After axonal IFN treatment for 24 h, axons were collected in NuPAGE lithium dodecyl sulfate sample buffer (Life Technologies). Ten chambers were pooled for each sample. Samples were loaded onto a 4 to 12% NuPAGE bis-Tris gels (Invitrogen) and stained with Coomassie blue. In-gel digestion with trypsin followed by liquid chromatography-tandem mass spectrometry analysis was performed as previously described (24) with some modifications (see also Text S1 in the supplemental material).
siRNA transfections.
Magnetofection SilenceMag (OZ Biosciences) was used for delivery of 50 nM On-TARGETplus rat nontargeting, Clip1 or Clip2 siRNA pools (Dharmacon) into neurons. At 96 h posttransfection, cells were harvested or infected for further analysis.
Statistical analysis.
Two-way analysis of variance (ANOVA) or Student’s t test was performed using GraphPad Prism 5.0. Values in the text, graphs, and figure legends throughout the manuscript are means ± standard errors of the means (SEMs).
SUPPLEMENTAL MATERIAL
Axonal IFN treatment induces changes in the axonal proteome. (A) Tabulation of proteins with significant changes in abundance in axons after IFN-γ treatment. Proteins were manually categorized based on their biological function or subcellular localization and color coded based on the changes in spectral counts following IFN-γ treatment. A purple ring around the protein indicates that similar changes were also seen in IFN-β-treated samples. Protein that was detected only in IFN-γ-treated sample and not in non-IFN-treated sample is indicated by an asterisk. (B) Axons in the N compartment were incubated with l-AHA and labeled with alkyne 488 in the presence or absence of IFN-β or IFN-γ for 2 h (bars, 10 µm). (C) Immunoblotting of p-STAT1 in axons of the N compartment with or without axonal emetine treatment for 1 h prior to IFN-β treatment for 6 h. (D) Immunoblotting of Clip2 and p-STAT1 in axons of the N compartment after axonal IFN-β or IFN-γ treatment for 24 h. Quantification of Clip2 bands are shown in the right panel. Band intensities were normalized to internal control β-actin of each sample and to the non-IFN-treated sample. (E) Immunoblotting of Clip1 and Clip2 in noncompartmentalized neurons that were transfected with the indicated siRNA (NT, nontargeting). Download
Axonal IFN pretreatment reduces the number of moving PRV particles in axons. IFN-β or IFN-γ was added to the N-compartment axons for 6 h (top panel) or 24 h (bottom panel) before infection with PRV-180. The movement of fluorescent particles in the N compartment was recorded at ~1 frame per second (fps). The playback time is 20 fps. Bars, 10 µm. Download
Signaling through IFN-β receptor is responsible for the defect in PRV retrograde transport in axons. Mouse SCG axons in the N compartment were not treated with antibody or treated with control antibody or IFNAR antibody prior to addition of IFN-β. Twenty-four hours after axonal IFN-β treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. The playback time is 30 fps. Bars, 10 µm. Download
Signaling through IFN receptors is responsible for the defect in PRV retrograde transport in axons. WT or IFNAR and IFNGR double knockout mouse SCG neuron axons in the N compartment were treated with IFN-β or IFN-γ. Twenty-four hours after axonal IFN treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. The playback time is 30 fps. Bars, 10 µm. Download
Axonal IFN pretreatment reduces the number of moving HSV-1 particles in axons. IFN-β or IFN-γ was added to the N-compartment axons for 24 h before infection with HSV-1 OK-14. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. The playback time is 30 fps. Bars, 10 µm. Download
Axonal IFN pretreatment does not affect transport of LysoTracker-positive organelles in axons. IFN-β or IFN-γ was added to the N-compartment axons for 24 h before staining with LysoTracker. Movement of fluorescent particles in the N compartment was recorded at ~5 fps. The playback time is 100 fps. Bars, 10 µm. Download
IFN-β-mediated inhibition of PRV transport does not require transcription in the nucleus. SCG neuron cell bodies in the S compartment were treated with DMSO (carrier) or actinomycin D (ActD), while axons in the N compartment were simultaneously treated with IFN-β (top panel) or IFN-γ (bottom panel) or without IFN. Twenty-four hours after treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~2 fps. The playback time is 40 fps. Bars, 10 µm. Download
Reducing the level of Clip1 or Clip2 reduces PRV particle transport. The cells in S compartments were transfected with nontargeting, Clip1, or Clip2 siRNA before PRV-180 infection (5 × 105 PFU) in the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~3.2 fps. The playback time is 65 fps. Bars, 10 µm. Download
Axonal IFN treatment of neurons induces changes in the axonal proteome. Proteins with significant changes in abundance in axons after IFN-β or IFN-γ treatment are listed. The numbers of biological replicates are 5, 4, and 3 for non-IFN-treated, IFN-β-treated, and IFN-γ-treated samples, respectively.
Supplemental experimental procedures. Download
ACKNOWLEDGMENTS
We thank Jian Song and the members of the Enquist lab, especially Ian Hogue and Margaret MacGibeny for their comments and critical reading of the manuscript.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW. 2016. Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. mBio 7(1):e02145-15. doi:10.1128/mBio.02145-15.
REFERENCES
- 1.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67:355–374. doi: 10.1146/annurev-micro-092412-155654. [DOI] [PubMed] [Google Scholar]
- 2.Koyuncu OO, Hogue IB, Enquist LW. 2013. Virus infections in the nervous system. Cell Host Microbe 13:379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lafaille FG, Ciancanelli MJ, Studer L, Smith G, Notarangelo L, Casanova JL, Zhang SY. 2015. Deciphering human cell-autonomous anti-HSV-1 immunity in the central nervous system. Front Immunol 6:208. doi: 10.3389/fimmu.2015.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lomniczi B. 1974. Factors influencing the interferon production of Aujeszky's disease (pseudorabies) virus. Arch Gesamte Virusforsch 44:150–152. [DOI] [PubMed] [Google Scholar]
- 7.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bigley NJ. 2014. Complexity of interferon-gamma interactions with HSV-1. Front Immunol 5:15. doi: 10.3389/fimmu.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lalli G. 2014. Regulation of neuronal polarity. Exp Cell Res 328:267–275. doi: 10.1016/j.yexcr.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 11.Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. 2008. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol 10:149–159. doi: 10.1038/ncb1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. 2003. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 40:1095–1104. doi: 10.1016/S0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Yaakov K, Dagan SY, Segal-Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, Blesch A, Pilpel Y, Twiss JL, Fainzilber M. 2012. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J 31:1350–1363. doi: 10.1038/emboj.2011.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holt CE, Schuman EM. 2013. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80:648–657. doi: 10.1016/j.neuron.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung H, Yoon BC, Holt CE. 2012. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13:308–324. doi: 10.1038/nrn3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curanovic D, Ch'ng TH, Szpara M, Enquist L. 2009. Compartmented neuron cultures for directional infection by alpha herpesviruses. Curr Protoc Cell Biol Chapter 26:Unit 26.4. doi: 10.1002/0471143030.cb2604s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Svennerholm B, Ziegler R, Lycke E. 1989. Herpes simplex virus infection of the rat sensory neuron. Effects of interferon on cultured cells. Arch Virol 104:153–156. doi: 10.1007/BF01313816. [DOI] [PubMed] [Google Scholar]
- 18.del Rio T, Ch'ng TH, Flood EA, Gross SP, Enquist LW. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J Virol 79:3903-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curanović D, Lyman MG, Bou-Abboud C, Card JP, Enquist LW. 2009. Repair of the UL21 locus in pseudorabies virus Bartha enhances the kinetics of retrograde, transneuronal infection in vitro and in vivo. J Virol 83:1173–1183. doi: 10.1128/JVI.02102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, Schreiber RD. 2006. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res 26:804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- 21.van den Broek MF, Müller U, Huang S, Aguet M, Zinkernagel RM. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J Virol 69:4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Souza KL, Elsner M, Mathias PC, Lenzen S, Tiedge M. 2004. Cytokines activate genes of the endocytotic pathway in insulin-producing RINm5F cells. Diabetologia 47:1292–1302. doi: 10.1007/s00125-004-1435-2. [DOI] [PubMed] [Google Scholar]
- 23.Miyake M, Fuchimoto S, Orita K. 1989. Differences in intracellular calcium mobilization by interferon-beta and interferon-gamma in RPMI-4788 cells. Exp Cell Biol 57:67–72. doi: 10.1159/000163510. [DOI] [PubMed] [Google Scholar]
- 24.Koyuncu OO, Song R, Greco TM, Cristea IM, Enquist LW. 2015. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. mBio 6:e00276-15. doi: 10.1128/mBio.00276-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koyuncu OO, Perlman DH, Enquist LW. 2013. Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host Microbe 13:54–66. doi: 10.1016/j.chom.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akhmanova A, Steinmetz MO. 2008. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol 9:309–322. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 27.Rosato PC, Leib DA. 2015. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog 11:e1005028. doi: 10.1371/journal.ppat.1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Leung S, Qureshi S, Darnell JE Jr, Stark GR. 1996. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-alpha. J Biol Chem 271:5790–5794. [DOI] [PubMed] [Google Scholar]
- 29.Spiekermann K, Biethahn S, Wilde S, Hiddemann W, Alves F. 2001. Constitutive activation of STAT transcription factors in acute myelogenous leukemia. Eur J Haematol 67:63–71. doi: 10.1034/j.1600-0609.2001.t01-1-00385.x. [DOI] [PubMed] [Google Scholar]
- 30.Gunaje JJ, Bhat GJ. 2001. Involvement of tyrosine phosphatase PTP1D in the inhibition of interleukin-6-induced Stat3 signaling by alpha-thrombin. Biochem Biophys Res Commun 288:252–257. doi: 10.1006/bbrc.2001.5759. [DOI] [PubMed] [Google Scholar]
- 31.Kristof AS, Marks-Konczalik J, Billings E, Moss J. 2003. Stimulation of signal transducer and activator of transcription-1 (STAT1)-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J Biol Chem 278:33637–33644. doi: 10.1074/jbc.M301053200. [DOI] [PubMed] [Google Scholar]
- 32.Fielhaber JA, Han YS, Tan J, Xing S, Biggs CM, Joung KB, Kristof AS. 2009. Inactivation of mammalian target of rapamycin increases STAT1 nuclear content and transcriptional activity in alpha4- and protein phosphatase 2A-dependent fashion. J Biol Chem 284:24341–24353. doi: 10.1074/jbc.M109.033530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Brien JJ, Nathanson NM. 2007. Retrograde activation of STAT3 by leukemia inhibitory factor in sympathetic neurons. J Neurochem 103:288–302. doi: 10.1111/j.1471-4159.2007.04736.x. [DOI] [PubMed] [Google Scholar]
- 34.Ohara R, Fujita Y, Hata K, Nakagawa M, Yamashita T. 2011. Axotomy induces axonogenesis in hippocampal neurons through STAT3. Cell Death Dis 2:e175. doi: 10.1038/cddis.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee N, Neitzel KL, Devlin BK, MacLennan AJ. 2004. STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J Comp Neurol 474:535–545. doi: 10.1002/cne.20140. [DOI] [PubMed] [Google Scholar]
- 36.Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI. 2004. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays 26:141–150. doi: 10.1002/bies.10402. [DOI] [PubMed] [Google Scholar]
- 37.Hoogenraad CC, Akhmanova A, Grosveld F, De Zeeuw CI, Galjart N. 2000. Functional analysis of CLIP-115 and its binding to microtubules. J Cell Sci 113:2285–2297. [DOI] [PubMed] [Google Scholar]
- 38.De Zeeuw CI, Hoogenraad CC, Goedknegt E, Hertzberg E, Neubauer A, Grosveld F, Galjart N. 1997. CLIP-115, a novel brain-specific cytoplasmic linker protein, mediates the localization of dendritic lamellar bodies. Neuron 19:1187–1199. doi: 10.1016/S0896-6273(00)80411-0. [DOI] [PubMed] [Google Scholar]
- 39.Moughamian AJ, Osborn GE, Lazarus JE, Maday S, Holzbaur EL. 2013. Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J Neurosci 33:13190–13203. doi: 10.1523/JNEUROSCI.0935-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jovasevic V, Naghavi MH, Walsh D. 2015. Microtubule plus end-associated CLIP-170 initiates HSV-1 retrograde transport in primary human cells. J Cell Biol 211:323–337. doi: 10.1083/jcb.201505123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoogenraad CC, Koekkoek B, Akhmanova A, Krugers H, Dortland B, Miedema M, van Alphen A, Kistler WM, Jaegle M, Koutsourakis M, Van Camp N, Verhoye M, van der Linden A, Kaverina I, Grosveld F, De Zeeuw CI, Galjart N. 2002. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet 32:116–127. doi: 10.1038/ng954. [DOI] [PubMed] [Google Scholar]
- 42.Hoogenraad CC, Akhmanova A, Howell SA, Dortland BR, De Zeeuw CI, Willemsen R, Visser P, Grosveld F, Galjart N. 2001. Mammalian Golgi-associated bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J 20:4041–4054. doi: 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Navarro-Lérida I, Martínez Moreno M, Roncal F, Gavilanes F, Albar JP, Rodríguez-Crespo I. 2004. Proteomic identification of brain proteins that interact with dynein light chain LC8. Proteomics 4:339–346. doi: 10.1002/pmic.200300528. [DOI] [PubMed] [Google Scholar]
- 44.Thiemann M, Schrader M, Völkl A, Baumgart E, Fahimi HD. 2000. Interaction of peroxisomes with microtubules. In vitro studies using a novel peroxisome-microtubule binding assay. Eur J Biochem 267:6264–6275. doi: 10.1046/j.1432-1327.2000.01713.x. [DOI] [PubMed] [Google Scholar]
- 45.Platt KB, Maré CJ, Hinz PN. 1979. Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky’s disease) virus: thermal sensitivity and rabbit virulence markers. Arch Virol 60:13–23. doi: 10.1007/BF01318093. [DOI] [PubMed] [Google Scholar]
- 46.Nagel CH, Pohlmann A, Sodeik B. 2014. Construction and characterization of bacterial artificial chromosomes (BACs) containing herpes simplex virus full-length genomes. Methods Mol Biol 1144:43–62. doi: 10.1007/978-1-4939-0428-0_4. [DOI] [PubMed] [Google Scholar]
- 47.Nagel CH, Döhner K, Binz A, Bauerfeind R, Sodeik B. 2012. Improper tagging of the non-essential small capsid protein VP26 impairs nuclear capsid egress of herpes simplex virus. PLoS One 7:e44177. doi: 10.1371/journal.pone.0044177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor MP, Kratchmarov R, Enquist LW. 2013. Live cell imaging of alphaherpes virus anterograde transport and spread. J Vis Exp 78:50723. doi: 10.3791/50723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Axonal IFN treatment induces changes in the axonal proteome. (A) Tabulation of proteins with significant changes in abundance in axons after IFN-γ treatment. Proteins were manually categorized based on their biological function or subcellular localization and color coded based on the changes in spectral counts following IFN-γ treatment. A purple ring around the protein indicates that similar changes were also seen in IFN-β-treated samples. Protein that was detected only in IFN-γ-treated sample and not in non-IFN-treated sample is indicated by an asterisk. (B) Axons in the N compartment were incubated with l-AHA and labeled with alkyne 488 in the presence or absence of IFN-β or IFN-γ for 2 h (bars, 10 µm). (C) Immunoblotting of p-STAT1 in axons of the N compartment with or without axonal emetine treatment for 1 h prior to IFN-β treatment for 6 h. (D) Immunoblotting of Clip2 and p-STAT1 in axons of the N compartment after axonal IFN-β or IFN-γ treatment for 24 h. Quantification of Clip2 bands are shown in the right panel. Band intensities were normalized to internal control β-actin of each sample and to the non-IFN-treated sample. (E) Immunoblotting of Clip1 and Clip2 in noncompartmentalized neurons that were transfected with the indicated siRNA (NT, nontargeting). Download
Axonal IFN pretreatment reduces the number of moving PRV particles in axons. IFN-β or IFN-γ was added to the N-compartment axons for 6 h (top panel) or 24 h (bottom panel) before infection with PRV-180. The movement of fluorescent particles in the N compartment was recorded at ~1 frame per second (fps). The playback time is 20 fps. Bars, 10 µm. Download
Signaling through IFN-β receptor is responsible for the defect in PRV retrograde transport in axons. Mouse SCG axons in the N compartment were not treated with antibody or treated with control antibody or IFNAR antibody prior to addition of IFN-β. Twenty-four hours after axonal IFN-β treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. The playback time is 30 fps. Bars, 10 µm. Download
Signaling through IFN receptors is responsible for the defect in PRV retrograde transport in axons. WT or IFNAR and IFNGR double knockout mouse SCG neuron axons in the N compartment were treated with IFN-β or IFN-γ. Twenty-four hours after axonal IFN treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. The playback time is 30 fps. Bars, 10 µm. Download
Axonal IFN pretreatment reduces the number of moving HSV-1 particles in axons. IFN-β or IFN-γ was added to the N-compartment axons for 24 h before infection with HSV-1 OK-14. Movement of fluorescent particles in the N compartment was recorded at ~1.5 fps. The playback time is 30 fps. Bars, 10 µm. Download
Axonal IFN pretreatment does not affect transport of LysoTracker-positive organelles in axons. IFN-β or IFN-γ was added to the N-compartment axons for 24 h before staining with LysoTracker. Movement of fluorescent particles in the N compartment was recorded at ~5 fps. The playback time is 100 fps. Bars, 10 µm. Download
IFN-β-mediated inhibition of PRV transport does not require transcription in the nucleus. SCG neuron cell bodies in the S compartment were treated with DMSO (carrier) or actinomycin D (ActD), while axons in the N compartment were simultaneously treated with IFN-β (top panel) or IFN-γ (bottom panel) or without IFN. Twenty-four hours after treatment, PRV-180 was added to the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~2 fps. The playback time is 40 fps. Bars, 10 µm. Download
Reducing the level of Clip1 or Clip2 reduces PRV particle transport. The cells in S compartments were transfected with nontargeting, Clip1, or Clip2 siRNA before PRV-180 infection (5 × 105 PFU) in the N compartment. Movement of fluorescent particles in the N compartment was recorded at ~3.2 fps. The playback time is 65 fps. Bars, 10 µm. Download
Axonal IFN treatment of neurons induces changes in the axonal proteome. Proteins with significant changes in abundance in axons after IFN-β or IFN-γ treatment are listed. The numbers of biological replicates are 5, 4, and 3 for non-IFN-treated, IFN-β-treated, and IFN-γ-treated samples, respectively.
Supplemental experimental procedures. Download