

Abstract
SNARE proteins catalyze many forms of biological membrane fusion, including Ca2+-triggered exocytosis. Although fusion mediated by SNAREs generally involves proteins anchored to each fusing membrane by a transmembrane domain (TMD), the role of TMDs remains unclear, and previous studies diverge on whether SNAREs can drive fusion without a TMD. This issue is important because it relates to the question of the structure and composition of the initial fusion pore, as well as the question of whether SNAREs mediate fusion solely by creating close proximity between two membranes versus a more active role in transmitting force to the membrane to deform and reorganize lipid bilayer structure. To test the role of membrane attachment, we generated four variants of the synaptic v-SNARE synaptobrevin-2 (syb2) anchored to the membrane by lipid instead of protein. These constructs were tested for functional efficacy in three different systems as follows: Ca2+-triggered dense core vesicle exocytosis, spontaneous synaptic vesicle exocytosis, and Ca2+-synaptotagmin-enhanced SNARE-mediated liposome fusion. Lipid-anchoring motifs harboring one or two lipid acylation sites completely failed to support fusion in any of these assays. Only the lipid-anchoring motif from cysteine string protein-α, which harbors many lipid acylation sites, provided support for fusion but at levels well below that achieved with wild type syb2. Thus, lipid-anchored syb2 provides little or no support for exocytosis, and anchoring syb2 to a membrane by a TMD greatly improves its function. The low activity seen with syb2-cysteine string protein-α may reflect a slower alternative mode of SNARE-mediated membrane fusion.
Keywords: exocytosis, lipid vesicle, membrane fusion, membrane trafficking, synapse
Introduction
Nearly all biological fusion mediated by SNARE proteins employs at least one t-SNARE and one v-SNARE anchored to the membrane by a transmembrane domain (TMD)3 (1–3). Ca2+-triggered exocytosis of neurotransmitter and hormone from many cells requires the vesicle-SNARE synaptobrevin 2 (syb2) and plasma membrane-SNARE syntaxin, both of which have a TMD (4, 5). Mutations in the TMD alter flux through exocytotic fusion pores in a manner consistent with structural models of fusion pores formed by the TMDs of syntaxin (6, 7) and syb2 (8). Yeast t- and v-SNAREs anchored to membranes by geranylgeranyl moieties instead of a TMD support docking but not fusion (9). SNAREs anchored to membranes by lipid instead of a TMD support lipid mixing of liposomes only when the lipid moiety is long enough to span a lipid bilayer or multiple lipid moieties are present (10) or an accessory protein is present (11).
Although early work supported a role of SNARE TMDs in membrane fusion, this issue has become controversial. Recent work on fusion of yeast vacuoles suggests that some SNAREs with lipid anchors can support lipid mixing, content mixing, or both, but other SNAREs cannot function when their TMD is replaced by a lipid anchor (12). Lipid-anchored forms of syntaxin and syb2 were reported to rescue neurotransmitter release at synapses as effectively as wild type (WT) SNAREs (13). This uncertainty about the role of membrane attachment raises important questions about the mechanism of membrane fusion. Many models of membrane fusion posit an active role for the TMD, beyond simply tethering the SNARE motif to the membrane. The initial fusion pore could not be proteinaceous if the TMD can be dispensed with (14, 15), and the transmission of force from the SNARE complex to the membrane is difficult to conceptualize without a TMD (16–19).
In this study, we sought to test the role of membrane attachment in the function of syb2 in diverse milieus. Four different lipid-anchored variants of syb2 were generated and assayed in Ca2+-triggered release of catecholamine from chromaffin cells, spontaneous release of excitatory neurotransmitter from synapses, and Ca2+-triggered fusion of reconstituted proteoliposomes. These lipid-anchored SNAREs supported little or no fusion above baseline levels. Only one construct harboring a cysteine string protein-α (CSPα) motif that can accept many acyl moieties supported a level of fusion significantly above background. None of the four lipid-anchored SNARE constructs supported fusion nearly as well as WT syb2. This work indicates that the form of membrane attachment of syb2 plays an important role in membrane fusion in synapses, endocrine cells, and proteoliposomes. A TMD may provide better support for fusion by forming a nascent fusion pore more readily or by more efficient force transmission to the lipid bilayer.
Experimental Procedures
Synaptobrevin 2 and Cellubrevin Double Knock-out Chromaffin Cells and Hippocampal Neurons
Syb2/cellubrevin double knock-out (DKO) mice were obtained by crossing syb2 heterozygous knock-out mice with cellubrevin homozygous knock-out mice (20). Litters were one-quarter DKO (expressing neither syb2 nor cellubrevin), whereas the remaining three-quarters, taken as littermate controls, had one or two WT syb2 alleles and no cellubrevin. Chromaffin cells were cultured from E17.5–18.5 DKO embryos; individual embryos were genotyped and cultured in separate dishes.
Hippocampi from DKO E17.5–18.5 embryos were dissected in DMEM with 5 mm HEPES, dissociated in 0.05% trypsin for 30 min at 37 °C, and homogenized by rapid pipetting with 1-ml pipette tips. Dissociated neurons were then plated on 12-mm coverslips coated with 0.2 mg/ml poly-l-lysine. Hippocampal neurons were cultured in medium containing Neurobasal-A, Glutamax, penicillin/streptomycin, and B-27.
Molecular Biology, Viral Infection, and Protein Purification
Mouse syb2 constructs were amplified by PCR adding XhoI 5′ and EcoRI 3′ sites. The codons encoding short lipid-anchoring motifs were added to the 3′ end primer right after residue 94. The construct containing residues 1–92 of syb2 and the C-terminal domain of CSPα (Syb2ΔTMR#1 (13)) was provided by T. Südhof and used as the template for PCR. The PCR fragments of syb2 constructs were digested with XhoI and EcoRI and ligated into the lentiviral vector pLox-CMV-EGFP to make EGFP-syb2 fusion constructs in which the sequence encoding EGFP was fused to the N terminus of syb2 by the linker sequence SGLRSRG. WT syb2, ΔTMD-CSPα, and ΔTMD-CIIL coding sequences without an N-terminal EGFP tag were subcloned into a bicistronic lentiviral vector pLox-CMV-IRES2-EGFP (modified from Ref. 21). To create syb2-RST-mVenus, a megaprimer containing the sequence of RST-mVenusand junction regions targeting to the vector pLox-CMV-syb2 was first amplified and then used for amplification and generation of pLox-CMV-syb2-RST-mVenus. All constructs were confirmed by sequencing.
Lentiviral particles were generated in HEK 293T cells as described previously (21). Chromaffin cells were infected by viral particles on the day of dissection, and hippocampal neurons were infected on the 7th day in vitro.
Bacterial expression vectors to generate full-length syb2 and its cytoplasmic domain were provided by J. E. Rothman. The cDNAs to generate full-length t-SNARE heterodimers were subcloned into pRSFDuet as described previously (22). The cDNAs for ΔTMD-LC, ΔTMD-CIIL, ΔTMD-CAC, and ΔTMD-CSPα described above were subcloned into pTrcHis for bacterial expression and Ni2+-Sepharose affinity purification. All proteins were expressed in Escherichia coli as His-tagged fusion proteins and purified as described previously (23).
Amperometry Recording and Analysis
To detect catecholamine release from chromaffin cells, a carbon fiber electrode (ALA Scientific Instruments, Westbury NY) was positioned at the surface of a cell under visual control and polarized to 650 mV. Amperometry current was recorded with a VA-10 amplifier (ALA Scientific Instruments). Chromaffin cells 1–3 days in vitro were bathed in a solution composed of (in mm) 150 NaCl, 4.2 KCl, 1 NaH2PO4, 0.7 MgCl2, 2 CaCl2, 10 HEPES, pH 7.4, at 22 °C. Transfected cells were visualized by EGFP fluorescence, and secretion was triggered by ejecting depolarizing solution (in mm: 105 KCl, 5 NaCl, 1 NaH2PO4, 0.7 MgCl2, 2 CaCl2, 10 HEPES, pH 7.4) from an ∼2-μm tipped glass pipette positioned 10–15 μm from the cell. Current was recorded in 23-s episodes, beginning with a 3-s baseline, a 6-s period of depolarization, and 14 s after the end of depolarization. Secreting cells were stimulated up to six times. Spike frequency was determined for each secreting cell as the mean number of events with amplitudes ≥4 pA in the first episode, divided by 20 s. To construct cumulative spike plots, the events following depolarization in the first episode from all cells expressing a given construct were pooled, binned in 1-s intervals, and divided by cell number. The initial segment of each cumulative plot between 10 and 70% of the plateau was fitted by linear regression to obtain the 10–70% rising slope. One-way analysis of variance was applied to frequency measurements (Fig. 2B) and slopes (Fig. 2D), and with p < 0.05, the Tukey post-test was used to identify mutants that differed from WT syb2 or DKO. Bar graphs display mean ± S.E.
FIGURE 2.

Amperometry recording from chromaffin cells. A, amperometry traces from chromaffin cells: DKO cells, littermate controls, and DKO cells expressing untagged WT syb2, EGFP-Syb2, Syb2-RST-mVenus, lipid-anchored mutants, and ΔTMD-94. Untransfected DKO cells and DKO cells expressing ΔTMD-94 exhibited low secretion in response to depolarization by a solution containing 105 mm KCl (indicated by bars), but littermate and DKO cells expressing untagged WT syb2 or N-terminal tagged EGFP-Syb2 produced robust trains of spikes. Expression of C-terminal tagged Syb2-RST-mVenus construct in DKO cells produced much less support for exocytosis compared with N-tagged and untagged syb2. DKO cells expressing ΔTMD-CSPα, ΔTMD-CAC, ΔTMD-LC, and ΔTMD-CIIL all secreted poorly. B, spike frequencies of cells expressing the indicated protein during the first depolarization. Syb2-RST-mVenus, the lipid-anchored constructs, and ΔTMD constructs produced frequencies statistically indistinguishable from untransfected DKO cells. n = 26–62 cells from 2 to 7 DKO embryos. C, cumulative spike counts versus time in the first response to depolarization (indicated by the bar) from chromaffin cells expressing the indicated proteins. D, slopes of the rises in cumulative spike count plots from 10 to 70% of the plateau. The slopes of littermate control cells and DKO cells expressing N-tagged and untagged WT syb2 were similar and significantly higher than those from Syb2-RST-mVenus, lipid-anchored mutants, ΔTMD, and untransfected DKO cells. All lipid-anchored syb2 constructs were statistically indistinguishable from DKO except ΔTMD-CSPα. The comparison with EGFP-syb2 or DKO for one-way analysis of variance with post test is indicated by bars. **, p < 0.01; ***, p < 0.001; n.s., not significant. Paired comparisons of slopes with a t test were also performed in GraphPad Prism, and the slopes of Syb2-RST-mVenus and all mutants (except ΔTMD-94) were significantly higher than DKO.
Patch Clamp Recording and Analysis
Cultured hippocampal neurons were bathed in a solution composed of (in mm) 140 NaCl, 5 KCl, 5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, with pH adjusted to 7.4 by NaOH. The bathing solution also contained 0.5 μm tetrodotoxin, 10 μm SR95531, and 50 μm amino phosphonovalerate to isolate mEPSCs mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptors. Patch pipettes were filled with internal solution containing (in mm) 120 cesium gluconate, 8 CsCl, 2 NaCl, 10 EGTA, 5 HEPES, 5 phosphocreatine, 2 Mg-ATP, and 0.3 Na-GTP, with pH adjusted to 7.2 by CsOH. Patch pipettes containing this solution had resistances of 3–8 megohms. All recordings were performed at room temperature. The voltage was held at −70 mV with an EPC-7 patch clamp amplifier (HEKA). Current was filtered at 3 kHz, digitized at 50 kHz, and read into a computer with PClamp software. Frequency was the mean of the average number of events in each cell divided by time. Differences in mEPSC frequencies were evaluated with the Mann-Whitney U test. Bar graphs display mean ± S.E.
Immunocytochemistry and Imaging
DKO chromaffin cells were transfected with neuropeptide Y-DsRed (NPY-DsRed) and WT or mutant EGFP-syb2 constructs. Three days after transfection, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized by 0.3% Triton X-100 for 10 min, blocked in 10% goat serum and 0.1% Triton X-100 for 1 h, and immunostained with mouse anti-syb2 antibody (104211, Synaptic Systems) and rabbit anti-RFP antibody (ab62341, Abcam) for 1 h. After washing three times with phosphate-buffered saline (PBS), cells were treated with Cy2-goat anti-mouse and Alexa Fluor 568-goat anti-rabbit secondary antibodies for 1 h, followed by three washes with PBS. All the above steps were carried out at room temperature. Images of immunostained chromaffin cells were acquired at room temperature on a Nikon Eclipse Ti confocal microscope with a ×100, 1.49 numerical aperture oil-immersion lens and Andor DV-897 camera. The laser intensity was set to 8% for both 488- and 561-nm illumination, and the exposure time was between 100 and 200 ms. Background subtraction was performed with ImageJ software. Pearson's coefficient and total fluorescence intensity (area of cell·(mean cell intensity − background)) were quantified using Nikon Eclipse Ti analysis software. One-way analysis of variance with Tukey post test was used for Pearson's coefficients and total fluorescence intensity to evaluate the significance of differences between syb2 mutants and WT syb2 or syb2 mutants and ΔTMD-94.
Neurons on coverslips were fixed using 4% paraformaldehyde in PBS for 20 min. After washing with PBS, the neurons were permeabilized, blocked in blocking buffer containing 0.3% Triton X-100, 3% goat serum, and 2% BSA at room temperature for 1 h, and incubated at 4 °C overnight with polyclonal guinea pig anti-synaptophysin 1 (1:1000) (101004, Synaptic Systems) and monoclonal mouse anti-syb2 (1:500) (104211, Synaptic Systems). After rinsing with PBS, the coverslips were incubated at room temperature with Cy3-conjugated rabbit anti-guinea pig IgG (1:300) and Cy2-conjugated goat anti-mouse IgG (1:500) for 45 min. All antibodies were diluted with blocking buffer. Fluorescent signals were detected using an Olympus IX83 microscope with 60 × 1.49 numerical apperture oil-immersion objective and ORCA flash 4.0 camera. Images were acquired with MetaMorph software and analyzed with Fiji ImageJ software.
Reconstituted Proteoliposomes
Proteoliposomes, generated as described previously (23), were composed of 25% phosphatidylserine, 45% phosphatidylcholine, 30% phosphatidylethanolamine and harbored the t-SNAREs - syntaxin-1A and SNAP-25B. Briefly, lipids were dried under a stream of nitrogen gas while rotating. The lipid film was further lyophilized for at least 1 h to remove any residual organic solvent. The lipid film was solubilized with proteins in 25 mm HEPES-KOH, pH 7.4, 100 mm KCl, 10% glycerol, 1% octyl glucoside, 1 mm DTT. Vesicles were formed by rapid dilution of the octyl glucoside with 2 volumes of reconstitution buffer (25 mm HEPES-KOH, 100 mm KCl, 10% glycerol, 1 mm DTT) and overnight dialysis against 4 liters of reconstitution buffer. Proteoliposomes were collected by density gradient centrifugation and analyzed by SDS-PAGE and Coomassie Blue staining. Vesicles harboring full-length syb2 were prepared as described above; however, the phosphatidylethanolamine content was reduced from 30 to 27% to accommodate the addition of 1.5% rhodamine-phosphatidylethanolamine and 1.5% NBD-phosphatidylethanolamine to the lipid mixture.
Lipid-anchored Proteoliposomes
Syb2 proteins lacking their TMD were attached to liposomes via cysteine residue(s). Briefly, the syb2 proteins were dialyzed overnight at 4 °C against cross-linking buffer (25 mm HEPES-KOH, pH 7.4, 200 mm NaCl, 5 mm tris(2-carboxyethyl)phosphine). The protein was quantified by SDS-PAGE and stained with Coomassie Blue; concentrations were determined using a BSA standard curve. Lipids stored in chloroform were mixed (45% palmitoyloleoylphosphatidylcholine, 25% POPE, 25% dioleoylphosphatidylserine, 5% POPE-maleimide) and dried under a stream of nitrogen and lyophilized as mentioned above. The lipid film was hydrated to 3 mm (total lipid) with cross-linking buffer and formed into vesicles by extrusion through a 100-nm filter (Avanti Polar Lipids). Extruded vesicles (500 μl) were incubated with 50 μm protein overnight at 4 °C; the reaction was quenched by addition of 5 mm DTT. Proteoliposomes were floated through an Accudenz (Accurate Chemical) density gradient to remove unincorporated proteins as described previously (23, 24). POPE-maleimide was purchased from NOF America Corp. All other lipids were purchased from Avanti Polar Lipids.
In Vitro Fusion Assay
Lipid mixing experiments were carried out as described previously (24). Briefly, 100-μl reactions containing 4.5 μl of t-SNARE liposomes, 0.5 μl of v-SNARE liposomes, 1 μm synaptotagmin 1 C2AB, and 0.2 mm EGTA were incubated at 37 °C for 20 min, followed by the addition of 1 mm Ca2+ (arrows in Fig. 5). Fluorescence signals were measured using a BioTek synergy plate reader equipped with 460/40 excitation and 530/25 emission filters. Addition of 25 μl of 2.5% n-dodecyl maltoside was used to obtain the maximal dequenching signal to normalize the data. ΔTMD-94 (5 μm) was included in a subset of reactions to inhibit SNARE-mediated fusion.
FIGURE 5.

Ca2+-dependent proteoliposome fusion in vitro. A, vesicles harboring t-SNAREs, WT syb2, POPE-linked syb2 ΔTMD constructs, or protein-free liposomes were separated by SDS-PAGE and stained with Coomassie Blue. Note that ΔTMD-CSPα has a higher molecular mass due to the CSPα motif and multiple acyl lipids. B, representative fusion traces from reactions involving v-SNARE vesicles harboring either WT or lipid-anchored syb2 and 1 μm of the cytosolic C2AB domain of synaptotagmin 1. Reactions were performed with additions of EGTA or Ca2+ at the arrow. Including ΔTMD-94 (5 μm) demonstrated SNARE dependence of fusion. Representative data are shown from 2 to 3 independent experiments.
Results
We tested four lipid-anchored syb2 constructs, two cytosolic syb2 constructs, full-length syb2, and constructs with fluorescent protein tags at either the C or N terminus (Fig. 1; “Experimental Procedures”). Three lipid-anchored constructs were truncated at the last residue of the linker before the TMD (residue 94) and had various cysteine-harboring sequences appended to the C terminus. ΔTMD-CIIL contains a geranylgeranylation site (9); ΔTMD-LC was used previously in tests of liposome fusion (10), and ΔTMD-CAC contains a well known prenylation motif. We also generated a CSPα construct designated ΔTMD-CSPα by subcloning the intact sequence of Syb2ΔTMR#1 (used by Zhou et al. (13)) into our expression vector. The connection between syb2 and the C terminus of CSPα was not modified. In this construct the first 92 residues of syb2 (from the N terminus) were followed by 81 residues of the CSPα C terminus, which includes a palmitoylation sequence, CCYCCCCLCCCFNCCCGKC. These proteins provide single (ΔTMD-CIIL and ΔTMD-LC), double (ΔTMD-CAC), and multiple (ΔTMD-CSPα) sites for post-translational acylation of long hydrocarbon chains.
FIGURE 1.
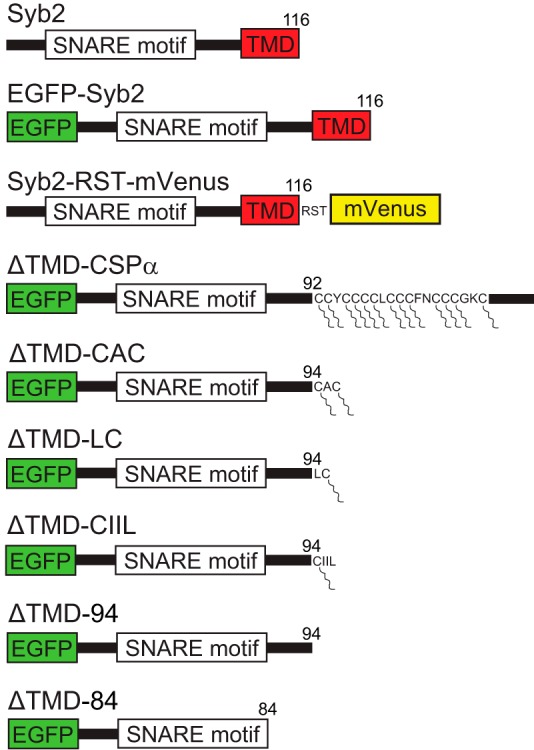
Synaptobrevin 2 constructs. Untagged WT full-length syb2 (Syb2) was expressed from a bicistronic vector with EGFP expressed from an IRES2. N-tagged Syb2 fused to EGFP at the N terminus. C-tagged syb2 with mVenus fused to the C terminus with RST as the amino acid sequence of the linker (Syb2-RST-mVenus). Lipid-anchored syb2, and ΔTMD syb2 constructs, with EGFP fused to the N terminus. In the ΔTMD-CSPα construct syb2 was truncated at residue 92 and extended with the CSPα lipid-anchoring sequence. In the ΔTMD-CAC, ΔTMD-LC, and ΔTMD-CIIL constructs, syb2 was truncated at residue 94 and extended with the indicated lipid-anchoring motifs. Constructs used in liposome fusion did not contain EGFP, and their cysteines were conjugated to POPE-maleimide (see under “Experimental Procedures”). Lipid moieties are indicated as segmented lines connected to cysteines. ΔTMD-94 and ΔTMD-84 contain sequences of WT syb2 residues 1–94 and 1–84, respectively. The numbers on the right indicate the last residue of syb2 in each construct.
We truncated syb2 at amino acids 84 and 94 to generate two cytosolic syb2 mutants (ΔTMD-84 and ΔTMD-94) that respectively omit or include the linker between the SNARE motif and TMD. We generated N-terminal EGFP-tagged constructs for all lipid-anchored syb2, cytosolic syb2 constructs, and WT to provide an expression marker. To control for the N-terminal EGFP tag, we subcloned WT syb2, ΔTMD-CSPα, and ΔTMD-CIIL without tags into a bicistronic vector from which EGFP was expressed from an IRES2. In addition to full-length syb2 with an N-terminal tag (EGFP-Syb2), we tested the control protein used by Zhou et al. (13), WT syb2 with a C-terminal mVenustag (Syb2-RST-mVenus) in which the sequence RST served as the linker.
Endocrine Release
To assay syb2 constructs in endocrine release, we prepared cultured chromaffin cells from double knock-out (DKO) mice lacking the major dense-core vesicle SNAREs syb2 and cellubrevin. In contrast to chromaffin cells from WT mice, DKO chromaffin cells display very low secretion (20). We used amperometry recording to assay catecholamine release from individual chromaffin cells in response to depolarization by local application of a solution containing 105 mm KCl. We confirmed the poor secretion of DKO chromaffin cells (Fig. 2A) and found that DKO cells expressing WT syb2, with or without an N-terminal EGFP tag, secreted vigorously at levels comparable with littermate control cells; these cells all produced similar vesicle fusion frequencies of nearly 1 event/s (Fig. 2B) compared with ∼0.1 events/s in DKO cells with no syb2. The residual release in this background likely reflects the action of a non-canonical v-SNARE, as has been found to be the case for residual spontaneous release at synapses (25). However, the molecules responsible for residual release in chromaffin cells lacking syb2 and cellubrevin have not been identified. These data thus indicate that DKO chromaffin cells provide a suitable background for testing syb2 mutants. A large number of syb2 mutants with TMD substitutions also rescue Ca2+-triggered exocytosis in this system (8). By contrast, the syb2 construct with a C-terminal mVenus tag, Syb2-RST-mVenus (13), only weakly rescued secretion, increasing the frequency of release events ∼2-fold over background in DKO cells (Fig. 2B).
Expressing lipid-anchored syb2 constructs resulted in little or no rescue of secretion in DKO chromaffin cells (Fig. 2A). DKO cells expressing ΔTMD-CIIL, ΔTMD-84, or ΔTMD-94 secreted as poorly as untransfected DKO cells (Fig. 2B). ΔTMD-CSPα, ΔTMD-CAC, and ΔTMD-LC produced very small apparent increases in spike frequency over untransfected DKO cells, but these changes were not statistically significant. Untagged ΔTMD-CSPα and ΔTMD-CIIL produced results indistinguishable from the corresponding EGFP-tagged constructs (data not shown). The fact that three untagged constructs (including WT) produced results similar to the corresponding N-tagged constructs indicates that fusing EGFP to the N terminus does not influence function.
More detailed analysis of secretion kinetics confirmed the lack of support for dense-core vesicle exocytosis by lipid-anchored syb2. Cumulative plots of spike number versus time from DKO cells expressing WT syb2 (N-tagged or untagged) exhibited a brief delay after the start of depolarization and then rose nearly linearly before reaching a plateau (Fig. 2C, bar indicates depolarization). However, the C-tagged syb2-RST-mVenusconstruct produced a slower rising phase. The slopes of the linear regions (between 10 and 70% of the plateau), determined by linear regression, were much lower for ΔTMD-CSPα, ΔTMD-CAC, ΔTMD-LC, and C-tagged syb2-RST-mVenus than for WT and N-tagged syb2, and only ∼2-fold greater than that for untransfected DKO cells (Fig. 2D). Slopes from DKO cells expressing ΔTMD-CIIL, ΔTMD-84, and ΔTMD-94 were indistinguishable from slopes from untransfected DKO cells; slopes from DKO cells expressing untagged ΔTMD-CSPα and ΔTMD-CIIL were indistinguishable from slopes obtained with the corresponding N-tagged proteins (data not shown). Only ΔTMD-CSPα produced a significant increase in slope over background. This slope was comparable with that with C-tagged syb2-RST-mVenus, and both were about one-third that seen in cells expressing untagged or N-tagged WT syb2.
We logged the number of cells responding with one or more spikes during the first two depolarizations (Table 1). Spikes were observed in 83 and 89% of DKO chromaffin cells expressing untagged and N-tagged WT syb2, respectively, versus 64% of untransfected DKO cells, and 68–72% for Syb2-RST-mVenusand lipid-anchored syb2 constructs. As noted above, the residual secretion in the DKO background probably reflects the presence of a non-canonical v-SNARE, which can produce a low level of secretion in over half the DKO cells. Expression of ΔTMD-84 and ΔTMD-94 reduced the number to 52 and 46%, respectively, suggesting that these constructs can interfere with the function of the putative non-canonical v-SNAREs.
TABLE 1.
The percent of secreting DKO chromaffin cells expressing the indicated construct, with the number of cells responding to depolarization over the number of cells tested in parentheses
| Syb2 | EGFP-syb2 | Syb2-RST-mVenus | ΔTMD-CSPα | ΔTMD-CAC | ΔTMD-LC | ΔTMD-CIIL | ΔTMD-94 | ΔTMD-84 | None | |
|---|---|---|---|---|---|---|---|---|---|---|
| Percent responding | 83% (44:53) | 89% (49:55) | 68% (26:38) | 67% (62:91) | 72% (52:72) | 67% (32:48) | 72% (34:47) | 46% (33:71) | 52% (46:88) | 64% (54:84) |
It is possible that the lack of activity observed with lipid-anchored constructs resulted from incorrect localization. To address this possibility, the expression of each construct was evaluated by confocal fluorescence microscopy and analyzed for colocalization with the dense-core vesicle cargo, NPY-DsRed (Fig. 3). All of the lipid-anchored ΔTMD constructs targeted to dense-core vesicles, but ΔTMD-CAC and ΔTMD-LC constructs, also displayed some diffuse cytosolic expression (Fig. 3A). The ΔTMD-CSPα and ΔTMD-CIIL constructs targeted vesicles as effectively as WT syb2. Consistent with the lack of a membrane-anchoring motif, ΔTMD-94 was entirely diffuse with no puncta. Syb2 staining could not be seen in untransfected DKO cells. Expression patterns revealed by line scans showed peaks for WT syb2 and all lipid-anchored constructs, but the line scan for ΔTMD-94 was relatively smooth. Thus, the lipid-anchored constructs were efficiently targeted to vesicle membranes and did not accumulate on the plasma membrane.
FIGURE 3.

Expression and vesicle targeting in chromaffin cells. A, confocal images of untransfected DKO cells and DKO cells transfected with NPY-DsRed (red) and either WT syb2, or lipid-anchored syb2, or ΔTMD-94 (green). The inset and line scan panels correspond to the white squares and dashed lines in the merged panels. In line scan panels, black traces indicate syb2 variants, and red indicates NPY-DsRed. B, Pearson's coefficient quantifies colocalization. Except for ΔTMD-CAC, lipid-anchored syb2 showed targeting comparable with WT syb2. WT and all lipid-anchored syb2 constructs targeted more efficiently than ΔTMD-94. p value for ΔTMD-LC is shown. The negative control construct ΔTMD-94 was well below all the other constructs (p value range 10−8–10−4). C, quantification of total fluorescence intensity indicates roughly similar expression levels for all constructs. n = 18–33 cells from three DKO embryos. **, p < 0.01; ***, p < 0.001. Scale bars, 3 μm.
The colocalization of syb2 variants with NPY-DsRed was quantified using the Pearson's coefficient (Fig. 3B). WT and lipid-anchored syb2 constructs targeted dense-core vesicles significantly better than ΔTMD-94 (p < 0.0001 for WT and all lipid-anchored constructs). Only ΔTMD-CAC showed significantly lower colocalization than WT syb2, but the Pearson's coefficient remained well above that for ΔTMD-94. Thus, even for the constructs that target vesicles slightly less efficiently than WT, targeting is still well above the negative control provided by ΔTMD-94. The expression levels were quantified by measuring total fluorescence intensity, and all the constructs had comparable expression levels (Fig. 3C). These results indicate that the poor performance of lipid-anchored syb2 constructs in secretion reflects a role for the TMD in fusion rather than poor expression or targeting.
Synaptic Release
The ability of syb2 constructs to mediate synaptic release was examined by patch clamp recording of mEPSCs in cultured hippocampal neurons. mEPSCs report the spontaneous fusion of single synaptic vesicles and can be studied in neurons after blocking action potentials with tetrodotoxin. DKO hippocampal neurons have been shown to present mEPSCs at a low frequency, with residual spontaneous release reflecting the activity of the non-canonical v-SNARE Vti1a (26). Spontaneous release in DKO neurons is dramatically enhanced by expression of WT syb2 (27). In this study control neurons presented mEPSCs at a frequency of ∼1/s, and in untransfected DKO neurons this frequency was 12-fold lower or ∼0.08/s (Fig. 4A). As with the evoked endocrine release just described, expression of WT EGFP-syb2 rescued spontaneous synaptic vesicle fusion to levels seen in littermate control neurons, but lipid-anchored and ΔTMD syb2 constructs did not (Fig. 4A). Individual events illustrate that regardless of the syb2 variant, mEPSCs had the characteristic shape of an excitatory synaptic current, with a rapid rise and slower decay (Fig. 4B). Expressing N-tagged WT syb2 in DKO neurons rescued spontaneous release to produce a frequency indistinguishable from the ∼1/s frequency of littermate control neurons (Fig. 4C).
FIGURE 4.

Spontaneous synaptic release in neurons. A, patch clamp recordings of mEPSCs from littermate control and DKO hippocampal neurons, either untransfected or expressing the indicated syb2 construct. B, individual mEPSCs from each trace of A (boxed). C, mEPSC frequencies from littermate neurons and DKO neurons untransfected or expressing syb2 constructs. Syb2-RST-mVenus, lipid-anchored syb2, ΔTMD-94, and untransfected DKO cells produced similar, statistically indistinguishable low mEPSC frequencies (n = 11–30 neurons; three DKO embryos for Syb2-RST-mVenus and five for ΔTMD-LC, >9 for others). The comparison with EGFP-Syb2 or DKO is indicated by bars. D, targeting syb2 constructs in neurons. EGFP-Syb2 and lipid-anchored syb2 (green) targeted synapses labeled with synaptophysin (red). E, Pearson's coefficients indicated colocalizations of WT syb2 and lipid-anchored syb2 were well above that for ΔTMD-94, but ΔTMD-CAC was significantly lower than WT syb2. The asterisks indicate comparisons with EGFP-Syb2. **, p < 0.01; ***, p < 0.001; n.s., not significant. n = 23–44 neurons from 2 to 4 DKO embryos.
DKO neurons expressing the four lipid-anchored syb2 variants secreted poorly, and the mEPSC frequency was indistinguishable from the background in untransfected DKO neurons. Expression of ΔTMD-CSPα, ΔTMD-CAC, and ΔTMD-84 slightly increased the mEPSC frequency ∼2.5-fold, but as with the other lipid-anchored syb2 constructs and ΔTMD-94, the frequencies were statistically indistinguishable from that in untransfected DKO neurons. mEPSC frequencies in DKO cells expressing lipid-anchored syb2 constructs were all much lower than frequencies seen in control cells and DKO cells expressing WT syb2. Untagged ΔTMD-CSPα and ΔTMD-CIIL expressed from a bicistronic vector also failed to rescue spontaneous release in DKO neurons (data not shown). Thus, lipid-anchored syb2 failed to support spontaneous synaptic vesicle fusion in neurons. Expression of the C-tagged Syb2-RST-mVenus construct produced an ∼2.5-fold increase over background that was similar to that seen with ΔTMD-CSPα and ΔTMD-CAC and was not statistically significant (Fig. 4, A–C).
To determine whether the poor rescue of synaptic release was due to mistargeting of the expressed proteins, we examined colocalization with synaptophysin, a synaptic vesicle marker. All syb2 constructs colocalized with synaptophysin, indicating that these molecules targeted synapses (Fig. 4D). However, the ΔTMD-CAC construct showed some diffuse expression in addition to the punctate, synaptic labeling. Expression of ΔTMD-94 was entirely diffuse with no puncta, and appeared uniform in cell bodies and processes. Quantification of colocalization between syb2 and syp indicated that the targeting of lipid-anchored syb2 was very similar to WT syb2, although the Pearson's coefficient for ΔTMD-CAC was slightly lower than that for WT syb2 it was still well above ΔTMD-94 (Fig. 4E). ΔTMD-CAC showed a similar targeting of dense-core vesicles that was slightly lower than WT but well above ΔTMD-94 (Fig. 3, A and B).
Liposome Fusion
To characterize the role of the syb2 TMD in membrane fusion in vitro, we assayed fusion of proteoliposomes harboring syntaxin-1A/SNAP-25B heterodimer with proteoliposomes harboring a WT or lipid-anchored syb2 construct (identical to those in cellular assays but without an EGFP tag). The lipid-anchored ΔTMD constructs were linked chemically by their cysteines to liposomes with 5% maleimide-POPE. Proteoliposome protein content was analyzed by SDS-PAGE with Coomassie Blue (Fig. 5A). Fusion of liposomes leads to mixing of the 1.5% NBD-PE and 1.5% rhodamine-PE in the v-SNARE vesicles with the nonfluorescent lipids in the t-SNARE vesicles. This dilutes the FRET pair and dequenches NBD fluorescence to give a signal that can be monitored over time (28). To enhance fusion rate and extent, the cytosolic domain of synaptotagmin 1 was included, and the reaction was triggered by addition of Ca2+ (arrows in Fig. 5B) (23). ΔTMD-94 was used as a control to block SNARE-mediated fusion. As observed previously with SNARE-mediated fusion (10), replacement of the syb2 TMD with one or two lipid anchors failed to support Ca2+-synaptotagmin enhancement of SNARE-mediated fusion (Fig. 5B). Interestingly, ΔTMD-CSPα, which harbors several potential lipid anchors (Fig. 1), supported a small but significant amount of SNARE-mediated lipid mixing. Overall, these data are consistent with the amperometry data from chromaffin cells and the patch clamp data from neurons and suggest that the TMD of syb2 is required for efficient membrane fusion in vitro as well as in vivo.
Discussion
This study tested the role of the syb2 TMD in three different assays as follows: Ca2+-triggered dense-core vesicle exocytosis from endocrine cells, spontaneous synaptic vesicle exocytosis from neurons, and liposome fusion. In each assay, four different lipid-anchored syb2 variants failed to function as effectively as the WT protein. Proteins that can accept one or two lipidic acyl moieties, ΔTMD-LC, ΔTMD-CIIL, and ΔTMD-CAC, clearly failed to support liposome fusion. Some of these constructs supported low levels of exocytosis that were statistically indistinguishable from background and 4–6-fold lower than the levels of exocytosis achieved with WT syb2. This failure to support exocytosis cannot be attributed to mislocalization, as the proteins correctly targeted dense-core vesicles in chromaffin cells and synapses in neurons. In contrast to these three lipid-anchored syb2 constructs, ΔTMD-CSPα displayed limited fusion activity. This construct produced low but statistically significant enhancement of dense-core vesicle and proteoliposome fusion, but this enhancement was well below that observed with WT syb2. This unique property of ΔTMD-CSPα may reflect the 13 cysteine residues of its lipid-anchor motif (Fig. 1), which enable it to accept many lipidic acyl moieties. SNAREs harboring multiple lipidic acyl moieties have been shown to support lipid mixing previously (10). With the exception of ΔTMD-CSPα, the present results indicate that attachment of syb2 to the membrane by a lipid anchor does not support biological fusion. Fusion requires more than just membrane proximity provided by the formation of a SNARE complex with lipid-anchored syb2.
The TMD of syb2 performs some additional functions in addition to anchoring to the membrane. It is unlikely that this additional function includes protein folding because the SNARE motif of syb2 is a random coil when not incorporated into a SNARE complex (29). The TMD may contribute to the availability of the SNARE motif and the manner in which the SNARE motif is presented. This could happen if the TMD influenced the conformation of the rest of the protein or if the TMD controlled membrane insertion of the juxtamembrane region (30), which has been shown to influence exocytosis (31). Micro-localization and dimerization are additional possible functional roles for the TMD, although the functional relevance of dimerization has been difficult to establish (32).
The conclusion of an important role for the TMD is consistent with reports that substitutions within the TMDs of t-SNAREs (6, 7) and v-SNAREs (8) alter flux through fusion pores. The gap junction-like structure composed of these TMDs can form a proteinaceous channel without bending the adjacent lipid bilayers. Only when the fusion pore expands does it incorporate lipid and bend lipid bilayers (33, 34). The important functional role for the syb2 TMD indicated by the present results is consistent with the virtually universal presence of a TMD in v-SNAREs (1–3).
Previous work has shown that lipid-anchored SNAREs can only drive lipid mixing in an in vitro fusion assay when the SNARES harbor either a lipid moiety long enough to span a lipid bilayer or multiple lipid moieties (35). Accessory proteins can also enable lipid-anchored SNAREs to drive fusion (11). A recent study of vacuole fusion indicated that yeast SNAREs differ in their requirement for a TMD for both lipid and content mixing (12). Another recent study reported that lipid-anchored SNAREs are fully functional in spontaneous synaptic release in cortical neurons (13), and this conclusion differs from that put forward here. These authors reported similar ∼3–4-fold increases in fusion over background with both ΔTMD-CSPα and their WT control, syb2-RST-mVenus. Here, we showed that the syb2-RST-mVenus construct functions poorly in the rescue of release, in contrast to untagged and N-tagged WT syb2 constructs, which rescue release to levels seen in littermate control cells. The mEPSC frequencies we reported here in hippocampal neurons for the DKO background, ΔTMD-CSPα and syb2-RST-mVenus (Fig. 4), were all similar to those reported previously in cortical neurons (13). Our replication of these results points to the poor function of the syb2-RST-mVenus construct as a WT control and thus indicates this as the basis for the discrepancy between our conclusion of poor function by lipid-anchored syb2, and the previous conclusion that they are fully functional (13). The poor function of the syb2-RST-mVenusconstruct probably results from the presence of arginine in the linker, as a previous study (36) showed that positive charge in precisely the same location immediately after the C terminus of the syb2 TMD impairs function to a degree similar to that found in this study with the syb2-RST-mVenus construct.
This study clarifies the role of the syb2 TMD in synaptic release and presents the first test of lipid-anchored SNAREs in Ca2+-triggered dense-core vesicle exocytosis, as well as Ca2+-synaptotagmin enhancement of SNARE-mediated liposome fusion. TMDs are essential elements in models that posit the initiation of SNARE-mediated membrane fusion through formation of a proteinaceous fusion pore (14, 15, 37). This study indicates that lipid-anchored syb2 variants lacking a TMD either fail to support fusion or support fusion very poorly. This was observed not only in two different forms of biological fusion but also in proteoliposome fusion in vitro. Because one or two lipidic linkages failed to provide significant support for fusion in any assay, this would suggest that proximity is not sufficient and that the TMD of syb2 plays an important role. This role may be formation of a proteinaceous fusion pore (14, 15, 37) or transmission of force to the lipid bilayer (16–19). Lipid anchors do not support these functions or support them poorly. Whether some other forms of attachment of proteins to a membrane can support such functions to achieve the levels of fusion seen with WT syb2 remains to be tested.
The low levels of fusion seen with ΔTMD-CSPα in dense-core vesicle release and liposome fusion support the idea that anchoring a SNARE protein to a lipid bilayer through a large number of lipid moieties provides limited support for fusion (10). Multiple lipidic linkages may enable SNAREs to create a defect and deform lipid bilayers sufficiently to drive fusion at a slow rate. Contacts between the syntaxin and syb2 TMDs (38) contribute to the stability of the SNARE complex (39), and removal of its TMD would prevent a SNARE from harnessing these contacts as a driving force for fusion. SNARE-mediated fusion can occur through hemifusion intermediates (40), presumably bypassing proteinaceous fusion pores. Further studies will be required to address the interesting question of whether the limited fusion seen with ΔTMD-CSPα reflects a different mechanism involving hemifusion.
The number of SNARE complexes needed to trigger fusion may be only one for liposomes (41, 42), three for dense-core vesicles (43), and two for synaptic vesicles (44). These numbers are well below the estimates of 5–10 based on pore conductance (7, 45), and it is difficult to envision how fewer SNARE complexes could form a purely proteinaceous fusion pore. Other proteins can act as accessories to enable fusion driven by lipid-anchored SNAREs (11), and it is possible that another integral vesicle membrane protein such as synaptophysin can donate TMDs to a fusion pore. The speed of SNARE-mediated membrane fusion differs dramatically between different cellular membrane compartments, and it is possible that SNAREs can fuse membranes by more than one mechanism. The speed with which SNAREs catalyze fusion may depend on whether the nascent fusion pore contains protein or lipid or both (46, 47). Thus, proteinaceous fusion pores may serve as an adaptation to provide speed and temporal control for synaptic transmission.
Author Contributions
Che-Wei Chang, Chung-Wei Chiang, and J. D. G. designed and executed experiments, analyzed data, and wrote the paper. E. R. C. and M. B. J. designed the experiments, interpreted the data, and wrote the paper.
Acknowledgment
We thank Dr. Thomas Südhof for providing the syb2-CSPα construct.
This work was supported by National Institutes of Health Grants NS44057 (to M. B. J.) and MH061876 (to E. R. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TMD
- transmembrane domain
- CSPα
- cysteine string protein-α
- DKO
- double knock-out
- EGFP
- enhanced GFP
- syb2
- synaptobrevin-2
- mEPSC
- miniature excitatory post-synaptic current
- POPE
- palmitoyloleoylphosphatidylethanolamine.
References
- 1.Weimbs T., Mostov K., Low S. H., and Hofmann K. (1998) A model for structural similarity between different SNARE complexes based on sequence relationships. Trends Cell Biol. 8, 260–262 [DOI] [PubMed] [Google Scholar]
- 2.Jahn R., and Südhof T. C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem. 68, 863–911 [DOI] [PubMed] [Google Scholar]
- 3.Ungar D., and Hughson F. M. (2003) SNARE protein structure and function. Annu. Rev. Cell Dev. Biol. 19, 493–517 [DOI] [PubMed] [Google Scholar]
- 4.Lin R. C., and Scheller R. H. (2000) Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 16, 19–49 [DOI] [PubMed] [Google Scholar]
- 5.Südhof T. C., and Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han X., and Jackson M. B. (2005) Electrostatic interactions between the syntaxin membrane anchor and neurotransmitter passing through the fusion pore. Biophys. J. 88, L20–L22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han X., Wang C. T., Bai J., Chapman E. R., and Jackson M. B. (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289–292 [DOI] [PubMed] [Google Scholar]
- 8.Chang C. W., Hui E., Bai J., Bruns D., Chapman E. R., and Jackson M. B. (2015) A structural role for the synaptobrevin 2 transmembrane domain in dense-core vesicle fusion pores. J. Neurosci. 35, 5772–5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grote E., Baba M., Ohsumi Y., and Novick P. J. (2000) Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J. Cell Biol. 151, 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNew J. A., Weber T., Parlati F., Johnston R. J., Melia T. J., Söllner T. H., and Rothman J. E. (2000) Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 150, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H., Zick M., Wickner W. T., and Jun Y. (2011) A lipid-anchored SNARE supports membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 108, 17325–17330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pieren M., Desfougères Y., Michaillat L., Schmidt A., and Mayer A. (2015) Vacuolar SNARE protein transmembrane domains serve as nonspecific membrane anchors with unequal roles in lipid mixing. J. Biol. Chem. 290, 12821–12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou P., Bacaj T., Yang X., Pang Z. P., and Südhof T. C. (2013) Lipid-anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson M. B., and Chapman E. R. (2008) The fusion pores of Ca2+-triggered exocytosis. Nat. Struct. Mol. Biol. 15, 684–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindau M., and Almers W. (1995) Structure and function of fusion pores in exocytosis and ectoplasmic membrane fusion. Curr. Opin. Cell Biol. 7, 509–517 [DOI] [PubMed] [Google Scholar]
- 16.Jackson M. B. (2010) SNARE complex zipping as a driving force in the dilation of proteinaceous fusion pores. J. Membr. Biol. 235, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kesavan J., Borisovska M., and Bruns D. (2007) v-SNARE actions during Ca2+-triggered exocytosis. Cell 131, 351–363 [DOI] [PubMed] [Google Scholar]
- 18.Lindau M., Hall B. A., Chetwynd A., Beckstein O., and Sansom M. S. (2012) Coarse-grain simulations reveal movement of the synaptobrevin C terminus in response to piconewton forces. Biophys. J. 103, 959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Risselada H. J., Kutzner C., and Grubmüller H. (2011) Caught in the act: visualization of SNARE-mediated fusion events in molecular detail. Chembiochem 12, 1049–1055 [DOI] [PubMed] [Google Scholar]
- 20.Borisovska M., Zhao Y., Tsytsyura Y., Glyvuk N., Takamori S., Matti U., Rettig J., Südhof T., and Bruns D. (2005) v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 24, 2114–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gascón S., Paez-Gomez J. A., Díaz-Guerra M., Scheiffele P., and Scholl F. G. (2008) Dual-promoter lentiviral vectors for constitutive and regulated gene expression in neurons. J. Neurosci. Methods 168, 104–112 [DOI] [PubMed] [Google Scholar]
- 22.Chicka M. C., Hui E., Liu H., and Chapman E. R. (2008) Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+. Nat. Struct. Mol. Biol. 15, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tucker W. C., Weber T., and Chapman E. R. (2004) Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438 [DOI] [PubMed] [Google Scholar]
- 24.Hui E., Gaffaney J. D., Wang Z., Johnson C. P., Evans C. S., and Chapman E. R. (2011) Mechanism and function of synaptotagmin-mediated membrane apposition. Nat. Struct. Mol. Biol. 18, 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez D. M., and Kavalali E. T. (2012) The role of non-canonical SNAREs in synaptic vesicle recycling. Cell. Logist. 2, 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez D. M., Khvotchev M., Trauterman B., and Kavalali E. T. (2012) Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron 73, 121–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzman R. E., Schwarz Y. N., Rettig J., and Bruns D. (2010) SNARE force synchronizes synaptic vesicle fusion and controls the kinetics of quantal synaptic transmission. J. Neurosci. 30, 10272–10281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., and Rothman J. E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 [DOI] [PubMed] [Google Scholar]
- 29.Fasshauer D., Otto H., Eliason W. K., Jahn R., and Brünger A. T. (1997) Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J. Biol. Chem. 272, 28036–28041 [DOI] [PubMed] [Google Scholar]
- 30.Kweon D. H., Kim C. S., and Shin Y. K. (2003) Regulation of neuronal SNARE assembly by the membrane. Nat. Struct. Biol. 10, 440–447 [DOI] [PubMed] [Google Scholar]
- 31.Borisovska M., Schwarz Y. N., Dhara M., Yarzagaray A., Hugo S., Narzi D., Siu S. W., Kesavan J., Mohrmann R., Böckmann R. A., and Bruns D. (2012) Membrane-proximal tryptophans of synaptobrevin II stabilize priming of secretory vesicles. J. Neurosci. 32, 15983–15997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fdez E., Martínez-Salvador M., Beard M., Woodman P., and Hilfiker S. (2010) Transmembrane-domain determinants for SNARE-mediated membrane fusion. J. Cell Sci. 123, 2473–2480 [DOI] [PubMed] [Google Scholar]
- 33.Chang C. W., and Jackson M. B. (2015) Synaptobrevin transmembrane domain influences exocytosis by perturbing vesicle membrane curvature. Biophys. J. 109, 76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z., and Jackson M. B. (2010) Membrane bending energy and fusion pore kinetics in Ca2+-triggered exocytosis. Biophys. J. 98, 2524–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNew J. A., Weber T., Engelman D. M., Söllner T. H., and Rothman J. E. (1999) The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol. Cell 4, 415–421 [DOI] [PubMed] [Google Scholar]
- 36.Ngatchou A. N., Kisler K., Fang Q., Walter A. M., Zhao Y., Bruns D., Sørensen J. B., and Lindau M. (2010) Role of the synaptobrevin C terminus in fusion pore formation. Proc. Natl. Acad. Sci. U.S.A. 107, 18463–18468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson M. B. (2011) Inferring structures of kinetic intermediates in Ca2+-triggered exocytosis. Curr. Top. Membr. 68, 185–208 [DOI] [PubMed] [Google Scholar]
- 38.Stein A., Weber G., Wahl M. C., and Jahn R. (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460, 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poirier M. A., Hao J. C., Malkus P. N., Chan C., Moore M. F., King D. S., and Bennett M. K. (1998) Protease resistance of syntaxin.SNAP-25.VAMP complexes. Implications for assembly and structure. J. Biol. Chem. 273, 11370–11377 [DOI] [PubMed] [Google Scholar]
- 40.Xu Y., Zhang F., Su Z., McNew J. A., and Shin Y. K. (2005) Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 12, 417–422 [DOI] [PubMed] [Google Scholar]
- 41.van den Bogaart G., Holt M. G., Bunt G., Riedel D., Wouters F. S., and Jahn R. (2010) One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol. 17, 358–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Y., Zorman S., Gundersen G., Xi Z., Ma L., Sirinakis G., Rothman J. E., and Zhang Y. (2012) Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337, 1340–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohrmann R., de Wit H., Verhage M., Neher E., and Sørensen J. B. (2010) Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330, 502–505 [DOI] [PubMed] [Google Scholar]
- 44.Sinha R., Ahmed S., Jahn R., and Klingauf J. (2011) Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc. Natl. Acad. Sci. U.S.A. 108, 14318–14323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Z., Zhang Z., and Jackson M. B. (2010) Synaptotagmin IV modulation of vesicle size and fusion pores in PC12 cells. Biophys. J. 98, 968–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackson M. B., and Chapman E. R. (2006) Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu. Rev. Biophys. Biomol. Struct. 35, 135–160 [DOI] [PubMed] [Google Scholar]
- 47.Fang Q., Berberian K., Gong L. W., Hafez I., Sørensen J. B., and Lindau M. (2008) The role of the C terminus of the SNARE protein SNAP-25 in fusion pore opening and a model for fusion pore mechanics. Proc. Natl. Acad. Sci. U.S.A. 105, 15388–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]