

Abstract
Cytosolic calcium influx activates signaling pathways known to support pancreatic beta cell function and survival by modulating gene expression. Impaired calcium signaling leads to decreased beta cell mass and diabetes. To appreciate the causes of these cytotoxic perturbations, a more detailed understanding of the relevant signaling pathways and their respective gene targets is required. In this study, we examined the calcium-induced expression of the cytoprotective beta cell transcription factor Npas4. Pharmacological inhibition implicated the calcineurin, Akt/protein kinase B, and Ca2+/calmodulin-dependent protein kinase signaling pathways in the regulation of Npas4 transcription and translation. Both Npas4 mRNA and protein had high turnover rates, and, at the protein level, degradation was mediated via the ubiquitin-proteasome pathway. Finally, beta cell cytotoxicity of the calcineurin inhibitor and immunosuppressant tacrolimus (FK-506) was prevented by Npas4 overexpression. These results delineate the pathways regulating Npas4 expression and stability and demonstrate its importance in clinical settings such as islet transplantation.
Keywords: beta cell, calcineurin, insulin secretion, pancreatic islet, transcription factor, MIN6 cell, Npas4, activity-regulated, calcium signaling
Introduction
Pancreatic beta cells are critical for the regulation of systemic glycemia because of their ability to sense ambient glucose levels and release the glucose-lowering hormone insulin. The triggering phase of insulin secretion involves glucose uptake into the cell via GLUT1/2, phosphorylation by glucokinase, and metabolism into ATP. This results in the closure of the ATP-sensitive potassium channel, followed by membrane depolarization, calcium influx through L-type voltage-dependent calcium channels and exocytosis of insulin granules (1, 2).
In addition to the direct stimulation of insulin secretion, calcium influx also triggers a number of calcium-dependent signaling pathways that promote beta cell function and survival (Fig. 2A) (3–13). Downstream of these signaling pathways, activation of transcription factors of the CREB,4 forkhead box protein O (FOXO), and NFAT families mediates gene expression. CREB mediates anti-apoptotic effects in beta cells involving the action of insulin (14) or the incretin hormones (e.g. gastric inhibitory polypeptide and glucagon-like peptide 1) (15, 16), whereas FOXO1 prevents stress-induced beta cell dedifferentiation (17) and reduces glucose-induced oxidative stress (18).
FIGURE 2.

Npas4 mRNA expression in MIN6 beta cells relies on the CaMK, Akt, and CaN signaling pathways. Pharmacological inhibitors for several calcium-dependent signaling pathways were tested for their effect on Npas4 induction. MIN6 cells were kept in either basal medium alone (white columns, Basal) or with 40 mm KCl (black columns, KCl) for 2 h in the presence of inhibitors or vehicle control. RNA was extracted and reverse-transcribed, and TaqMan PCR was carried out for Npas4 using GusB as a reference gene and normalizing to control KCl stimulation. A, simplified schematic of calcium-dependent signaling pathways targeted by pharmacological inhibitors (red). Glucose-induced depolarization and ligand-receptor interaction lead to a rise in cytosolic calcium levels ([Ca2+]cyt) via calcium influx through the L-type voltage-dependent calcium channel (L-VDCC) and ER calcium release. The rise in cytosolic calcium levels leads to activation of kinase or phosphatase activity, which, in turn, results in the recruitment of transcription factors to regulatory elements and changes in gene transcription. GIPR, glucose-dependent insulinotropic polypeptide receptor; CaM, calmodulin; ψm, mitochondrial membrane potential; InsR, insulin receptor; PLC, phospholipase C; IP3, inositol trisphosphate; DAG, diacylglycerol; BIM-XI, bisindolylmaleimide XI hydrochloride. B, Npas4 induction was prevented completely with the CaMK inhibitor KN-93 at either 3 or 30 μm (n = 3). C, using 4 μm AIP partially reduced Npas4 induction (n = 7). D, Npas4 is regulated by the Akt pathway because treatment with 10 μm Akti-1/2 partially reduces Npas4 mRNA induction, but treatment with 25 nm Rapa had no effect (n = 3). E, treatment with 20 nm human insulin in low-glucose KRBH did not induce Npas4 expression (n = 3). F, the CaN inhibitor FK-506 (tacrolimus) significantly reduced Npas4 induction at doses of 10 and 100 nm (n = 5). Error bars represent mean ± S.E., and significance was determined using a two-tailed Student's t test or a one-way ANOVA with Dunnett post hoc analysis and Bartlett's test for equal variances where applicable. *, p ≤ 0.05; **, p ≤ 0.01.
In further support of the functional importance of calcium signaling pathways in beta cells, knock out of the regulatory subunit calcineurin b1 of the phosphatase CaN resulted in hypoinsulinemia and hyperglycemia because of reduced beta cell proliferation and mass in aged mice (5). Conditional expression of active NFATc1 (which localizes to the nucleus independently of CaN) rescued the knockout phenotype.
The importance of calcineurin activity within human β cells has also been demonstrated during transplantation. Use of the CaN inhibitor tacrolimus (FK-506) as an immunosuppressant results in early graft failure of human islets transplanted into diabetic mice (19), causes beta cell toxicity, and is linked to new-onset diabetes (20–26). Given the importance of calcium signaling in the maintenance of beta cell function and viability, a more thorough understanding of the relevant signaling pathways and their respective gene targets could offer further insights into the nature of the cytotoxicity caused by impaired calcium signaling.
We have previously identified Npas4 (27–29) as a calcium-regulated, cytoprotective beta cell transcription factor (12, 30). Npas4 was induced rapidly and dramatically in beta cells by membrane depolarization and calcium influx. Expression of Npas4 protected beta cells from thapsigargin- and palmitate-induced ER stress and prevented apoptotic cell death (12). However, the beta cell signaling pathways that couple elevations in intracellular calcium with Npas4 induction have not been described previously.
In this report, we demonstrated that Npas4 mRNA expression in beta cells relies on signaling pathways downstream of the kinases Akt and CaMK and the phosphatase CaN. Npas4 was regulated dynamically at the transcriptional and translational levels via rapid degradation. At the protein level, this was mainly achieved via ubiquitin-dependent proteasomal degradation. Furthermore, Npas4 overexpression prevented FK-506-induced cytotoxicity. On the basis of our findings, we speculate that some of the beta cell cytotoxic effects of FK-506 are due to suppression of CaN-dependent Npas4 induction. Therefore, restoring Npas4 expression may have therapeutic potential for islet transplantation.
Experimental Procedures
Chemicals
AIP, FK-506, InSolutionTM AMPK inhibitor (compound C), InSolutionTM MEK1/2 inhibitor III (PD0325901),KN-93, and STO-609 were purchased from EMD Millipore. Akti-1/2, bisindolylmaleimide XI hydrochloride, and Rapa were from Sigma-Aldrich. All other chemicals were purchased from Fisher Scientific or Sigma-Aldrich. Cell culture reagents and disposables were obtained from BD Falcon, Corning, HyClone, and LifeTech.
Animal Care and Procedures
All procedures were approved by the University of British Columbia Animal Care Committee. Islets were isolated from male CD-1® IGS mice (Charles River Laboratories) between 10–20 weeks of age through standard collagenase digestion.
Cell Culture
MIN6 cells (passages 28–36) were maintained in DMEM with high glucose (25 mm) supplemented with 10% FBS, 2 mm l-glutamine, and 100 units/ml, 100 μg/ml penicillin/streptomycin. Cells were passaged once a week and fed every other day. Islets were cultured in 11 mm glucose RPMI medium supplemented with 10% FBS, 2 mm l-glutamine, and 100 units/ml, 100 μg/ml penicillin/streptomycin.
Stimulation of Islets and MIN6 Cells
Following isolation, mouse islets recovered overnight in Petri dishes containing RPMI medium. Afterward, islets were directly transferred onto 12-well plates and preincubated in KRBH (114 mm NaCl, 20 mm HEPES, 4.7 mm KCl, 1.2 mm KH2PO4, 2.5 mm CaCl2, 1.2 mm MgSO4, and 0.2% BSA (pH 7.4)) supplemented with 2.8 mm glucose for 30 min. Vehicle (DMSO or distilled H2O) or drug (10 μm Akti-1/2, 10 nm FK-506, or 3 μm KN-93) was added to the culture medium, and preincubation continued for 30 min. The medium was then replaced, and islets were stimulated for 2 h in KRBH containing 2.8 or 25 mm glucose and either vehicle or drug, respectively.
MIN6 cells were seeded at 0.2 × 106 cells/well into 24-well plates, 0.8 × 106 cells/well into 12-well plates, or 2 × 106 cells/well into 6-well plates. After 30 h, cells were transferred to DMEM with low glucose (5.5 mm) overnight.
For analysis of Npas4 mRNA induction, cells were pretreated with pharmacological inhibitors or vehicle (DMSO or distilled H2O) for 60 min (AIP, bisindolylmaleimide XI hydrochloride, KN-93, MEK1/2, or STO-609), 30 min (compound C, FK-506) or 15 min (Akti-1/2, EGTA, or Rapa). Because it has been observed previously that elevated glucose alone does not maximally induce Npas4 expression in MIN6 cells (12), MIN6 cells were depolarized with 40 mm KCl (in high-glucose medium) for 2 h. For assessment of insulin signaling on Npas4 expression, MIN6 cells were kept in serum-free KRBH with 2.8 mm glucose for 2 h, followed by stimulation with or without 20 nm of fast-acting human insulin (Novo Nordisk Canada) for 2 h.
For Npas4 stability studies, cells were stimulated with 40 mm KCl for 2 h and then either subjected to continued KCl exposure or transferred to basal medium for 1–4 h, respectively. To study mRNA decay, ActD was used to block transcription at a concentration of 5 μg/ml. Npas4 protein half-life was assessed using 10 μm of the proteasome inhibitor MG132. Alternatively, MIN6 cells were subjected to 2 h of 40 mm KCl stimulation followed by 1–2 h of KCl treatment with or without 1.5 μg/ml of the translational blocker cycloheximide.
Lysates were collected in TRIzol (LifeTech) for real-time quantitative PCR or in non-reducing sample buffer-minus lysis buffer (62.5 mm Tris (pH 6.8), 1 mm Na3VO4, 1 mm NaF, 2% SDS, and 10% glycerol in distilled H2O) for Western blot analysis. Alternatively, lysates for Western blot were collected in Nonidet P-40 lysis buffer (150 mm NaCl, 20 mm Tris (pH 7.4), 1 mm EDTA, 0.5 mm EGTA, 2% SDS, 5% glycerol, and 0.5% Nonidet P-40 in distilled H2O). Protein concentration was determined via BCA assay.
Adenoviral Infection and Cell Death Assay
Npas4 and eGFP adenovirus were constructed using the AdEasy system. Prior to infection, MIN6 cells were seeded at 1.0 × 106 cells/well into 6-well plates. Islets were dispersed by 0.25% trypsin digestion and seeded into 12-well plates. All cells were infected with a multiplicity of infection of 20:1 in 800 μl of medium (MIN6 cells) or 10:1 in 250 μl of medium (islets) for 2 h. Afterward, the medium was topped up to 2 or 1 ml, respectively, and infection continued overnight. The following day, the medium was removed, and cells were washed with PBS and recovered for 6 h in standard culture medium prior to treatment.
Cells infected with Ad-Npas4 or Ad-eGFP were transferred to culture medium containing vehicle (DMSO) or FK-506 (10 or 37 nm) and cultured for 24 h. Protein lysates were obtained in Nonidet P-40 lysis buffer (MIN6 cells) or non-reducing sample buffer-minus lysis buffer (islets). Cell death was assayed via Western blot analysis against cleaved caspase-3.
Real-time Quantitative PCR
RNA was extracted and purified with TRIzol according to the protocol of the manufacturer. Reverse transcription was performed using Superscript II (LifeTech). For quantitative PCR, samples were run in triplicate using 40 ng of cDNA and PrimeTime primers/probes (Integrated DNA Technologies) in a ViiA7 real-time PCR system (ABI). Expression levels were quantified with the ΔΔCt method using GusB as a reference gene. The probe/primer1/primer2 used were as follows: GusB, TCTAGCTGGAAATGTTCACTGCCCTG/CACCCCTACCACTTACATCG/ACTTTGCCACCCTCATCC; Npas4, TGCATCAACTCCAGAGCCAAGTTCA/GTCTCAACATTCCCCTACGAAG/CCCTCCACTTCCATCTTCATG.
Co-immunoprecipitation
MIN6 cells were cultured in basal medium, treated for 2 h with 40 mm KCl, or treated with KCl followed by a 1-h washout in basal medium. The proteasomal inhibitor MG132 (10 μm) was used to prevent degradation of ubiquitinated protein. Cells were then washed twice with ice-cold PBS, and lysates were obtained on ice using precooled Nonidet P-40 buffer (Nonidet P-40 lysis buffer without SDS, supplemented with 1 mm Na3VO4, 50 mm NaF, 50 mm β-glycerophosphate, and protease inhibitor mixture (Roche)). To remove insoluble material, lysates were spun down at 14,000 × g for 10 min at 4 °C and stored at −80 °C until use. Protein concentration was determined via BCA assay.
10% of lysate (30 μg) was set aside as input. Lysate containing 300 μg of total protein was incubated with 1 μg of mouse anti-ubiquitin (Enzo Life Sciences, catalog no. BML-PW8810) at 4 °C overnight with gentle agitation. The next day, 0.75 mg of protein G Dynabeads (LifeTech) were suspended in Nonidet P-40 buffer and added to the lysate-antibody mixture. Incubation continued with agitation for 4 h at 4 °C.
Afterward, bead-antibody-antigen complexes were separated magnetically, washed three times with 0.02% Tween-PBS, and transferred to a new tube. Complexes were then disrupted by resuspension in Nonidet P-40 buffer containing diluted 10× SDS loading dye (625 mm Tris-HCl (pH 6.8), 10% SDS, 0.02% bromophenol blue, 30% β-mercaptoethanol, and 50% glycerol in H2O) and through boiling. Half of the volume of the resulting immunoprecipitation fraction was used for Western blotting.
Western Blot Analysis
For Western blot analysis, crude lysates obtained in non-reducing sample buffer-minus or Nonidet P-40 lysis buffer were sonicated for 2 min at 85% (S-4000 with cup horn, Misonix). 10× SDS loading dye was added, and samples were denatured by boiling. Proteins were separated using standard SDS-PAGE and blotted to PVDF membrane (Bio-Rad). After blocking with 5% milk powder in Tris-buffered saline with 0.1% Tween, membranes were probed with 1:1000 rabbit anti-NPAS4 (Sigma, catalog no. HPA039255), 1:1000 rabbit anti-cleaved caspase-3 (Cell Signaling Technology, catalog no. 9661), or 1:100,000 mouse anti-GAPDH (Sigma-Aldrich, catalog no. G8795) overnight at 4 °C. Membranes were then probed with HRP-conjugated secondary antibodies used at a dilution of 1:10,000 (goat anti-mouse, Jackson ImmunoResearch Laboratories, catalog no. 115-035-174) or 1:5000 (goat anti-rabbit, Jackson ImmunoResearch Laboratories, catalog no. 111-035-047) and visualized using Luminata Crescendo Western HRP substrate (EMD Millipore). Relative protein immunoreactivity was measured using densitometry (ImageJ).
Statistics
Prism 5 (GraphPad) was used for statistical analysis. Student's t test and one-way ANOVA with Dunnett post hoc analysis or Bonferroni's multiple comparison test were used. p ≤ 0.05 was considered significant.
Results
Npas4 Expression Depends on CaMK, Akt, and CaN Signaling in Beta Cells
Because Npas4 is induced by depolarization and dependent on calcium (Fig. 1) (12), we first aimed to understand which of the major calcium signaling pathways regulate Npas4 transcription using pharmacological inhibitors (Fig. 2A) (31). These kinase and phosphatase inhibitors were verified using Western blot analysis for phosphoproteins.5 Using the above approach, the CaMK inhibitor KN-93 completely prevented Npas4 mRNA induction (Fig. 2B). To verify a role of CaMKII for Npas4 induction, the peptide inhibitor AIP (32) was used and significantly reduced the induction of Npas4 expression under stimulatory conditions (Fig. 2C), although to a lesser degree than with KN-93. This is likely because AIP is less effective at suppressing CaMKII activity in beta cells than KN-93.5
FIGURE 1.
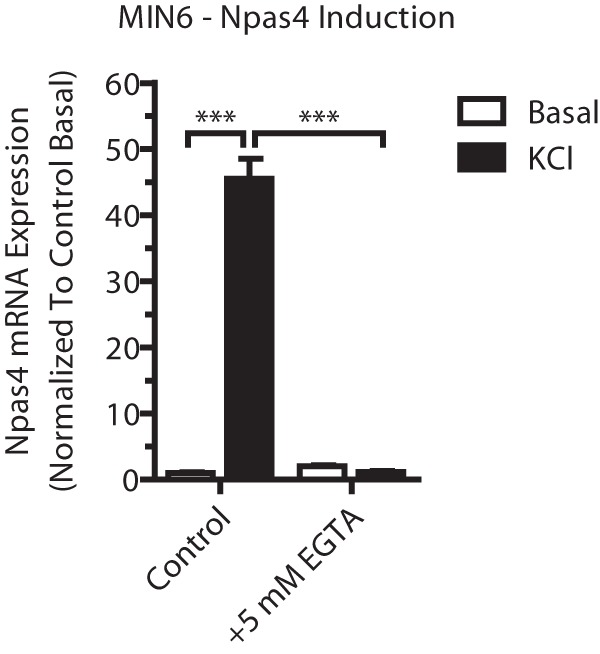
Npas4 induction is calcium-dependent. MIN6 cells were incubated in basal medium alone (white columns, Basal) or with 40 mm KCl (black columns, KCl) for 2 h before treatment with 5 mm EGTA (a calcium chelator). RNA was extracted and reverse-transcribed, and TaqMan PCR was carried out for Npas4 using GusB as a reference gene and normalizing to control basal (n = 23). Error bars represent mean ± S.E., and significance was determined using a one-way ANOVA with Dunnett post hoc analysis and Bartlett's test for equal variances. ***, p ≤ 0.001.
Because insulin has autocrine effects on beta cells, including increases in cytosolic calcium (through effects on sarco/endoplasmic reticulum Ca2+-ATPase) and regulation of gene transcription (33), the role of insulin receptor signaling in Npas4 expression was assessed using the Akt inhibitor Akti-1/2 and the mTOR inhibitor Rapa. Npas4 mRNA levels were reduced mildly with a high dose of the Akt inhibitor but not Rapa (Fig. 2D). To understand whether autocrine insulin action is sufficient to induce Npas4 expression, as reported for other beta cell genes (34), serum-starved MIN6 cells were treated with 20 nm human insulin in low-glucose, serum-free KRBH. No induction of Npas4 mRNA was seen compared with the control (Fig. 2E), suggesting that autocrine-paracrine regulation of Npas4 expression via insulin receptor signaling is dependent on depolarization.
Finally, the CaN inhibitor FK-506 significantly reduced Npas4 mRNA induction at concentrations as low as 10 nm (Fig. 2F), demonstrating that CaN signaling is a major pathway through which Npas4 transcription is activated. Notably, pharmacological inhibition of CaMKK, AMPK, PKC, or MEK1/2 did not affect Npas4 induction (Fig. 3, A–D). These results show that the CaMK, Akt, and CaN signaling pathways are important in the regulation of Npas4 transcription in beta cells.
FIGURE 3.
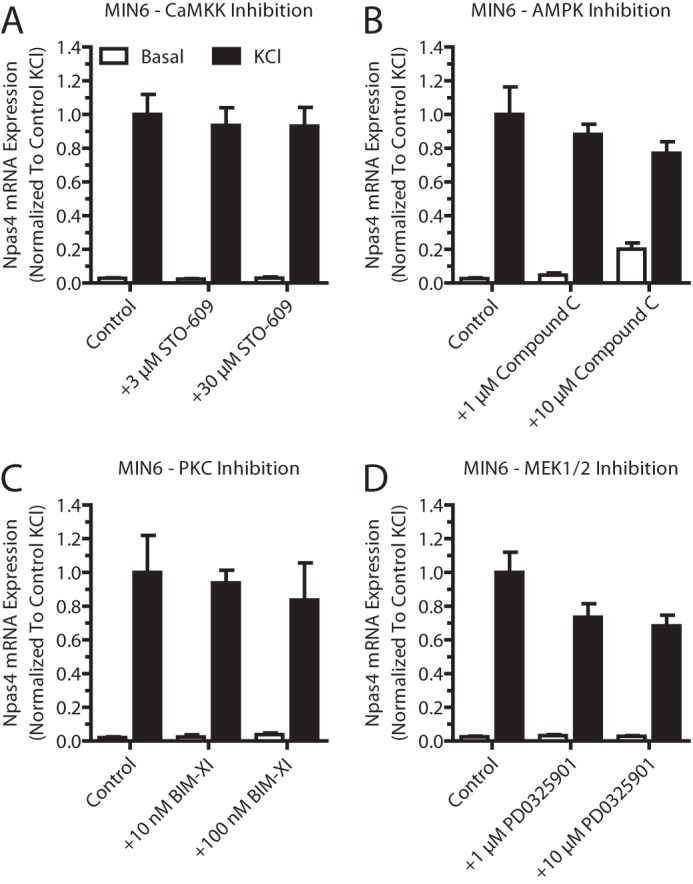
Pharmacological inhibition of several calcium signaling pathways did not affect Npas4 mRNA induction. Pharmacological inhibitors for several calcium-dependent signaling pathways were tested for their effect on Npas4 induction. MIN6 cells were kept in either basal medium alone (white columns, Basal) or with 40 mm KCl (black columns, KCl) for 2 h in the presence of inhibitors or vehicle control. RNA was extracted and reverse-transcribed, and TaqMan PCR was carried out for Npas4 using GusB as a reference gene and normalizing to control KCl stimulation. A—D, 3 and 30 μm STO-609 (A, n = 3–4), 1 and 10 μm compound C (B, n = 3–4), 10 and 100 nm bisindolylmaleimide XI hydrochloride (BIM-XI) (C, n = 3), or 1 and 10 μm PD0325901 (D, n = 5) did not significantly reduce induction of Npas4 expression. Error bars represent mean ± S.E., and significance was determined using a one-way ANOVA with Dunnett post hoc analysis and Bartlett's test for equal variances where applicable.
Next, we determined whether the transcriptional changes seen with KN-93, FK-506, and Akti-1/2 were reflected at the protein level. Although blocking CaMKs with KN-93 prevented expression of Npas4 in MIN6 cells and mouse islets (Fig. 4, A–D), AIP failed to do so (Fig. 5, G and H). It is likely that AIP did not work efficiently in beta cells because positive controls showed reduced CREB phosphorylation after KN-93 but not AIP treatment.5 CaN inhibition with FK-506 in MIN6 cells and islets (Fig. 4, I–L) resulted in a 60% and 74% decrease in the induction of Npas4 protein, respectively, compared with the 37% inhibition of transcript induction with 10 nm FK-506 (Fig. 2F), raising the possibility that CaN has additional effects on Npas4 translation or stability. Strikingly, although Akt inhibition only modestly decreased Npas4 transcription, protein expression was reduced by ∼75% when using Akti-1/2, independent of mTOR signaling (Fig. 4, E–H).
FIGURE 4.

Npas4 protein expression in beta cells depends on the CaMK, Akt, and CaN signaling pathways. MIN6 cells (A, B, E, F, I, and J) were kept in either basal medium alone (Basal) or stimulated with 40 mm KCl (KCl) for 2 h in the presence of inhibitors or vehicle control. Mouse islets (C, D, G, H, K, and L) were treated in KRBH containing 2.8 mm (2.8G) or 25 mm (25G) glucose for 2 h in the presence of inhibitors or vehicle control. A–D, under stimulatory conditions with KCl in MIN6 cells (A and B, n = 4) or high glucose in islets (C and D, n = 3), Npas4 protein was not induced when CaMKs were inhibited with 3 μm KN-93. E–H, 10 μm of the Akt inhibitor Akti-1/2 prevented Npas4 induction in response to KCl in MIN6 cells (E and F, n = 3) and high glucose in islets (G and H, n = 3) whereas mTOR inhibition with 25 nm Rapa treatment did not. I–L, a dose of 10 nm FK-506 significantly reduced the induction of Npas4 protein levels following KCl treatment in MIN6 cells (I and J, n = 3) and high glucose in islets (K and L, n = 4). Error bars represent mean ± S.E., and significance was determined using a two-tailed Student's t test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; N.D., not detectable.
FIGURE 5.

The MEK1/2, CaMKK, PKC, and CaMKII signaling pathways do not affect Npas4 protein expression. MIN6 cells were kept in either basal medium alone (Basal) or with 40 mm KCl (KCl) for 2 h in the presence of inhibitors or vehicle control. A–H, no decrease in Npas4 protein could be observed using inhibitors of the MEK1/2 (A and B, PD0325901 = 1 μm, n = 4), CaMKK (C and D, STO-609 = 3 μm, n = 4), PKC (E and F, bisindolylmaleimide XI hydrochloride (BIM-XI) = 100 nm, n = 4), or CaMKII (G and H, AIP = 4 μm, n = 4) signaling pathways. The asterisk in A indicates a mobility shift of the NPAS4 band when using the MEK1/2 inhibitor. I and J, unexpectedly, 10 μm compound C caused a decrease in Npas4 protein induction (n = 3). Error bars represent mean ± S.E., and significance was determined using a two-tailed Student's t test with Welch's correction. *, p ≤ 0.05.
Inhibitors that had no significant effect on Npas4 transcription also failed to affect protein levels (Fig. 5, A–F). However, inhibition of MEK1/2 resulted in a mobility shift of the Npas4 band from ∼120 to 110 kDa (Fig. 5A). This suggested that Npas4 was phosphorylated by a member of the MAPK family. Lastly, although the AMPK inhibitor was not effective at preventing Npas4 induction at the mRNA level (Fig. 3B), Npas4 protein expression was reduced after application of compound C (Fig. 5, I and J).
Together, our findings demonstrate that Npas4 protein expression is highly regulated and demonstrate a role for CaN, CaMK, Akt, and AMPK signaling in Npas4 translation and stability. The results also implicate the MAPK pathway in posttranslational modification of Npas4.
Cellular Activity Dynamically Regulates Npas4 Expression in MIN6 Beta Cells
We next sought to understand how Npas4 mRNA and protein are regulated when induced. Following a 2-h KCl stimulation, additional exposure to stimulatory conditions (+KCl) or transfer to non-stimulatory conditions (+Basal) both resulted in a gradual, time-dependent decrease of Npas4 expression (Fig. 6A). Continued exposure to KCl slowed the return of Npas4 levels to baseline (t½, 55.7 ± 12.3 min for +KCl versus 24.8 ± 2.5 min for +Basal), likely through predominant effects on Npas4 transcription. To test this, de novo transcription was blocked with ActD, and, following 2-h KCl treatment, Npas4 mRNA was stabilized modestly after 1 h in stimulatory medium (+KCl +ActD, t½ = 28.1 ± 3.1 min) compared with basal medium (+Basal +ActD, t½ = 12.6 ± 4.1 min) (Fig. 6B). In addition, following KCl stimulation, MIN6 cells remained refractory to further Npas4 induction for up to 5 h (Fig. 7). Together, this suggests that Npas4 mRNA has a high turnover rate that is decreased in the presence of a stimulus and that a recovery period is required before Npas4 expression can be restimulated.
FIGURE 6.

Cellular activity dynamically regulates Npas4 expression. MIN6 cells were kept in basal medium (Basal) or stimulated with 40 mm KCl (KCl) for 2 h. A and B, afterward, cells were stimulated further with KCl (+KCl) or transferred to basal medium (+Basal) in the absence (A) or presence (B) of the transcriptional blocker actinomycin D (+ActD, 5 μg/ml). Npas4 mRNA levels declined gradually during both +KCl and +Basal treatments (A), with a t½ of 55.7 (±12.3) min and 24.8 (±2.5) min, respectively. With addition of ActD (B), Npas4 expression returned to basal levels more rapidly under both +KCl and +Basal conditions. KCl treatment had a stabilizing effect, with a t½ of 28.1 (±3.1) min versus 12.6 (±4.1) min under basal treatment. No Npas4 induction was seen when ActD was added from the start of the experiment together with KCl (6 h KCl +ActD). t½ was determined via nonlinear regression for one-phase exponential decay. n = 4–5. C–H, NPAS4 expression was assessed via Western blot analysis and quantified using densitometry, with GAPDH as a reference and normalizing to 2-h KCl treatment. Over the course of 4 h, continued exposure to KCl (C and D, n = 5) or transfer to basal medium (E and F, n = 5) led to a gradual decline in Npas4 protein levels. MIN6 cells in stimulatory KCl medium treated with the translational inhibitor cycloheximide (CHX) for 1 or 2 h exhibited a rapid decline in NPAS4 levels compared with no cycloheximide (G and H, n = 3). Error bars represent mean ± S.E., and significance was determined using a two-tailed Student's t test with Welch's correction or a one-way ANOVA with Dunnett post hoc analysis where applicable. *, p ≤ 0.05; **, p ≤ 0.01.
FIGURE 7.
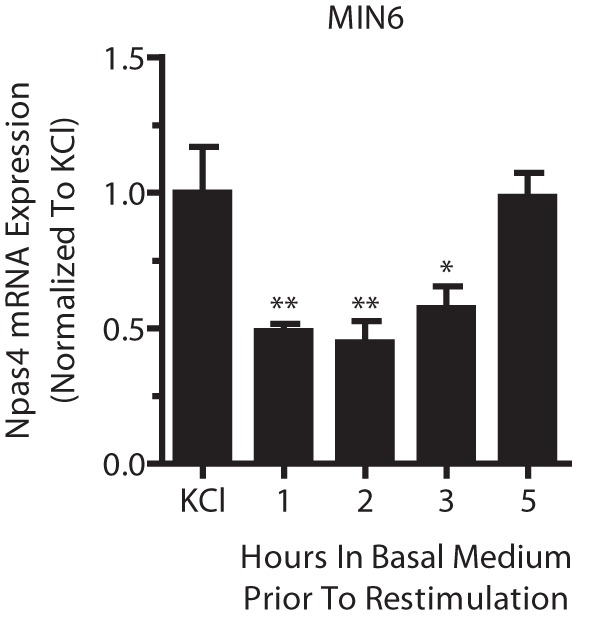
Beta cell Npas4 expression remains refractory for several hours after depolarization. MIN6 cells were stimulated for 2 h with 40 mm KCl (KCl) and transferred to basal medium for 1, 2, 3, or 5 h before restimulation with 40 mm KCl for 2 h. RNA was extracted and reverse-transcribed, and TaqMan PCR was carried out for Npas4 using GusB as a reference gene and normalizing to the initial 2-h KCl treatment. MIN6 cells remained refractory to KCl restimulation for up to 5 h before Npas4 induction, comparable with the initial KCl treatment. Error bars represent mean ± S.E., and significance was determined using a one-way ANOVA with Dunnett post hoc analysis (n = 4). *, p ≤ 0.05; **, p ≤ 0.01.
The dynamic changes in mRNA levels were also reflected at the protein level (Fig. 6, C–F). Under continuous KCl treatment, Npas4 protein levels declined modestly by 10–38% for the first 1–3 h and had dropped by 55% 4 h after the initial stimulus (Fig. 6, C and D). Cells transferred to basal medium demonstrated a more rapid decay of Npas4 protein, reaching 50% after 1 h and close to baseline levels (90% reduction) after 4 h in basal medium (Fig. 6, E and F). Moreover, following the initial 2-h KCl treatment, Npas4 protein levels were reduced significantly by 86% after 1-h co-treatment with KCl and the translational blocker cycloheximide (Fig. 6, G and H), suggesting that depolarization alone has no effect on protein stability. In conclusion, Npas4 protein has a short half-life and is degraded continuously, and stimulatory conditions do not prevent degradation.
Npas4 Is Degraded via the Ubiquitin-Proteasome Pathway
Although the above studies demonstrate that Npas4 protein expression decays rapidly, how it is degraded remains unclear. The ubiquitin-dependent proteasomal pathway is the primary pathway for protein degradation and may also play a role in preventing ER stress through clearance of protein aggregates in the beta cell (35). Its role in regulating Npas4 protein in MIN6 beta cells was studied with the use of the proteasomal inhibitor MG132. In the presence of MG132, the previously observed decreases in Npas4 protein levels (Fig. 6, C–F) were prevented or reduced following continued stimulation in KCl or transfer to basal medium (Fig. 8, A–D). These findings were corroborated by co-immunoprecipitation of Npas4 with ubiquitin (Fig. 8E). Of note, there was a trend toward Npas4 protein accumulation in KCl-stimulated cells treated with MG132 (Fig. 8, A and B), suggesting that proteasomal repression during beta cell activity might result in Npas4 stabilization (36). In summary, the above findings demonstrate that Npas4 protein is ubiquitinated and degraded via the proteasome.
FIGURE 8.

Npas4 is degraded via the ubiquitin-proteasome pathway. MIN6 cells were kept in either basal medium alone (Basal) or with 40 mm KCl (2 h KCl) for 2 h in the presence of 10 μm MG132 or vehicle control. A–D, MIN6 cells were subjected to either stimulation in KCl medium (+ KCl, A and B) or basal medium washout (+Basal, C and D). Blocking the proteasome with MG132 prevented the gradual degradation of Npas4 protein after 3 and 6 h of KCl treatment (A and B, conditions 3 and 4 + MG132). Addition of MG132 also prevented Npas4 degradation following 1 h in basal medium, but NPAS4 levels declined modestly after 3 h of basal washout (C and D, conditions 3 and 4 + MG132). E, MIN6 cells were cultured in basal medium (1), treated for 2 h with 40 mm KCl (2) or KCl, followed by a 1-h washout in basal medium (3), all in the presence of 10 μm MG132. Cells were lysed and immunoprecipitated with an ubiquitin-specific antibody (IP α Ub) and then Western-blotted. Npas4 immunoreactivity was observed in KCl-treated (2′ and 3′) but not unstimulated (1′) IP fractions. n = 3. Error bars represent mean ± S.E., and significance was determined using a one-way ANOVA with Bonferroni's multiple comparison test. *, p ≤ 0.05; ***, p ≤ 0.001.
Npas4 Prevents FK-506 Induced Beta Cell Death
Because Npas4 is regulated by calcineurin (Figs. 2 and 4) and the immunosuppressant FK-506 (a CaN inhibitor, Fig. 2A) is toxic for beta cells (6) and has been implicated in new-onset diabetes (25), we hypothesized that loss of Npas4 induction in FK-506-treated beta cells may cause beta cell cytotoxicity. Furthermore, restoration of Npas4 expression would be protective against the detrimental effects of FK-506 treatment. To bypass the inhibitory effects of FK-506 on Npas4 expression, MIN6 and dispersed islet cells were transduced with an Npas4 or eGFP control adenovirus, followed by treatment with 10 or 37 nm FK-506 for 24 h. In agreement with previous findings, FK-506 increased levels of cleaved caspase-3 in eGFP-transduced MIN6 cells, but overexpression of Npas4 completely prevented the FK-506-mediated induction of cleaved caspase-3 (Fig. 9, A and B). A similar trend was also observed in dispersed murine islet cells (Fig. 9, C and D). This indicates that Npas4 may be able to protect beta cells from the detrimental effects of clinically relevant FK-506 regimens.
FIGURE 9.

FK-506-induced beta cell death is prevented by overexpression of Npas4. MIN6 cells or dispersed mouse islet cells were infected with control virus (Ad-eGFP) or Ad-Npas4 overnight at a multiplicity of infection of 20:1 or 10:1, respectively. The medium was replaced, and cells were given a 1-day recovery period. Following 24-h treatment with vehicle (DMSO) or clinically relevant doses of FK-506 (10 or 37 nm), expression of the apoptosis marker cleaved caspase-3 was assessed via Western blot analysis and quantified using densitometry, with GAPDH as a reference and normalizing to eGFP-transduced, DMSO-treated cells. A and B, Ad-eGFP infected MIN6 had significantly increased levels of cleaved caspase-3 after treatment with 10 and 37 nm FK-506 whereas Ad-Npas4-transduced MIN6 cells did not. C and D, after 24-h treatment with vehicle or 37 nm FK-506, dispersed islet cells infected with eGFP showed increased cleaved caspase-3 expression, whereas Npas4 transduction prevented it. Error bars represent mean ± S.E., and significance was determined using a one-way ANOVA with Bonferroni's multiple comparison test and Bartlett's test for equal variances (n = 5). ***, p ≤ 0.001.
Discussion
In our previous report, we provided evidence for the beneficial effects of calcium-dependent Npas4 expression in beta cells (12) but had not identified the relevant calcium signaling pathways involved in its induction, dynamics of expression, or stability. In this report, we demonstrate that glucose-stimulated Npas4 expression in beta cells is dependent on CaMK, Akt, and CaN signaling and that Npas4 protein is posttranslationally modified by MAPK signaling. Moreover, Npas4 message and protein levels decay rapidly after the removal of a stimulus, and protein stability is mainly regulated via ubiquitin-proteasomal degradation. Finally, evidence supporting a calcineurin-dependent cytoprotective role for Npas4 is provided, which demonstrates that inhibition of Npas4 expression is a potential mechanism through which the immunosuppressant FK-506 exerts cytotoxicity on islet cells and suggests that expressing Npas4 during islet transplantation might prove to be protective.
Npas4 mRNA and protein levels appear to be regulated tightly, induced rapidly upon depolarization, and degraded quickly after removal of such stimulatory conditions (Fig. 6). Posttranscriptional regulation of mRNA stability and translation is known to be regulated by miRs (37) that might be involved in the rapid degradation of Npas4 mRNA. As Bersten et al. (38) have shown, neuronal Npas4 mRNA stability was regulated negatively by miR-224 and miR-203 targeting the 3′ UTR. It remains to be determined whether miR-224/miR-203 expression contributes to the regulation of Npas4 mRNA stability in beta cells.
Additionally, cellular activity (i.e. depolarization) of MIN6 cells increased Npas4 t½ 2.2-fold, independent of de novo transcription (Fig. 6B). A similar stabilizing effect of elevated glucose was observed by Welsh et al. (39) on preproinsulin t½ (77 h in high glucose versus 29 h in low glucose). It was later discovered that high glucose and cAMP-dependent stabilization of preproinsulin mRNA are likely mediated via translocation of polypyrimidine tract binding protein from the nucleus to the cytoplasm, where it binds the 3′ UTR of preproinsulin RNA and stabilizes it as part of a multiprotein complex (40). It will be interesting to determine whether a similar mechanism governs Npas4 mRNA stability in beta cells in response to cellular activity.
We demonstrated that Npas4 protein degradation mainly depends on the ubiquitin-proteasome pathway (Fig. 8). This posttranslational control ensures that, in the absence of stimuli, Npas4 protein is degraded efficiently. Notably, during periods of beta cell activity, several genes of the ubiquitin-proteasome pathway are down-regulated (36). This pathway may be advantageous in the case of Npas4 because it would result in decreased degradation and, possibly, a greater cytoprotective effect. Previous studies have implicated the von Hippel-Lindau tumor suppressor, which mediates substrate specificity as part of an ubiquitin E3 ligase complex, in the degradation of another PAS (Per-Arnt-Sim) domain transcription factor, Hif1α (30, 41, 42). However, we saw no interaction between Npas4 and von Hippel-Lindau tumor suppressor.5 Furthermore, a gradual reduction in Npas4 protein after transfer to non-stimulatory conditions in the presence of MG132 (Fig. 8, C and D) suggested the contribution of other degradation mechanisms, such as lysosomal cathepsins or calpain-mediated decay (43, 44). Their involvement remains to be established. Nonetheless, these findings have implicated the relevance of ubiquitination in the regulation of Npas4 and warrants studies of the contributions of other posttranslational modifications on Npas4 stability, activity, and subcellular localization, such as SUMOylation and phosphorylation.
In agreement with what has been observed during hippocampal Npas4 induction (45), we confirmed that PKC was not involved in beta cell Npas4 expression. In the same study, a MEK inhibitor only modestly decreased hippocampal Npas4 induction, which is comparable with our observations (Fig. 3D). Notably, Ooe et al. (45) determined that Npas4 protein phosphorylation and transcriptional activity were MAPK-dependent. Here we also observed a mobility shift of the Npas4 band. It may be informative to follow up on this observation and understand whether phosphorylation or another posttranslational modification is driven by MAPK signaling and to establish the physiological role of such modification.
Npas4 expression had been previously linked to CaMKs (46), and CaMKIV is the main isoform regulating Npas4 in the brain (47). Notably, CaMKI and CaMKIV depend on phosphorylation events by CaMKKα/β, and we observed inhibition of Npas4 induction by KN-93 but not STO-609, a specific CaMKK inhibitor (Figs. 2B and 3A). This indicated that Npas4 is regulated by CaMKII but not CaMKK-dependent CaMKI/IV or AMPK signaling (31). The latter was supported at the RNA level by using the AMPK inhibitor compound C, which had no effect on Npas4 mRNA, and the CaMKII inhibitor AIP, which caused modest reductions in Npas4 mRNA induction. Paradoxically, we observed reductions in Npas4 protein with compound C but not AIP.
AMPK has diverse biological roles (8, 48, 49) and can be activated by KCl-induced calcium influx in beta cells (50). Although AMPK is not involved in regulating Npas4 transcription, it is possible that its activity controls Npas4 translation or protein turnover through an unknown mechanism. Compound C, however, has documented off-target effects (51), warranting cautious interpretation of these results and requiring further studies with more specific inhibitors.
As for the CaMKII inhibitor AIP, it is likely that low cell permeability prevented it from having a significant effect on Npas4 protein levels. It has been noted that the CaMK inhibitor KN-93 may inhibit L-type voltage-dependent calcium channel currents through blocking of its α1C and α1D subunits (CaV1.2 and CaV1.3) (10, 52). Nonetheless, using the ratiometric calcium probe Fura-2, we did not observe a reduction in calcium influx in MIN6 cells following application of KN-93 with KCl.5
Recently, Santos et al. (53) described a “metabolic memory” of human islet cells, accomplished through consecutive high-glucose pulses interspersed by low-glucose breaks to human islets. Following a 24-h consolidation period, pulse-treated islets had higher levels of phosphorylated (active) CaMKII and mounted a stronger insulin response to a glucose stimulus compared with control or KN-93 treated islets. The metabolic memory was CaMKII-dependent and involved up-regulation of key proteins for glucose sensing and insulin release and production. Here we used KN-93 to prevent Npas4 RNA and protein induction. Additionally, we gathered preliminary evidence that beta cells “remember” prior induction of Npas4 expression, requiring up to 5 h before the same stimulus can induce equal levels of Npas4 again (Fig. 7). Therefore, we propose that Npas4 could contribute to the CaMKII-induced metabolic memory.
In islet cells, PI3K/Akt signaling has been shown to promote protein synthesis, survival, and proliferation and is a common downstream signaling cascade for many growth factors, including insulin (54–57). Neuronal Npas4 mRNA expression had been linked previously to stimulation by growth factors (45, 58), although this may not translate to changes at the protein level (29). Specifically, Ooe et al. (45) have reported that Npas4 expression downstream of NGF stimulation was dependent on PI3K/Akt. The MIN6 and islet in vitro model used in our study likely features insulin as a predominant growth factor because it is readily secreted into the culture medium and can signal through the insulin receptor in a paracrine-autocrine fashion (33, 59, 60). However, gene expression analysis for Npas4 in MIN6 cells treated with insulin showed no induction compared with controls (Fig. 2E), likely because depolarization and calcium influx are crucial to Npas4 expression (12), and Akt signaling may not trigger these events alone. Because glucose stimulation or depolarization of beta cells results in insulin secretion and, likely, Akt activation, a role for Akt signaling on Npas4 expression in beta cells was studied via pharmacological inhibition of Akt under stimulatory conditions. Although the effects on Npas4 transcription were modest, Npas4 translation or protein stability in beta cells seems to be critically dependent on Akt signaling (Fig. 4, E–H). The Rapa-sensitive mTOR complex 1 is downstream of Akt and positively controls cap-dependent translation initiation (61–63), but we did not observe any effects on Npas4 protein levels with Rapa. Akt-dependent and mTOR-independent regulation of translation has been reported for Hif1α, another PAS domain protein (64). Consequently, the main effect of Akt signaling may occur through direct or indirect effects on Npas4 translation. However, an effect on protein stability cannot be ruled out without additional experiments. Finally, constitutively activated Akt signaling has been shown to reduce beta cell susceptibility to free fatty acid-induced apoptosis and protect from ER stress (65, 66). Because similar observations have been made for Npas4 (12), it is conceivable that some of the prosurvival effects of Akt signaling are mediated through improved translation of Npas4.
CaN signaling is known to be important for islet function and survival, as demonstrated using a murine model with absence of nuclear NFATc1, a transcription factor downstream of CaN. Mice lacking nuclear NFATc1 develop hyperglycemia, hypoinsulinemia, and significant decreases in key beta cell genes (5). In support of this prosurvival role, the CaN inhibitor FK-506, used for immunosuppressive maintenance therapy following organ transplantation, has well documented cytotoxic effects on beta cells, such as an increased incidence of new-onset diabetes or graft failure following islet transplantation (19–26, 67). Here we provide evidence that FK-506 treatment significantly inhibits both Npas4 mRNA and protein expression at a therapeutically relevant concentration (Figs. 2F and 4, I–L). Furthermore, we confirm the effect of FK-506 on the induction of cleaved caspase-3 in beta cells. In the absence of stimulation or with inhibition of calcineurin via FK-506, Npas4 is not expressed, and, therefore, cytoprotection against FK-506 is not expected. To bypass one of the normal induction pathways (i.e. through calcineurin), Npas4 was adenovirally overexpressed, which reduced cleaved caspase-3 levels (Fig. 9, A–D). A possible explanation is that, although FK-506 increases cell death independently of Npas4, its cytotoxic effects are exacerbated by the inability to induce Npas4.
Notably, CaN signaling has recently been linked to islet survival via the PI3K/Akt pathway (6). The cross-talk between the two pathways provides further support for our observation that Npas4 expression is driven by both CaN and Akt and potentially mediates some of the effects on islet survival.
In summary, we demonstrate the involvement of the calcium-dependent CaN, Akt, and CaMK signaling pathways in the regulation of Npas4 expression and protein stability in pancreatic beta cells. Npas4 expression was regulated tightly in accordance with its role as an immediate early gene. Inhibition of CaN using the beta cell toxic immunosuppressant FK-506 increased beta cell apoptotic signaling, an effect prevented by Npas4 overexpression. This further establishes a beneficial role for Npas4 in enhancing islet survival, which may hold therapeutic potential in enhancing graft survival during islet transplantation.
Author Contributions
T. S., P. V. S., C. N., and R. G. S. designed and performed the experiments. F. C. L. directed the research. T. S., P. V. S., and F. C. L. wrote the manuscript. All authors subsequently edited the manuscript. F. C. L. is the guarantor of this work, had full access to all data, and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Acknowledgments
We thank Dr. Megan K. Levings, Dr. Brad G. Hoffman, and Dr. Christopher A. Maxwell (University of British Columbia, Vancouver, British Columbia, Canada) for the reagents that made this work possible. MIN6 cells were provided by Dr. Jun-Ichi Miyazaki (Osaka University, Japan). Finally, we thank all members of the Lynn laboratory for thoughtful reading of the manuscript.
This work was supported by Canadian Institutes of Health Research Grant MOP 343144 (to F. C. L.).
T. Speckmann, unpublished observation.
- CREB
- cAMP response element-binding protein
- NFAT
- nuclear factor of activated T cells
- CaN
- calcineurin
- ER
- endoplasmic reticulum
- CaMK
- Ca2+/calmodulin-dependent protein kinase
- AIP
- autocamtide-2-related inhibitory peptide
- AMPK
- 5′ AMP-activated protein kinase
- Akti-1/2
- Akt inhibitor VIII trifluoroacetate salt hydrate
- Rapa
- rapamycin
- KRBH
- Krebs-Ringer solution buffered with HEPES
- ActD
- actinomycin D
- eGFP
- enhanced GFP
- DMSO
- dimethyl sulfoxide
- ANOVA
- analysis of variance
- mTOR
- mechanistic target of rapamycin
- CaMKK
- Ca2+/calmodulin-dependent protein kinase kinase
- miR
- microRNA.
References
- 1.Rorsman P., Braun M., and Zhang Q. (2012) Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 51, 300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilon P., Chae H. Y., Rutter G. A., and Ravier M. A. (2014) Calcium signaling in pancreatic β-cells in health and in type 2 diabetes. Cell Calcium 56, 340–361 [DOI] [PubMed] [Google Scholar]
- 3.Srinivasan S., Bernal-Mizrachi E., Ohsugi M., and Permutt M. A. (2002) Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. Am. J. Physiol. Endocrinol. Metab. 283, E784–793 [DOI] [PubMed] [Google Scholar]
- 4.Kim S. J., Winter K., Nian C., Tsuneoka M., Koda Y., and McIntosh C. H. (2005) Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic β-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J. Biol. Chem. 280, 22297–22307 [DOI] [PubMed] [Google Scholar]
- 5.Heit J. J., Apelqvist A. A., Gu X., Winslow M. M., Neilson J. R., Crabtree G. R., and Kim S. K. (2006) Calcineurin/NFAT signalling regulates pancreatic β-cell growth and function. Nature 443, 345–349 [DOI] [PubMed] [Google Scholar]
- 6.Soleimanpour S. A., Crutchlow M. F., Ferrari A. M., Raum J. C., Groff D. N., Rankin M. M., Liu C., De León D. D., Naji A., Kushner J. A., and Stoffers D. A. (2010) Calcineurin signaling regulates human islet β-cell survival. J. Biol. Chem. 285, 40050–40059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suefuji M., Furukawa N., Matsumoto K., Oiso H., Shimoda S., Yoshinaga T., Matsuyama R., Miyagawa K., Kondo T., Kawashima J., Tsuruzoe K., and Araki E. (2012) The impact of Ca2+/calmodulin-dependent protein kinase II on insulin gene expression in MIN6 cells. Biochem. Biophys. Res. Commun. 421, 801–807 [DOI] [PubMed] [Google Scholar]
- 8.Fu A., Eberhard C. E., and Screaton R. A. (2013) Role of AMPK in pancreatic β cell function. Mol. Cell Endocrinol. 366, 127–134 [DOI] [PubMed] [Google Scholar]
- 9.Dadi P. K., Vierra N. C., Ustione A., Piston D. W., Colbran R. J., and Jacobson D. A. (2014) Inhibition of pancreatic β-cell Ca2+/calmodulin-dependent protein kinase II reduces glucose-stimulated calcium influx and insulin secretion, impairing glucose tolerance. J. Biol. Chem. 289, 12435–12445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhatt H. S., Conner B. P., Prasanna G., Yorio T., and Easom R. A. (2000) Dependence of insulin secretion from permeabilized pancreatic β-cells on the activation of Ca2+/calmodulin-dependent protein kinase II: a re-evaluation of inhibitor studies. Biochem. Pharmacol. 60, 1655–1663 [DOI] [PubMed] [Google Scholar]
- 11.Dixit S. S., Wang T., Manzano E. J., Yoo S., Lee J., Chiang D. Y., Ryan N., Respress J. L., Yechoor V. K., and Wehrens X. H. (2013) Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PLoS ONE 8, e58655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabatini P. V., Krentz N. A., Zarrouki B., Westwell-Roper C. Y., Nian C., Uy R. A., Shapiro A. M., Poitout V., and Lynn F. C. (2013) Npas4 is a novel activity-regulated cytoprotective factor in pancreatic β-cells. Diabetes 62, 2808–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Silva Xavier G., Leclerc I., Varadi A., Tsuboi T., Moule S. K., and Rutter G. A. (2003) Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem. J. 371, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B., Barbosa-Sampaio H., Jones P. M., Persaud S. J., and Muller D. S. (2012) The CaMK4/CREB/IRS-2 cascade stimulates proliferation and inhibits apoptosis of β-cells. PLoS ONE 7, e45711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin S., Le Lay J., Everett L. J., Gupta R., Rafiq K., and Kaestner K. H. (2014) CREB mediates the insulinotropic and anti-apoptotic effects of GLP-1 signaling in adult mouse β-cells. Mol. Metab. 3, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynn F. C., Pamir N., Ng E. H., McIntosh C. H., Kieffer T. J., and Pederson R. A. (2001) Defective glucose-dependent insulinotropic polypeptide receptor expression in diabetic fatty Zucker rats. Diabetes 50, 1004–1011 [DOI] [PubMed] [Google Scholar]
- 17.Talchai C., Xuan S., Lin H. V., Sussel L., and Accili D. (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitamura Y. I., Kitamura T., Kruse J. P., Raum J. C., Stein R., Gu W., and Accili D. (2005) FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metab. 2, 153–163 [DOI] [PubMed] [Google Scholar]
- 19.Johnson J. D., Ao Z., Ao P., Li H., Dai L. J., He Z., Tee M., Potter K. J., Klimek A. M., Meloche R. M., Thompson D. M., Verchere C. B., and Warnock G. L. (2009) Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 18, 833–845 [DOI] [PubMed] [Google Scholar]
- 20.Krentz A. J., Dousset B., Mayer D., McMaster P., Buckels J., Cramb R., Smith J. M., and Nattrass M. (1993) Metabolic effects of cyclosporin A and FK 506 in liver transplant recipients. Diabetes 42, 1753–1759 [DOI] [PubMed] [Google Scholar]
- 21.Pirsch J. D., Miller J., Deierhoi M. H., Vincenti F., and Filo R. S. (1997) A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression after cadaveric renal transplantation: FK506 Kidney Transplant Study Group. Transplantation 63, 977–983 [DOI] [PubMed] [Google Scholar]
- 22.Mayer A. D., Dmitrewski J., Squifflet J. P., Besse T., Grabensee B., Klein B., Eigler F. W., Heemann U., Pichlmayr R., Behrend M., Vanrenterghem Y., Donck J., van Hooff J., Christiaans M., Morales J. M., Andres A., Johnson R. W., Short C., Buchholz B., Rehmert N., Land W., Schleibner S., Forsythe J. L., Talbot D., and Pohanka E. (1997) Multicenter randomized trial comparing tacrolimus (FK506) and cyclosporine in the prevention of renal allograft rejection: a report of the European Tacrolimus Multicenter Renal Study Group. Transplantation 64, 436–443 [DOI] [PubMed] [Google Scholar]
- 23.Hirano Y., Fujihira S., Ohara K., Katsuki S., and Noguchi H. (1992) Morphological and functional changes of islets of Langerhans in FK506-treated rats. Transplantation 53, 889–894 [DOI] [PubMed] [Google Scholar]
- 24.Vincenti F., Friman S., Scheuermann E., Rostaing L., Jenssen T., Campistol J. M., Uchida K., Pescovitz M. D., Marchetti P., Tuncer M., Citterio F., Wiecek A., Chadban S., El-Shahawy M., Budde K., Goto N., and DIRECT (Diabetes Incidence after Renal Transplantation: Neoral C Monitoring Versus Tacrolimus) Investigators (2007) Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am. J. Transplant. 7, 1506–1514 [DOI] [PubMed] [Google Scholar]
- 25.Cotovio P., Neves M., Rodrigues L., Alves R., Bastos M., Baptista C., Macário F., and Mota A. (2013) New-onset diabetes after transplantation: assessment of risk factors and clinical outcomes. Transplant. Proc. 45, 1079–1083 [DOI] [PubMed] [Google Scholar]
- 26.Demirci M. S., Toz H., Yilmaz F., Ertilav M., Asci G., Ozkahya M., Zeytinoglu A., Nart D., and Ok E. (2010) Risk factors and consequences of post-transplant diabetes mellitus. Clin. Transplant. 24, E170–177 [DOI] [PubMed] [Google Scholar]
- 27.Moser M., Knoth R., Bode C., and Patterson C. (2004) LE-PAS, a novel Arnt-dependent HLH-PAS protein, is expressed in limbic tissues and transactivates the CNS midline enhancer element. Brain Res. Mol. Brain Res. 128, 141–149 [DOI] [PubMed] [Google Scholar]
- 28.Ooe N., Saito K., Mikami N., Nakatuka I., and Kaneko H. (2004) Identification of a novel basic helix-loop-helix-PAS factor, NXF, reveals a Sim2 competitive, positive regulatory role in dendritic-cytoskeleton modulator drebrin gene expression. Mol. Cell Biol. 24, 608–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Y., Bloodgood B. L., Hauser J. L., Lapan A. D., Koon A. C., Kim T. K., Hu L. S., Malik A. N., and Greenberg M. E. (2008) Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabatini P. V., and Lynn F. C. (2015) All-encomPASsing regulation of β-cells: PAS domain proteins in beta-cell dysfunction and diabetes. Trends Endocrinol. Metab. 26, 49–57 [DOI] [PubMed] [Google Scholar]
- 31.Means A. R. (2008) The year in basic science: calmodulin kinase cascades. Mol. Endocrinol. 22, 2759–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishida A., Kameshita I., Okuno S., Kitani T., and Fujisawa H. (1995) A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun. 212, 806–812 [DOI] [PubMed] [Google Scholar]
- 33.Leibiger I. B., Leibiger B., and Berggren P. O. (2008) Insulin signaling in the pancreatic β-cell. Annu. Rev. Nutr. 28, 233–251 [DOI] [PubMed] [Google Scholar]
- 34.da Silva Xavier G., Varadi A., Ainscow E. K., and Rutter G. A. (2000) Regulation of gene expression by glucose in pancreatic β-cells (MIN6) via insulin secretion and activation of phosphatidylinositol 3′-kinase. J. Biol. Chem. 275, 36269–36277 [DOI] [PubMed] [Google Scholar]
- 35.Hartley T., Brumell J., and Volchuk A. (2009) Emerging roles for the ubiquitin-proteasome system and autophagy in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 296, E1–10 [DOI] [PubMed] [Google Scholar]
- 36.López-Avalos M. D., Duvivier-Kali V. F., Xu G., Bonner-Weir S., Sharma A., and Weir G. C. (2006) Evidence for a role of the ubiquitin-proteasome pathway in pancreatic islets. Diabetes 55, 1223–1231 [DOI] [PubMed] [Google Scholar]
- 37.Lynn F. C. (2009) Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol. Metab. 20, 452–459 [DOI] [PubMed] [Google Scholar]
- 38.Bersten D. C., Wright J. A., McCarthy P. J., and Whitelaw M. L. (2014) Regulation of the neuronal transcription factor NPAS4 by REST and microRNAs. Biochim. Biophys. Acta 1839, 13–24 [DOI] [PubMed] [Google Scholar]
- 39.Welsh M., Nielsen D. A., MacKrell A. J., and Steiner D. F. (1985) Control of insulin gene expression in pancreatic β-cells and in an insulin-producing cell line, RIN-5F cells: II: regulation of insulin mRNA stability. J. Biol. Chem. 260, 13590–13594 [PubMed] [Google Scholar]
- 40.Fred R. G., and Welsh N. (2009) The importance of RNA binding proteins in preproinsulin mRNA stability. Mol. Cell Endocrinol. 297, 28–33 [DOI] [PubMed] [Google Scholar]
- 41.Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., and Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 42.Cantley J., Selman C., Shukla D., Abramov A. Y., Forstreuter F., Esteban M. A., Claret M., Lingard S. J., Clements M., Harten S. K., Asare-Anane H., Batterham R. L., Herrera P. L., Persaud S. J., Duchen M. R., Maxwell P. H., and Withers D. J. (2009) Deletion of the von Hippel-Lindau gene in pancreatic β cells impairs glucose homeostasis in mice. J. Clin. Invest. 119, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sreenan S. K., Zhou Y. P., Otani K., Hansen P. A., Currie K. P., Pan C. Y., Lee J. P., Ostrega D. M., Pugh W., Horikawa Y., Cox N. J., Hanis C. L., Burant C. F., Fox A. P., Bell G. I., and Polonsky K. S. (2001) Calpains play a role in insulin secretion and action. Diabetes 50, 2013–2020 [DOI] [PubMed] [Google Scholar]
- 44.Jung M., Lee J., Seo H. Y., Lim J. S., and Kim E. K. (2015) Cathepsin inhibition-induced lysosomal dysfunction enhances pancreatic β-cell apoptosis in high glucose. PLoS ONE 10, e0116972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ooe N., Kobayashi K., Motonaga K., Saito K., and Kaneko H. (2009) Dynamic regulation of bHLH-PAS-type transcription factor NXF gene expression and neurotrophin dependent induction of the transcriptional control activity. Biochem. Biophys. Res. Commun. 378, 761–765 [DOI] [PubMed] [Google Scholar]
- 46.Qiu J., Tan Y. W., Hagenston A. M., Martel M. A., Kneisel N., Skehel P. A., Wyllie D. J., Bading H., and Hardingham G. E. (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang S. J., Zou M., Lu L., Lau D., Ditzel D. A., Delucinge-Vivier C., Aso Y., Descombes P., and Bading H. (2009) Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 5, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mihaylova M. M., and Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salt I. P., Johnson G., Ashcroft S. J., and Hardie D. G. (1998) AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic β cells, and may regulate insulin release. Biochem. J. 335, 533–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leclerc I., and Rutter G. A. (2004) AMP-activated protein kinase: a new β-cell glucose sensor? Regulation by amino acids and calcium ions. Diabetes 53, S67–74 [DOI] [PubMed] [Google Scholar]
- 51.Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao L., Blair L. A., and Marshall J. (2006) CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem. Biophys. Res. Commun. 345, 1606–1610 [DOI] [PubMed] [Google Scholar]
- 53.Santos G. J., Ferreira S. M., Ortis F., Rezende L. F., Li C., Naji A., Carneiro E. M., Kaestner K. H., and Boschero A. C. (2014) Metabolic memory of β-cells controls insulin secretion and is mediated by CaMKII. Mol. Metab. 3, 484–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vasavada R. C., Gonzalez-Pertusa J. A., Fujinaka Y., Fiaschi-Taesch N., Cozar-Castellano I., and Garcia-Ocaña A. (2006) Growth factors and β cell replication. Int. J. Biochem. Cell Biol. 38, 931–950 [DOI] [PubMed] [Google Scholar]
- 55.Elghazi L., Rachdi L., Weiss A. J., Cras-Méneur C., and Bernal-Mizrachi E. (2007) Regulation of β-cell mass and function by the Akt/protein kinase B signalling pathway. Diabetes Obes. Metab. 9, 147–157 [DOI] [PubMed] [Google Scholar]
- 56.Assmann A., Hinault C., and Kulkarni R. N. (2009) Growth factor control of pancreatic islet regeneration and function. Pediatr. Diabetes 10, 14–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang Y., and Chang Y. (2014) Regulation of pancreatic islet β-cell mass by growth factor and hormone signaling. Prog. Mol. Biol. Transl. Sci. 121, 321–349 [DOI] [PubMed] [Google Scholar]
- 58.Srimontri P., Hirota H., Kanno H., Okada T., Hirabayashi Y., and Kato K. (2014) Infusion of growth hormone into the hippocampus induces molecular and behavioral responses in mice. Exp. Brain Res. 232, 2957–2966 [DOI] [PubMed] [Google Scholar]
- 59.Luciani D. S., and Johnson J. D. (2005) Acute effects of insulin on β-cells from transplantable human islets. Mol. Cell Endocrinol. 241, 88–98 [DOI] [PubMed] [Google Scholar]
- 60.Wang M., Li J., Lim G. E., and Johnson J. D. (2013) Is dynamic autocrine insulin signaling possible? A mathematical model predicts picomolar concentrations of extracellular monomeric insulin within human pancreatic islets. PLoS ONE 8, e64860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brunn G. J., Hudson C. C., Sekulić A., Williams J. M., Hosoi H., Houghton P. J., Lawrence J. C. Jr., and Abraham R. T. (1997) Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277, 99–101 [DOI] [PubMed] [Google Scholar]
- 62.Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., and Sabatini D. M. (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U.S.A. 95, 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hatanaka M., Maier B., Sims E. K., Templin A. T., Kulkarni R. N., Evans-Molina C., and Mirmira R. G. (2014) Palmitate induces mRNA translation and increases ER protein load in islet β-cells via activation of the mammalian target of rapamycin pathway. Diabetes 63, 3404–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pore N., Jiang Z., Shu H. K., Bernhard E., Kao G. D., and Maity A. (2006) Akt1 activation can augment hypoxia-inducible factor-1α expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol. Cancer Res. 4, 471–479 [DOI] [PubMed] [Google Scholar]
- 65.Srinivasan S., Ohsugi M., Liu Z., Fatrai S., Bernal-Mizrachi E., and Permutt M. A. (2005) Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3β in mouse insulinoma cells. Diabetes 54, 968–975 [DOI] [PubMed] [Google Scholar]
- 66.Wrede C. E., Dickson L. M., Lingohr M. K., Briaud I., and Rhodes C. J. (2002) Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic β-cells (INS-1). J. Biol. Chem. 277, 49676–49684 [DOI] [PubMed] [Google Scholar]
- 67.Nishimura R., Nishioka S., Fujisawa I., Shiku H., Shimada M., Sekiguchi S., Fujimori K., Ushiyama A., Matsue T., Ohuchi N., Satomi S., and Goto M. (2013) Tacrolimus inhibits the revascularization of isolated pancreatic islets. PLoS ONE 8, e56799. [DOI] [PMC free article] [PubMed] [Google Scholar]