

Abstract
The ability of Mycobacterium tuberculosis to resist intraphagosomal stresses, such as oxygen radicals and low pH, is critical for its persistence. Here, we show that a cytoplasmic redox sensor, WhiB3, and the major M. tuberculosis thiol, mycothiol (MSH), are required to resist acidic stress during infection. WhiB3 regulates the expression of genes involved in lipid anabolism, secretion, and redox metabolism, in response to acidic pH. Furthermore, inactivation of the MSH pathway subverted the expression of whiB3 along with other pH-specific genes in M. tuberculosis. Using a genetic biosensor of mycothiol redox potential (EMSH), we demonstrated that a modest decrease in phagosomal pH is sufficient to generate redox heterogeneity in EMSH of the M. tuberculosis population in a WhiB3-dependent manner. Data indicate that M. tuberculosis needs low pH as a signal to alter cytoplasmic EMSH, which activates WhiB3-mediated gene expression and acid resistance. Importantly, WhiB3 regulates intraphagosomal pH by down-regulating the expression of innate immune genes and blocking phagosomal maturation. We show that this block in phagosomal maturation is in part due to WhiB3-dependent production of polyketide lipids. Consistent with these observations, MtbΔwhiB3 displayed intramacrophage survival defect, which can be rescued bypharmacological inhibition of phagosomal acidification. Last, MtbΔwhiB3 displayed marked attenuation in the lungs of guinea pigs. Altogether, our study revealed an intimate link between vacuolar acidification, redox physiology, and virulence in M. tuberculosis and discovered WhiB3 as crucial mediator of phagosomal maturation arrest and acid resistance in M. tuberculosis.
Keywords: lysosomal acidification, Mycobacterium tuberculosis, pH regulation, redox signaling, virulence factor
Introduction
Mycobacterium tuberculosis causes a chronic persistent infection that affects one-third of the world's population (see the World Health Organization website). Macrophage is the major human host cell for growth, survival, and persistence of M. tuberculosis. Studies indicate that M. tuberculosis continuously senses the phagosomal environment and modulates genetic pathways to regulate intramacrophage growth for long term persistence (1–3). In this regard, acidic pH has recently been appreciated as an important intraphagosomal signal sensed by M. tuberculosis to regulate gene expression and establish chronic infection (3). The importance of pH emerged from several studies showing that pathogenic mycobacteria successfully restrict fusion of phagosomes with acidic lysosomes and therefore multiply in a growth-permissive vacuolar compartment with a pH of ∼6.2 (4–6). However, upon activation of macrophages by interferon-γ (IFN-γ) and Escherichia coli-derived lipopolysaccharide (LPS), the pH of the M. tuberculosis phagosome drops to <5.0, resulting in M. tuberculosis growth restriction (6–8). Hence, the inhibition of phagosomal maturation during early stages of infection and induction of acid resistance mechanisms later during immune-activation are considered major virulence strategies adopted by M. tuberculosis to establish chronic infection (9). Despite the recognized role of acidic pH in regulating M. tuberculosis pathogenesis, the mechanisms by which M. tuberculosis responds to fluctuations in phagosomal pH and calibrates its gene expression for intracellular growth and persistence remain poorly characterized.
Recent studies indicate that M. tuberculosis maintains a neutral intrabacterial pH (∼7.2) after exposure to a range of acidic pH levels (from 6.2 to 4.5) in vitro, and inside the acidic phagosomal milieu of resting or activated macrophage (10). This indicates that M. tuberculosis stably maintains intrabacterial pH homeostasis during infection and, therefore, that the intrabacterial pH per se is unlikely to be the signal that triggers alterations in M. tuberculosis gene expression in response to changes in phagosomal acidity. Therefore, novel insights are needed to discover which aspects of mycobacterial physiology are modulated by phagosomal acidity and what are the bacterial sensors of phagosomal pH. In this context, an Fe-S cluster containing putative transcription factor WhiB3 is induced in response to acidic pH in medium, inside macrophages, and in the lungs of infected animals (11, 12). In fact, whiB3 was the only transcription factor whose expression was found to be pH-responsive inside macrophages. In vitro studies have shown that WhiB3 responds to dormancy signals, such as O2 and nitric oxide (NO), via its 4Fe-4S cluster (13). However, phenotypic experiments revealed no role of WhiB3 in controlling mycobacterial survival in response to NO or hypoxia (13). Therefore, how WhiB3 regulates mycobacterial persistence remained uncharacterized. Consistent induction of whiB3 in acidic environments in vitro and inside macrophages (12) implicated WhiB3 in regulating adaptation of M. tuberculosis in response to phagosomal pH. A fundamentally important question remains yet unanswered. What is the mechanism by which WhiB3 mediates the acid response of M. tuberculosis during infection?
In this study, we performed global microarray analysis to identify genes regulated by WhiB3 in response to acid stress. More importantly, we measured the dynamic changes in mycothiol redox potential (EMSH)3 of WT M. tuberculosis and MtbΔwhiB3 in response to acidic pH in vitro and inside naive and activated macrophages. Confocal studies and host microarray studies were performed to examine the function of WhiB3 in regulating phagosomal maturation. Last, the physiological importance of whiB3-mediated effects on gene expression, redox homeostasis, and phagosomal maturation were investigated by performing survival studies in macrophages and guinea pigs. Our study, for the first time, demonstrates how M. tuberculosis recalibrates its redox physiology in response to vacuolar pH during infection and identifies a major role of WhiB3 in responding to acidic stress.
Experimental Procedures
Bacterial Strains and Growth Conditions
Wild type M. tuberculosis H37Rv (WT M. tuberculosis), MtbΔwhiB3, and whiB3-comp strains were cultivated as described (13). E. coli cultures were grown in LB medium. When required, culture medium was supplemented with kanamycin (25 μg/ml) or hygromycin (50 μg/ml). For acid stress, the pH of 7H9 broth was adjusted using hydrochloric acid (HCl) and buffered using 100 mm MES. Approximately 1 × 107 cells/ml were exposed to various pH-adjusted media, and survival was monitored at day 0 and day 4 by serially diluting the culture and enumerating colony-forming units (cfu). For carbonyl cyanide m-chlorophenylhydrazone (CCCP) treatment, bacteria were treated with 500 μm CCCP for 4 h.
Generation of MtbΔwhiB3 and whiB3 Complemented Strains
For constructing MtbΔwhiB3, 1-kb left flanking and right flanking regions of whiB3 (Rv3416) were cloned upstream and downstream of the loxP-gfp-hygromycin-loxP cassette in a mycobacterial sacB-based suicide vector, pML523 (a kind gift from Michael Neiderwies, University of Alabama at Birmingham). The construct was digested with SpeI and NsiI to release left flanking and right flanking regions of whiB3 along with the loxP-gfp-hygromycin-loxP cassette, blunt-ended, and cloned into EcoRV/PstI-digested pRSF-duet vector (Clontech). The pRSF-whiB3 clone was pretreated with UV as described previously (14)and electroporated into WT M. tuberculosis for allelic exchange. The resulting MtbΔwhiB3 (HygRKanSGFP+) colonies were screened by antibiotic selection and verified by PCR. To unmark MtbΔwhiB3 strain, pCRE-ZEO-SacB (a gift from Dr. Amit Pandey, Translational Health Science and Technology Institute, Haryana, India) was electroporated into MtbΔwhiB3, and the loxP-gfp-hygromycin-loxP cassette was released from the genome. The resulting unmarked MtbΔwhiB3 strain was confirmed by the absence of antibiotic selection marker. Disruption of whiB3 was confirmed by quantitative RT-PCR (qRT-PCR).
For constructing the whiB3-comp strain, the whiB3 gene along with its promoter region was amplified from the M. tuberculosis genome and inserted into an E. coli-mycobacterial shuttle vector, pSD5. The pSD-whiB3 clone was electroporated into unmarked MtbΔwhiB3 strain to generate the whiB3-comp strain. Expression of whiB3 in the whiB3-comp strain was confirmed by qRT-PCR.
Cell Lines
The human monocytic cell line THP-1 and mice RAW 264.7 macrophages were cultivated as described previously (15). RAW 264.7 macrophages were activated using 100 ng/ml IFN-γ and 20 ng/ml LPS.
Antibodies and Reagents
Antibodies for mammalian markers (CD63 and vacuolar H+-ATPase (V-ATPase)) were purchased from Santa Cruz Biotechnology. Lysotracker® DND-red99 was purchased from Thermo Fisher Scientific. Secondary antibody Alexa Fluor 568 was purchased from Thermo Fisher Scientific. Fluorescein 5(6)-isothiocyanate (FITC) was purchased from Sigma-Aldrich. Bafilomycin A1 was purchased from Invivogen, and stocks were made in dimethyl sulfoxide (DMSO), aliquoted, and stored at −20 °C.
Growth Conditions for qRT-PCR and Microarray Analysis
For analyzing the influence of acid stress on gene expression, M. tuberculosis strains (M. tuberculosis, MtbΔwhiB3, and MtbΔmshA) were grown until an A600 nm of 0.3–0.4 and exposed to 7H9 medium adjusted to a range of acidic pH 4.5, 5.5, 6.2, and 7.0 and normal 7H9 (pH 6.6) for 2 h at 150 rpm at 37 °C. Total RNA from WT M. tuberculosis and MtbΔwhiB3 (pH 4.5 and 6.6) was processed and hybridized to the M. tuberculosis whole genome gene expression profiling microarray G2509F (AMADID: G2509F_034585, Agilent Technologies PLC). DNA microarrays were provided by the University of Delhi South Campus MicroArray Centre. RNA amplification, cDNA labeling, microarray hybridization, scanning, and data analysis were performed at the University of Delhi South Campus MicroArray Centre as described (16). Slides were scanned on a microarray scanner (Agilent Technologies) and analyzed using GeneSpring software. Results were analyzed in MeV with significance analysis of microarrays considered significant at p ≤ 0.05. The normalized data from the microarray gene expression experiment have been submitted to the NCBI Gene Expression Omnibus and can be queried via Gene Expression Omnibus series accession number GSE61579.
qRT-PCR
First-strand cDNA synthesis was performed using 500 ng of the total RNA with the iScript Select cDNA synthesis kit (Bio-Rad) using random oligonucleotide primers. PCR was performed using gene-specific primers (Table 1). Gene expression was analyzed with real-time PCR using iQTM SYBR Green Supermix (Bio-Rad) and a CFX96 RT-PCR system (Bio-Rad). Data analysis was performed with CFX ManagerTM software (Bio-Rad). For comparison between WT M. tuberculosis, MtbΔwhiB3, and the whiB3-comp strains, the induction ratio for each gene was normalized to WT M. tuberculosis 16S rRNA expression.
TABLE 1.
Oligonucleotides used in the study
| Target | Primer sequence |
|---|---|
| whiB3 RT F | 5′-AACGCAGACATCTGGAACTG-3′ |
| whiB3 RT R | 5′-TAGGGCTCACCGACCTCTAA-3′ |
| pks2 RT F | 5′-AAGTGTCTCCGAGGTGTATG-3′ |
| pks2 RT R | 5′-CGAGTGAAGTGCAGATTACG-3′ |
| pks3 RT F | 5′-GGCTGAGATTGACACTGAAC-3′ |
| pks3 RT R | 5′-TACCCGACATTCCATACGAG-3′ |
| papA1 RT F | 5′-ATCCGCTAAGTACGATGGTC-3′ |
| papA1 RT R | 5′-ACCGATCTAATTGCCCTCAG-3′ |
| Rv3616c RT F | 5′-GAGCAGAGCGTTCATCATCG-3′ |
| Rv3616c RTR | 5′-CGAACCTAACCAGCCATCAC-3′ |
| Rv2390c RT F | 5′-CCGGCGAGTTCAAAGATAAG-3′ |
| Rv2390c RT R | 5′-TGAACATCAGGACCACTACC-3′ |
| 16S rRNA RT F | 5′-GCCGTAAACGGTGGGTACTA-3′ |
| 16S rRNA RT R | 5′-TGCATGTCAAACCCAGGTAA-3′ |
| MtbΔwhiB3 clone check F1 | 5′-GTGGCATCGAGAGCCTCTTCAC-3′ |
| MtbΔwhiB3 clone check R1 | 5′-GACGCGTTGATCCTGCTGCAC-3′ |
Measurement of Intramycobacterial EMSH in Vitro and during Infection
For intramycobacterial EMSH determination, various strains expressing Mrx1-roGFP2 were cultured and exposed to pH stress as indicated above. At the indicated time points, cells were treated with 10 mm N-ethylmaleimide for 5 min at room temperature and fixed with 4% paraformaldehyde for 15 min at room temperature. After washing three times with 1× phosphate-buffered saline (PBS), bacilli were analyzed using a FACS Verse Flow cytometer (BD Biosciences). The biosensor response was measured by analyzing the ratio at a fixed emission (510 nm) after excitation at 405 and 488 nm. Data were analyzed using FACSuite software. Intramycobacterial EMSH was measured using the Nernst equation as described earlier (15).
For measuring intramycobacterial EMSH during infection, PMA-differentiated THP-1 cells were infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains expressing Mrx1-roGFP2 at a multiplicity of infection (MOI) of 10. Infected macrophages were treated with N-ethylmaleimide/paraformaldehyde, washed with 1× PBS, and analyzed by flow cytometry as described previously (15). In the case of bafilomycin A1 (BafA1) treatment, THP-1 cells were treated with 10 nm BafA1 (Invivogen) or DMSO (vehicle control) 1 h before infection and processed for infection and sample preparation as described above.
Survival of M. tuberculosis Strains in Macrophages
PMA-differentiated THP-1 monocytes and IFNγ + LPS-activated RAW 264.7 macrophages were infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains at an MOI of 2 for 4 h, followed by treatment with 200 μg/ml amikacin to remove extracellular bacteria. For BafA1 experiments, THP-1 cells were treated with 10 nm BafA1 or DMSO (vehicle control) 1 h prior to infection. Cells were maintained in BafA1 throughout the experiment. After infection, cells were washed thoroughly with warm RPMI medium and resuspended in 10% RPMI medium containing BafA1 or DMSO as per requirements. Samples were collected at the indicated time points, lysed using 0.06% SDS-7H9, serially diluted in 7H9, plated on OADC-7H11 agar medium, and incubated at 37 °C incubator. Colonies were counted 3 weeks after plating.
Isolation of WT M. tuberculosis Surface-exposed Polyketide Lipids
50 ml of WT M. tuberculosis was grown in a 37 °C shaker incubator until mid-log phase (A600 nm = 0.5–0.8). Surface-exposed extractable total lipids were isolated as described previously (17). For lipid complementation assays, 100 μg/ml lipids were coated on the coverslips, and THP-1 cells were seeded, PMA-differentiated, and infected with MtbΔwhiB3 as done above.
Confocal Microscopy
Logarithmically grown M. tuberculosis strains were stained with FITC as described earlier (18). PMA-differentiated THP-1 cells (0.25 × 106) were seeded, and infection was done with WT M. tuberculosis, MtbΔwhiB3, whiB3-comp strains at an MOI of 10 as described elsewhere (19). 1 h prior to the time point, the medium of the cells was replaced with complete medium containing 100 nm Lysotracker® Red DND-99. Staining for CD63 and V-ATPase was started with fixative treatment (4% paraformaldehyde in 1× PBS) for 15 min, followed by permeabilization (0.2% Triton X-100 in 1× PBS) and blocking (3% BSA and 0.5% Tween 80 in 1× PBS). Samples were stained with primary antibodies (anti-CD63 and anti-V-ATPase) followed by secondary antibodies, followed by mounting using ProLong® antifade reagent (Thermo Fisher Scientific). The stained cells were visualized under a Leica TCS SP5 confocal microscope. z stacks were taken, collapsed into a two-dimensional image, and analyzed by LAS AF version 2.6.0, build 7266. All of the z stacks were taken at 63× oil immersion objective (with zoom). A minimum of five fields were captured, and at least 50 macrophages were analyzed for all of the phagosomal markers, accounting for an approximate analysis of ∼100–200 bacteria/well. For each experimental group, a minimum of three replicates were scored.
THP-1 Microarrays
The infection of THP-1 monocytic cells, followed by RNA isolation, sample processing, and hybridization (Illumina Human-Ht-12 BeadChip), was performed as described earlier (19). Array data processing was performed on Illumina Bead Studio software. Expression analysis was done using a volcano plot-based approach using -fold change >1.5 and p value >0.05 as cut-off. GO-Elite (version 1.2.5) was used for significant biological analysis of differentially expressed genes with z-score > 1.96 and p value < 0.05 set as cut-offs. The normalized data from the microarray gene expression experiment can be queried via Gene Expression Omnibus series accession number GSE65714.
Aerosol Infection of Guinea Pigs
Outbred Hartley guinea pigs (∼300–400-g body weight) were given a low dose of logarithmic phase-grown cultures of WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp, using a Madison chamber aerosol generation instrument calibrated to deliver 50–100 cfu/animal lung. Animals were sacrificed (n = 5) at 1, 30, and 60 days postinfection for determination of organ bacterial burden and histopathology analysis. Histopathology analysis was performed as described previously (14). A blinded examination of at least three serial sections from each guinea pig was carried out.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism software, and values were presented as mean ± S.D. The statistical significance of the differences between experimental groups was determined by two-tailed, unpaired Student's t test. Differences with a p value of <0.05 were considered significant.
Results
M. tuberculosis WhiB3 Regulates Survival and Gene Expression in Response to Acidic pH
Recent studies indicated that intraphagosomal pH might be one of the earliest cues to which M. tuberculosis responds and realigns its transcriptional programming (12). Microarray studies have revealed that the expression of whiB3 was induced early in macrophages in a pH-dependent manner (12). We also found an ∼2-, 4-, and 35-fold increased expression of whiB3 as compared with 16S rRNA at pH 6.2, 5.5, and 4.5, respectively.
Given the sustained induction of whiB3 in response to acidic pH, we reasoned that WhiB3 might play an important role in tolerating acid stress. We generated an unmarked strain of M. tuberculosis lacking the entire open reading frame (ORF) of whiB3, and putative MtbΔwhiB3 clones were verified by PCR (Fig. 1, A–C). We monitored the survival of WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp in 7H9-tyloxapol medium adjusted to pH 6.6 (normal 7H9), 5.5, and 4.5. At day 4 post-treatment, survival at various pH stress conditions was monitored by enumerating cfu. We observed that MtbΔwhiB3 survived to a level comparable with WT M. tuberculosis at pH 6.6 and 5.5 (Fig. 1D). However, it displayed an ∼55-fold reduction in its survival at pH 4.5 as compared with WT M. tuberculosis (p = 0.007; Fig. 1D). Stable expression of whiB3 in MtbΔwhiB3 resulted in a significant complementation of this survival defect (Fig. 1D). These observations confirmed that not only is whiB3 expression induced by acidic pH; it is also required for growth in acidic environments in vitro.
FIGURE 1.

WhiB3 modulates survival of M. tuberculosis at acidic pH. A, schematic representation of disrupted whiB3 allele (Rv 3416) in the genome of M. tuberculosis. The entire whiB3 ORF was replaced by 1-kb right and 1-kb left flanking regions of whiB3 along with the loxP-hyg-gfp-loxP cassette. B, genomic DNA was isolated from putative KanSHygRGFP+ MtbΔwhiB3 colonies (26, 30, and 34), and replacement of whiB3 allele with Hyg-GFP cassette was confirmed using PCR with F1 and R1 primers (Table 1). An increase in amplicon size from 2.4 to 4.5 kb due to insertion of the loxP-hyg-gfp-loxP cassette was observed in case of mutant clones, confirming the double crossover event. C, RNA was isolated from logarithmically grown WT M. tuberculosis and the putative MtbΔwhiB3 clones. qRT-PCR for whiB3 was done using whiB3-specific oligonucleotides (Table 1), and Ct values were plotted to assess the expression. D, for assessing survival of M. tuberculosis strains at acidic pH, WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains were grown in 7H9-tyloxapol medium at the indicated pH values for 4 days at 37 °C, and survival was assessed by enumerating cfu. Data shown are the average of two independent experiments done in triplicate. Error bars, S.D. **, p < 0.01 (as compared with WT M. tuberculosis); +, p < 0.05 (as compared with whiB3-comp). RFU, relative fluorescence units.
Next, we sought to determine the role of WhiB3 in controlling pH-specific gene expression. We minimized any influence of pH-induced cell death on gene expression by performing microarrays at an early time point (2 h) after exposure to pH 4.5. Expression data revealed differential regulation of several genes involved in secretion, central metabolism/oxidative phosphorylation, lipid metabolism, amino acid metabolism, cell wall biosynthesis/membrane transporters, and gene regulation at pH 4.5 as compared with pH 6.6 (Fig. 2, A and B, and supplemental Table S1).
FIGURE 2.

WhiB3 regulates gene expression in response to acidic pH in vitro. WT M. tuberculosis and MtbΔwhiB3 strains were grown to an A600 nm of ∼0.3 and exposed to acidic pH of 4.5 for 2 h at 37 °C. Total RNA was isolated and subjected to microarray analysis as described under “Experimental Procedures.” A, genes with >2-fold (p < 0.05) up- or down- regulation by acid stress relative to neutral pH were classified in 14 classes based on the annotation given in TubercuList. The pie chart represents the relative fraction of various pathways affected by acid stress in WT M. tuberculosis. B, heat map comparing WT M. tuberculosis and MtbΔwhiB3 genes induced or repressed significantly (>2-fold, p < 0.05) at pH 4.5 relative to neutral pH. C, heat map showing the overlap of genes differentially regulated in WT M. tuberculosis and MtbΔwhiB3 by in vitro acid stress (pH 4.5) with concanamycin A-sensitive phagosome-induced genes (12). Highlighted areas represent genes affected by phagosomal acidification in WT M. tuberculosis in a WhiB3-dependent manner at pH 4.5 in vitro (1.5-fold up- and down-regulated, p < 0.05).
Surprisingly, genes directly implicated in mitigating redox stress in M. tuberculosis were also influenced by acid stress. For example, expression of major antioxidant genes, including thioredoxins (trxB1, trxB2, and trxC), superoxide dismutase (sodA), MSH synthesis (mshB and mca), and rubredoxin (rubA and rubB), was elevated by acidic conditions (supplemental Table S1). Genes associated with biosynthesis of redox-active amino acids (cysteine and methionine), DNA repair (recR, dnaK, dnaJ, and ogt), and NAD+/NADH balance (ndh) were also up-regulated (Fig. 2B and supplemental Table S1). Noticeably, the expression of whiB3 was induced to a higher degree than other transcriptional regulators (supplemental Table S1). To understand the physiological basis of our findings, we performed a comparative gene expression analysis at pH 4.5 in vitro with the transcriptional changes in WT M. tuberculosis in response to early phagosomal acidity (12). We observed that of 22 genes that were induced by early phagosomal acidity (12), 17 were also induced by pH 4.5 (≥2-fold, p < 0.05) (Fig. 2C).
Subsequently, we examined the role of WhiB3 in regulating pH-induced changes in gene expression. In MtbΔwhiB3, the expression of 70 pH-induced and 27 pH-repressed genes showed 2-fold (p < 0.05) differential expression as compared with WT M. tuberculosis (supplemental Table S1). Several genes exhibited pH-specific induction only in WT M. tuberculosis, whereas they were constitutively expressed in MtbΔwhiB3. This includes genes involved in the biosynthesis of complex polyketide lipids (sulfolipid-1 (pks2), polyacyltrehalose/diacyltrehalose (pks3-pks4)), and cysteine metabolism (cysW, cysN, cysA1, and metZ) (Fig. 2B and supplemental Table S1). Genes involved in amino acid biosynthesis (metH, metK, and sahH), ESX-1 secretion (Rv3614c-Rv3616c), MSH antioxidant system (mca and mtr), nitrite transport (narK1), and leucine biosynthesis (leuB) were up-regulated significantly more in WT M. tuberculosis as compared with MtbΔwhiB3 at pH 4.5 (Fig. 2B and supplemental Table S1). A large subset of genes was up-regulated to a higher degree in MtbΔwhiB3 as compared with WT M. tuberculosis at acidic pH, indicating the role of WhiB3 in fine tuning the expression of pH-inducible genes. This includes the PE-PPE family (PE-24, PE-8, PE-32, PPE-65, and PPE-31), ribosomal proteins (rpmH, rplU, and rplY), and transcriptional regulators (whiB7, Rv0827c, and Rv3183) (supplemental Table S1). Because induction of whiB3 was responsive to early phagosomal acidity (12), we checked the expression status of other phagosomal pH-responsive genes in MtbΔwhiB3. We discovered that of 22 phagosomal pH-responsive genes, expression of 16 was controlled by WhiB3 (Fig. 2C, ≥1.5-fold, p ≤0.05). Last, microarray data were validated by measuring the expression of a selected set of pH- and whiB3-dependent genes by qRT-PCR (Table 2). Taken together, our results implicate WhiB3 in controlling the survival and expression of genes involved in altering cell wall lipid composition, secretion, and redox balance in response to acid stress.
TABLE 2.
qRT-PCR analysis of a select set of pH-specific genes regulated by WhiB3 and mycothiol under acidic stress (pH 4.5)
Expression is compared within various treatment groups, and data are represented as -fold change in gene expression ± S.D. ND, not determined.
| Gene | WT M. tuberculosis pH 4.5/WT M. tuberculosis pH 6.6 | MtbΔwhiB3 pH 4.5/WT Mtb pH 6.6 | WT M. tuberculosis pH 4.5/MtbΔwhiB3 pH 4.5 | WT M. tuberculosis pH 7.0/MtbΔmshA pH 7.0 | MtbΔmshA pH 4.5/MtbΔmshA pH 7.0 |
|---|---|---|---|---|---|
| whiB3 | 26 ± 1.6 | ND | ND | −1.1 ± 0.47 | −37.63 ± 13.11 |
| pks2 | 12 ± 0.8 | 1.5 ± 0.24 | 2 ± 0.31 | −1.66 ± 0.805 | −4.9 ± 1.91 |
| pks3 | 5.17 ± 2.8 | 0.55±.0.16 | 9.3 ± 0.62 | −1.15 ± 0.17 | −5.11 ± 2.46 |
| papA1 | 4.6 ± 2.25 | 0.71 ± 0.07 | 6.1 ± 0.53 | −0.2 ± 1.93 | −12.43 ± 4.76 |
| Rv3616c | 14.8 ± 4.1 | 2.3 ± 1 | 8 ± 0.52 | −1.46 ± 2.48 | 1.96 ± 0.26 |
| Rv2390c | 54 ± 26 | 14.6 ± 3.1 | 3.7 ± 0.53 | 1.19 ± 0.36 | 3.03 ± 0.75 |
Acidic pH Induces Dynamic Changes in EMSH of M. tuberculosis in a WhiB3-dependent Manner
Because WhiB3 is believed to serve as an intracellular redox sensor in M. tuberculosis, we hypothesized that it might regulate gene expression by responding to pH-induced changes in intramycobacterial redox physiology. Therefore, we precisely measured the dynamic changes in EMSH of WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains in response to a range of pH conditionsin vitro. We exploited a highly sensitive, specific, and non-invasive biosensor of EMSH (Mrx1-roGFP2) in mycobacteria (15). MSH is the most abundant low molecular weight thiol produced by mycobacteria (15). Therefore, EMSH measurement provides a reliable and sensitive numerical evaluation of cytoplasmic redox state of mycobacteria. The biosensor shows an increase in the fluorescence excitation ratio at 405/488 nm upon oxidative stress, whereas a ratiometric decrease is associated with reductive stress (15).
WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp expressing Mrx1-roGFP2 were exposed to a gradient of pH conditions (pH 7.0, 6.2, 5.5, and 4.5), and the ratiometric response was measured by flow cytometry at various time points. By fitting ratiometric intensities into the Nernst equation, we precisely measured the EMSH of M. tuberculosis strains at various pH conditions (see “Experimental Procedures”). We found the steady-state EMSH of WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains at neutral pH to be approximately −275 mV (Fig. 3A). The comparable EMSH of MtbΔwhiB3 and WT M. tuberculosis indicates that WhiB3 is not required for maintaining ambient EMSH of M. tuberculosis at neutral pH. Interestingly, 24-h exposure of WT M. tuberculosis to either pH 6.2, pH 5.5, or pH 4.5 resulted in a significant decrease in intramycobacterial EMSH (approximately −305 ± 0.7 mV), indicating that a transition from neutral to either milder or harsher acidic pH conditions uniformly induces reductive EMSH in M. tuberculosis (Fig. 3, B–D). Moreover, pH-exposed WT M. tuberculosis largely maintained EMSH reduced throughout the course of the experiment. We noted a very modest recovery from reductive EMSH (i.e. approximately −295 mV) at pH 6.2 and 5.5 at later time points, whereas no such effect was observed at pH 4.5 (Fig. 3, B–D). Importantly, whereas changes in intramycobacterial EMSH for MtbΔwhiB3 mostly followed the WT M. tuberculosis pattern at pH 6.2 (Fig. 3B), distinct redox deviations were observed at pH 5.5 and 4.5. For example, at pH 5.5, MtbΔwhiB3 displayed a relatively lesser decrease in intramycobacterial EMSH (−297 ± 0.7 mV) at 24 h followed by a slightly better recovery at 48–72 h (approximately −287 ± 0.7 mV) as compared with WT M. tuberculosis (Fig. 3C). Noticeably, exposure of MtbΔwhiB3 to pH 4.5 displayed only a marginal decrease in EMSH (−287 ± 1.5 mV) at 24 h, followed by a significant increase in intramycobacterial EMSH at 48 h (−260.5 ± 3.5 mV) and at 72 h (−266 ± 0.7 mV) as compared with WT M. tuberculosis, suggesting an overall oxidative shift in EMSH of MtbΔwhiB3 (Fig. 3D). The complemented strain showed changes in intramycobacterial EMSH that were comparable with WT M. tuberculosis (Fig. 3, A–D). These results indicate that acidic pH perturbs the redox physiology of M. tuberculosis by inducing a reductive shift in intramycobacterial EMSH and that the loss of WhiB3 impaired the ability of M. tuberculosis to orchestrate an efficient and dynamic MSH-specific reductive response upon acid stress.
FIGURE 3.

WhiB3 regulates dynamic changes in intramycobacterial EMSH of M. tuberculosis in response to acid stress. WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains expressing Mrx1-roGFP2 were grown in 7H9-tyloxapol medium at a pH of 7.0 (A), 6.2 (B), 5.5 (C), and 4.5 (D). At the indicated time points, EMSH of 30,000 bacterial cells was measured by flow cytometry as described under “Experimental Procedures.” At 72 h postincubation, 500 μm CCCP was added to dissipate the pH gradient, and intramycobacterial EMSH was measured 4 h post-CCCP treatment (i.e. 76 h). **, p < 0.01; ***, p < 0.005 (as compared with WT M. tuberculosis); +, p < 0.05; ++, p < 0.01; +++, p < 0.005 (as compared with whiB3-comp). Shown is the viability of WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp upon CCCP treatment at pH 7.0 (E) or pH 4.5 (F and G) for 4 h, as determined by enumerating cfu. Data shown in each panel are the result of at least two independent experiments performed in triplicate. Error bars, S.D. **, p < 0.01; ***, p < 0.005.
The above results point toward a pH-mediated reductive shift in EMSH of M. tuberculosis that might act as a signal for WhiB3 to regulate gene expression. We reasoned that disruption of the MSH reductive pathway would provide an ideal opportunity to study, in parallel, the effect of reductive EMSH on pH-specific gene expression. Therefore, we expressed Mrx1-roGFP2 in an MSH-deficient M. tuberculosis strain (MtbΔmshA) and found that EMSH of the strain remained oxidized (∼−240 mV) at various pH conditions (7.0, 6.2, 5.5, and 4.5). Next, we analyzed the expression of a set of pH-inducible genes in a MSH-deficient M. tuberculosis strain (MtbΔmshA) by qRT-PCR. At pH 7.0, expression of pH-inducible genes in MtbΔmshA is comparable with WT M. tuberculosis (Table 2). In contrast, expression of pH-inducible genes did not show any up-regulation in MtbΔmshA at pH 4.5 (Table 2). More importantly, the expression of whiB3 was ∼37-fold down-regulated in MtbΔmshA at pH 4.5 (Table 2). These results, along with our microarray data showing whiB3-dependent expression of MSH biosynthetic genes, suggest that MSH and WhiB3 are the components of a regulatory circuit mediating gene expression upon acid stress.
Dissipation of pH Gradient Perturbs pH-specific Induction of Reductive EMSH in M. tuberculosis
Excess increase in intracellular acidity can damage DNA, proteins, and lipids to exert bacterial killing. However, whether acid is the main effector of bacterial killing in response to pH stress is not clear. Because M. tuberculosis maintains cytoplasmic pH homeostasis even under severe pH stress, how an increase in internal acidity will affect mycobacterial redox physiology and survival has not been studied. To examine the influence of elevated cytoplasmic acidity on intramycobacterial redox potential, we cultured M. tuberculosis strains at various pH levels for 72 h, as described above, and disrupted pH homeostasis by discharging the pH gradient (ΔpH) using a well known protonophore, CCCP. Treatment with 500 μm CCCP for 4 h is sufficient to permeate the cell wall of mycobacteria and equilibrates cytoplasmic pH with the external pH (20). We followed intramycobacterial EMSH and viability of M. tuberculosis strains at 4 h post-treatment with 500 μm CCCP. Expectedly, at pH 7.0, treatment with CCCP has no effect on either intramycobacterial EMSH or viability of M. tuberculosis (Fig. 3, A and E). At pH 6.2, CCCP treatment triggers a moderate recovery from reductive shift in EMSH of WT M. tuberculosis (−284 mV), MtbΔwhiB3 (−288 mV), and whiB3-comp (−280 mV) strains (Fig. 3B). However, under these experimental conditions, viability of M. tuberculosis strains was not significantly compromised (data not shown).Consistent with this pattern, CCCP treatment of M. tuberculosis strains at pH 5.5 and 4.5 completely reversed the pH-specific increase in reductive EMSH, as indicated by an increased shift in intramycobacterial EMSH toward oxidizing (ranging from −260 to −250 mV) (Fig. 3, C and D). Although an increase in oxidative stress upon CCCP treatment does not significantly influence the viability of M. tuberculosis strains at pH 5.5 (data not shown), an ∼3–4 fold reduction in cfu was observed at pH 4.5 (Fig. 3, F and G). Importantly, because a CCCP-mediated skew toward EMSH-oxidized was activated at moderate pH values of 5.5, at which ionophore showed no mycobactericidal effect, our results indicate that dissipation of ΔpH and consequent oxidative shift in intramycobacterial EMSH precedes bacterial death. Together, these results suggest that the maintenance of pH homeostasis and the induction of reductive EMSH are effective and overlapping mycobacterial strategies to avoid oxidative stress-mediated death caused by increased internal acidity.
WhiB3 Regulates Intramycobacterial EMSH and Survival during Infection
We next determined whether WhiB3 regulates intramycobacterial EMSH and survival in the natural context of infection. To investigate this issue, we infected THP-1 macrophages with M. tuberculosis strains expressing Mrx1-roGFP2 at an MOI of 10 and monitored intramycobacterial EMSH and survival, as described previously (15).
As expected, M. tuberculosis strains displayed subpopulations with oxidized (−240 ± 3 mV), reduced (−300 ± 6 mV), and basal (−275 ± 5 mV) EMSH inside THP-1 cells (15). The WT M. tuberculosis displayed a gradual increase in population with EMSH reduced (∼30–65%) at initial time points (0–24 h postinfection), followed by an increase in EMSH-oxidized population (∼20%) at the intermediate period (48 h postinfection) and recovery from oxidative stress at 72 h postinfection. (Fig. 4A). In contrast to what was observed with whiB3-sufficient strains (WT M. tuberculosis and whiB3-comp), MtbΔwhiB3 showed negligible proportions of EMSH-reduced bacteria and a greater fraction of bacteria with EMSH oxidized, at each time point examined (Fig. 4, A–C). The defective ability of MtbΔwhiB3 to maintain reductive EMSH correlated with a significant growth defect in its survival inside THP-1 macrophages as compared with WT M. tuberculosis and whiB3-comp (Fig. 4D).
FIGURE 4.

Regulation of intramycobacterial EMSH and survival of M. tuberculosis in a WhiB3-dependent manner inside macrophages. THP-1 cells were infected with Mrx1-roGFP2 expressing WT M. tuberculosis (A), MtbΔwhiB3 (B), and whiB3-comp (C) strains at an MOI of 10. At the indicated time points, ∼30,000 infected macrophages were analyzed by flow cytometry, intramycobacterial EMSH was measured, and the percentage of bacilli in each subpopulation was determined as described earlier (15). The percentage of bacilli in each subpopulation (EMSH-oxidized, EMSH-basal, and EMSH-reduced) was plotted as a bar graph as described earlier (15). The 0 h time point refers to time immediately after infection with M. tuberculosis strains for 6 h (4 h of internalization followed by 2 h of amikacin treatment to remove any extracellular bacilli). D, THP-1 macrophages were infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains as described above, and intramacrophage survival was monitored by enumerating cfu at the indicated time points. IFN-γ- and LPS-treated (activated) RAW 264.7 macrophages were infected with Mrx1-roGFP2 expressing WT M. tuberculosis (E), MtbΔwhiB3 (F), and whiB3-comp strains (MOI of 10) (G). At the indicated time points, intramycobacterial EMSH of M. tuberculosis subpopulations was measured as described for A–C. H, activated RAW 264.7 macrophages were infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains (MOI of 2), and intramacrophage survival was monitored as described for D. The data in all panels are representative of three independent experiments performed in quadruplicate. Error bars, S.D. *, p < 0.05 (EMSH-oxidized subpopulation of MtbΔwhiB3 as compared with WT M. tuberculosis); +, p < 0.05 (EMSH-oxidized subpopulation of MtbΔwhiB3 as compared with whiB3 Comp) in experiments related to measurements of EMSH. **, p < 0.01; ***, p < 0.005 (as compared with WT M. tuberculosis); ++, p < 0.01; +++, p < 0.005 (as compared with whiB3-comp) in intramacrophage survival experiments.
Activated murine macrophages are known to control mycobacterial survival by generating excessive acid, ROS, and RNS stress (1). To investigate the role of WhiB3 in controlling EMSH of M. tuberculosis upon stimulation of antimycobacterial stresses, we infected IFN-γ- and LPS-activated RAW264.7 cells with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp at an MOI of 10 and measured the intramycobacterial EMSH. In a parallel experiment, intramacrophage survival of M. tuberculosis strains was also compared upon immune activation. We observed a substantial shift in subpopulation with EMSH oxidized in all three strains (Fig. 4, E–G). However, MtbΔwhiB3 displayed the highest proportion of cells with EMSH oxidized (Fig. 4F). Inside immune activated macrophages, WT M. tuberculosis showed a decline in survival at 48 h postinfection, followed by a significant recovery at 96 h postinfection (Fig. 4H). However, although initial killing of MtbΔwhiB3 was comparable with WT M. tuberculosis, the mutant showed a 2-fold difference in survival at 96 h postinfection (p = 0.0018) (Fig. 4H). The whiB3-comp strain grew equivalent to WT M. tuberculosis levels inside activated macrophages (Fig. 4H).
An oxidative shift in the EMSH of WT M. tuberculosis inside activated macrophages, where phagolysosomal pH drops to 4.5, is in contrast with the reductive shift in intramycobacterial EMSH at pH 4.5 in 7H9 culture medium. However, these counterintuitive findings can be reconciled by the synergistic effect of vacuolar acidification with host ROS and RNS production. In particular, low pH of phagolysosomes allows dismutation of nitrous acid (generated via autoxidation of NO) to generate highly toxic nitrogen dioxide (21). Hence, the increase in EMSH-oxidized subpopulations is most likely due to composite response of M. tuberculosis toward multiple stresses encountered inside activated macrophages. Altogether, using in vitro and macrophage-based assays, we demonstrated the role of WhiB3 in mounting an efficient antioxidant response to promote intramacrophage survival.
Effect of Phagosomal Acidification on Intramycobacterial EMSH and Survival
Because phagosomal pH synergizes with multiple intramacrophage cues, such as ROS and RNS, determination of the specific effect of acidic pH on mycobacterial physiology, gene expression, and survival remains challenging. This has been extremely difficult inside activated macrophages, where several mycobactericidal mechanisms (e.g. lysosomal hydrolases, ROS, and RNS) are likely to be pH-dependent (21). Therefore, to begin delineating the role of vacuolar acidification on intramycobacterial EMSH, we decided to examine the effect of limited acidification encountered inside THP-1 cells (22). To block phagosomal acidification, we treated THP-1 macrophages with a specific inhibitor of V-ATPase, BafA1, and then infected them with M. tuberculosis strains expressing Mrx1-roGFP2 at an MOI of 10. Treatment with 10 nm BafA1 is known to effectively block phagosomal acidification without affecting macrophage viability during infection with M. tuberculosis (23). We also observed no influence of 10 nm BafA1 on THP-1 viability (data not shown). Given that M. tuberculosis phagosomes acidify to pH 6.2–6.4 within minutes of infection (24), we measured intramycobacterial EMSH at initial time points (0 and 24 h postinfection).
In case of WT M. tuberculosis, BafA1 treatment significantly diminished the proportion of bacilli with EMSH reduced as compared with untreated macrophages at 0 and 24 h postinfection (Fig. 5, A and B). The consequent increase in M. tuberculosis subpopulations with basal EMSH (similar to 7H9, pH 7.0, grown bacteria) indicates that cells are experiencing nearly neutral pH in vacuoles upon BafA1 treatment (Fig. 5, A and B). Importantly, these results indicate that limited acidification encountered inside macrophages is sufficient to induce reductive shift and heterogeneity in EMSH of M. tuberculosis during infection. In contrast to WT M. tuberculosis, MtbΔwhiB3 showed no significant changes in subpopulations with basal, oxidized, and reduced EMSH at 0 and 24 h postinfection upon BafA1 treatment (Fig. 5, A and B). The whiB3-comp strain showed intramycobacterial EMSH changes comparable with WT M. tuberculosis upon BafA1 treatment (Fig. 5, A and B). These results suggest that the lack of WhiB3 impaired the ability of M. tuberculosis to dynamically modulate cytoplasmic EMSH in response to vacuolar pH.
FIGURE 5.

Phagosomal acidification modulates EMSH and survival of M. tuberculosis in a WhiB3-dependent manner. A and B, THP-1 cells treated with 10 nm BafA1 or vehicle control (DMSO) were infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains (MOI of 10), and at the indicated time points, 30,000 infected cells were analyzed by flow cytometry, and relative distribution of M. tuberculosis subpopulations with varying intramycobacterial EMSH was measured as described earlier (15). The data are represented as a percentage of bacilli in each subpopulation ± S.D. *, p < 0.05; ***, p < 0.005 (EMSH-reduced subpopulation in BafA1-untreated as compared with BafA1-treated cells). C, THP-1 cells treated with BafA1 or vehicle control (DMSO) were infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains, and intramacrophage survival at the indicated time points was monitored by enumerating cfu. The data in all panels are representative of three independent experiments performed in quadruplicate. Error bars, S.D. ***, p < 0.005 (BafA1-treated MtbΔwhiB3-infected THP-1 macrophages as compared with untreated control).
Next, we examined whether the inability of MtbΔwhiB3 to maintain mycothiol redox homeostasis in response to vacuolar acidification was the reason underlying the intramacrophage survival defect of the mutant. To do this, we monitored the survival of WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains inside untreated and BafA1-treated THP-1 cells by enumerating cfu at 48 and 96 h postinfection. In contrast to defective intramacrophage growth of MtbΔwhiB3 observed earlier, treatment with BafA1 completely rescued its survival to WT M. tuberculosis and whiB3-comp levels (Fig. 5C). Together, these results indicate the importance of mycothiol redox buffer and WhiB3 in responding to phagosomal acidification and maintaining intramacrophage survival of M. tuberculosis.
WhiB3 Is Required to Subvert Phagosomal Acidification
One of the main mechanisms exploited by M. tuberculosis to resist acid stress is by blocking the normal process of phagosomal maturation to acidified phagolysosomes (4). In this regard, we have shown that the majority of bacilli within acidified phagolysosomal fractions display oxidative EMSH (15). Because a relatively higher proportion of EMSH-oxidized subpopulations of MtbΔwhiB3 were detected inside THP-1 cells, we hypothesized that increased fusion of phagosomes containing MtbΔwhiB3 with acidified lysosomes may be one of the factors that underlies the observed redox variability and intramacrophage survival defect. We therefore sought to determine the acidification status of phagosomes containing MtbΔwhiB3. To do this, THP-1 macrophages were infected with M. tuberculosis strains labeled with FITC at an MOI of 10, and localization of M. tuberculosis bacilli was assessed using well established markers of phagosomal maturation. Localization of M. tuberculosis bacilli was examined using confocal microscopy. For measurements, a minimum of five fields/well were captured, and ∼100–200 bacterium-containing phagosomes were scored per well. For each test group, three replicate wells were scored per experiment.
Staining with Lysotracker revealed that WT M. tuberculosis largely remained in non-acidified phagosomes, with only 25 ± 4% of bacilli colocalized to acidified phagosomes at 24 h postinfection (Fig. 6A). In contrast, a significantly greater fraction of MtbΔwhiB3 (∼70%, p = 0.0026) was found in acidified phagosomes, and this phenotype was significantly reversed in the complemented strain (Fig. 6A). It has been shown that M. tuberculosis actively inhibits phagosome acidification by preventing recruitment and/or inducing degradation of a molecular proton motor, V-ATPase (24, 25). Therefore, we questioned whether the increased association of MtbΔwhiB3 with acidified phagosomes correlates with greater V-ATPase association. Infected THP-1 macrophages were immunostained for human V-ATPase, and colocalization was measured at 24 h postinfection. A significantly greater percentage of phagosomes containing MtbΔwhiB3 were found to be positive for V-ATPase (∼43 ± 10%) as compared with WT M. tuberculosis (∼11 ± 12%) or whiB3-comp (∼25 ± 10%) (Fig. 6B). Because phagosome acidification is a relatively early step in phagosomal maturation, we next analyzed the status of MtbΔwhiB3-containing phagosomes for the late endosome-lysosome fusion marker, CD63. It has been reported that WT M. tuberculosis prevents phagosomes from maturing into the CD63-positive state (26). Consistent with our earlier results, ∼53 ± 2% of MtbΔwhiB3-containing phagosomes acquired CD63 as compared with ∼8 ± 3% and 31 ± 4% in the case of WT M. tuberculosis and whiB3-comp strains, respectively (Fig. 6C). The observed differences in the phagosomes of MtbΔwhiB3 were confirmed by repeating experiments at least three times in triplicate.
FIGURE 6.

MtbΔwhiB3 is enriched in phagosomes positive for late endosomal/lysosomal markers inside THP-1 macrophages. THP-1 cells were infected with FITC-labeled WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains (MOI of 10). At 24 h postinfection, cells were independently stained with Lysotracker (A) or immunofluorescent antibodies for markers of phagosome maturation (i.e. V-ATPase (B) and CD63 (C), as described under “Experimental Procedures”). A representative result of three independent experiments is shown. In the images, green indicates FITC-labeled bacteria; red indicates Lysotracker or V-ATPase or CD63; differential interference contrast (DIC) indicates cell morphology; and yellow indicates merged images of two signals (Merge I, FITC/phagosomal markers) and three signals (Merge II, FITC/phagosomal markers/DIC). Scale bar, 5 μm. The bar graphs represent mean percentages of bacterium-containing phagosomes that stain positive for markers. Error bars, S.D. of three replicate wells, with each well having ∼100–200 phagosomes scored. *, p < 0.05; ***, p < 0.005 (as compared with WT M. tuberculosis); +, p < 0.05; +++, p < 0.005 (as compared with whiB3-comp).
Our data show that WhiB3 positively regulates the pH-specific expression of various genes involved in producing secretory proteins and lipids (e.g. sulfolipid-1 (pks2, papA1, and mmpL8), TDM (fabD, acpP, and kasA), and ESX-1 system (Rv3615c, Rv3870, and Rv3871)), which are well known to restrict phagosomal maturation (27–29). Hence, the inability of MtbΔwhiB3 to block phagosomal maturation could be a consequence of defective polyketide surface lipid anabolism. To investigate this possibility, total surface exposed lipids were extracted from WT M. tuberculosis as described earlier (17). Moreover, pretreatment of macrophages with the surface-exposed lipids of mycobacteria has been shown to modulate cellular processes, such as phagosomal maturation and autophagy (30, 31). Based on these studies, THP-1 cells were pretreated with the surface lipids and subsequently infected with WT M. tuberculosis or MtbΔwhiB3 at an MOI of 10. Infected macrophages were assessed for the colocalization of Lysotracker with MtbΔwhiB3 at 0 and 24 h postinfection. Assessment of more than 100 phagosomes revealed that pretreatment with the WT M. tuberculosis lipids resulted in a significant inhibition of Lysotracker staining of MtbΔwhiB3-containing phagosomes as compared with untreated controls at each time point tested (Fig. 7, A–C). Importantly, the percentage of Lysotracker-positive phagosomes containing MtbΔwhiB3 upon pretreatment with total lipids was comparable with WT M. tuberculosis at 0 h postinfection (Fig. 7, A and C). These results suggest that the altered composition of surface-associated polyketide lipids is likely to be one of the factors responsible for the defective ability of MtbΔwhiB3 to block phagosomal maturation. In sum, the data generated from macrophages and in vitro experiments clearly suggest that WhiB3 protects M. tuberculosis from acid stress by regulating gene expression, dynamic changes in EMSH of M. tuberculosis, and polyketide-mediated restriction of phagosomal acidification.
FIGURE 7.

Treatment with surface-exposed lipids derived from WT M. tuberculosis reduced the enrichment of MtbΔwhiB3 in acidified phagosomes. Total surface-exposed lipids were extracted from WT M. tuberculosis and coated onto coverslips, followed by seeding of THP-1 cells as described under “Experimental Procedures.” Lipid- pretreated macrophages were infected with FITC-labeled MtbΔwhiB3 (MOI of 10). As a control, untreated THP-1 cells were infected with FITC-labeled WT M. tuberculosis and MtbΔwhiB3 (MOI of 10). At 0 h (A) and 24 h (B) postinfection, cells were stained with Lysotracker and analyzed by confocal microscopy. Representative microscopy images from at least three independent experiments for each time point are shown. In the images, green indicates FITC-labeled bacteria, red indicates Lysotracker, differential interference contrast (DIC) indicates cell morphology, and yellow indicates merged images of two signals (Merge I, FITC/phagosomal markers) and three signals (Merge II, FITC/phagosomal markers/DIC). Scale bar, 5 μm. C, mean percentages of bacterium-containing phagosomes that stain positive for Lysotracker. Error bars, S.D. of three replicate wells, with each well having >100 phagosomes scored. *, p < 0.05; ***, p < 0.005 (as compared with WT M. tuberculosis); +, p < 0.05; ++, p < 0.01 (as compared with whiB3-comp).
WhiB3 Modulates the Expression of Host Innate Response Genes
We examined whether WhiB3-mediated regulation of bioactive lipids and secretory proteins modulates expression of host transcriptome. Using microarrays, we compared the expression of THP-1 cells infected with WT M. tuberculosis and MtbΔwhiB3 at various time points. We found that major innate immune mechanisms normally suppressed by pathogenic M. tuberculosis strains, such as phagosomal maturation/endocytosis, apoptosis, and TLR signaling, were up-regulated in MtbΔwhiB3 (1.5-fold, p < 0.05), whereas genes that negatively regulate autophagy, such as mTOR signaling, were largely down-regulated in the mutant as compared with WT M. tuberculosis (1.5-fold, p < 0.05) (Fig. 8 and supplemental Table S2). In agreement with the role of WhiB3 in responding to early increase in phagosomal acidity, we observed a significantly greater impact of WhiB3 loss on host transcriptome at an early time point (i.e. 12 h postinfection) (Fig. 8). Finally, using qRT-PCR, we validated microarray data by measuring the expression of a selected set of genes involved in phagosomal maturation in a whiB3-dependent manner (data not shown). Altogether, the data indicate that WhiB3 plays an important role in influencing expression of host-directed mechanisms associated with controlling intraphagosomal survival of M. tuberculosis.
FIGURE 8.
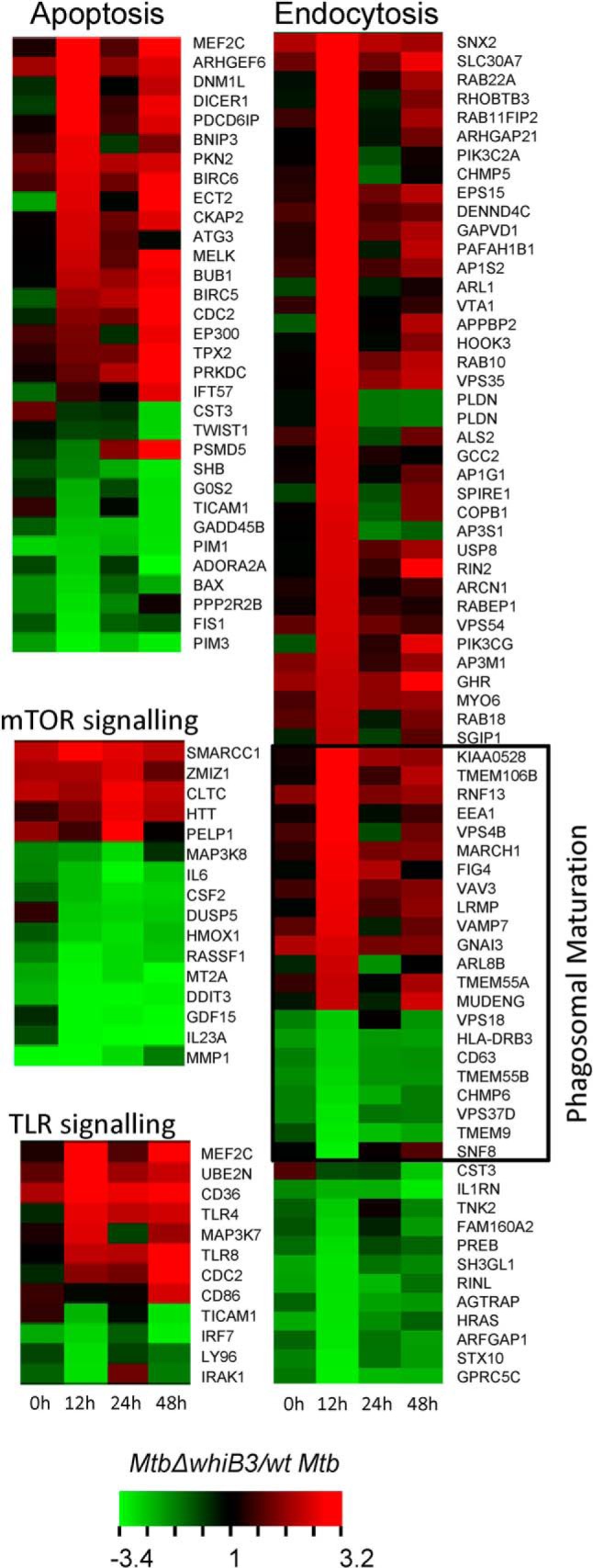
Regulation of host transcriptome by M. tuberculosis WhiB3. PMA-differentiated THP-1 cells were infected with WT M. tuberculosis and MtbΔwhiB3. At the indicated time points, total RNA from THP-1 was isolated and transcriptomic analysis was done, as described under “Experimental Procedures.” The figure shows a heat map of major pathways differentially regulated in MtbΔwhiB3 as compared with M. tuberculosis, >1.5-fold (p < 0.05).
WhiB3 Regulates Survival of M. tuberculosis in Vivo
Given that WhiB3 regulates pH-dependent modulation of expression and redox signaling, resulting in differential intracellular trafficking and survival inside macrophages, we hypothesize that WhiB3 plays an important role during M. tuberculosis infection. To examine this, we assessed the in vivo phenotype of MtbΔwhiB3 in guinea pigs. Aerosol infection of out-bred Hartley guinea pigs showed a clear growth attenuation of MtbΔwhiB3 as compared with WT M. tuberculosis in the lungs of animals. At day 1 postinfection, cfu analysis showed that nearly identical numbers of bacteria were implanted in the lungs of guinea pigs infected with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strain (Fig. 9A). At days 30 and 60 postinfection, the number of bacteria present in lungs of animals infected with MtbΔwhiB3 was ∼70 (p = 0.0073) and ∼200-fold (p = 0.0007) lower than in those infected with WT M. tuberculosis, respectively (Fig. 9A). Interestingly, in contrast to our lung data, bacterial numbers in the spleen at days 30 and 60 postinfection were comparable in WT M. tuberculosis and MtbΔwhiB3 (Fig. 9B). The attenuated virulence phenotype exhibited by MtbΔwhiB3 was partially restored in the animals infected with whiB3-comp strain (Fig. 9A). Histopathological analysis of lungs from infected animals further validated the requirement of WhiB3 during infection. The lungs of MtbΔwhiB3-infected animals showed less severe pathology as indicated by decreased tissue consolidation, smaller granulomas, and open alveolar space as compared with the WT M. tuberculosis and whiB3-comp strains (Fig. 9C).
FIGURE 9.

WhiB3 regulates survival of M. tuberculosis in vivo. Outbred Hartley guinea pigs (n = 5) were given an aerosol challenge with WT M. tuberculosis, MtbΔwhiB3, and whiB3-comp strains and assessed for survival in lungs (A) and spleen (B). Error bars, S.D. Statistical significance for the pulmonic and splenic bacterial load was obtained by comparing different strains: ***, p < 0.001 (as compared with WT M. tuberculosis); ++, p < 0.005 (as compared with whiB3Comp). C, hematoxylin and eosin-stained lung sections (30 and 60 days postinfection) from guinea pigs infected with WT M. tuberculosis, MtbΔwhiB3, and the whiB3-comp strains. The pathology sections show granulomas (G) and alveolar space (AS). All images were taken at ×40 magnification.
Discussion
In 1905, Metchnikoff (32) reported the presence of acidic milieu within the phagosomes of macrophages infected with pathogens. Despite this early observation, how M. tuberculosis bacilli respond, resist, and persist in response to a gradient of acidic pH during infection remains poorly characterized. Here, we identified acidic pH as a physiological stimulus to which WhiB3 regulates (i) gene expression, (ii) mycothiol redox homeostasis, (iii) phagosomal maturation, and (iv) virulence.
Our microarray data highlight the role of WhiB3 in regulating acid stress response in M. tuberculosis. Differential regulation of several genes involved in redox metabolism of M. tuberculosis in response to acid stress in a WhiB3-dependent manner and acute sensitivity displayed by MtbΔwhiB3 at pH 4.5 suggest that WhiB3 facilitates M. tuberculosis persistence in response to acid stress by maintaining intramycobacterial redox homeostasis. Until now, direct evidence linking acidic stress encountered in phagosomes to internal redox balance of M. tuberculosis was lacking. A recent in vitro study, using a genetically encoded redox biosensor (roGFP-R12), has demonstrated that the cytoplasmic redox state of M. tuberculosis shifts to reductive when bacilli are cultured under specific carbon sources at pH 5.5 (33). Because conventional roGFPs, such as roGFP-R12, predominantly interact with glutathione redox buffer (34, 35), which is absent in mycobacteria, the utility of roGFPs in M. tuberculosis is limited by unknown specificity and poor response to changes in redox potential (15). Moreover, reliable measurement range for roGFPs (i.e. between 10 and 90% of sensor oxidation) covers about ±30 mV from the standard midpoint potential (34). Therefore, roGFP-R12 with a less negative midpoint potential (−265 mV) cannot accurately measure a reductive shift in redox potential beyond −295 mV. In this context, Mrx1-roGFP2 with a midpoint potential of −280 mV allowed dynamic and precise imaging of the EMSH of M. tuberculosis, in response to both oxidative and reductive stresses, with high sensitivity and temporal resolution (15, 36). Using this bioprobe, we provide accurate numerical evidence that acidic pH promotes reductive shift in EMSH of M. tuberculosis in vitro and inside phagosomes in a WhiB3-dependent manner. The reductive shift in EMSH at initial phases of intramacrophage growth is consistent with the rapid drop in vacuolar pH within minutes of infection with M. tuberculosis (24), which also serves as an early cue to induce expression of genes linked to reductive stress (e.g. whiB3, whiB7, and dosR) (29, 37). More importantly, inhibition of vacuolar acidification resulted in both the loss of gene expression (28) and a substantial decrease in the M. tuberculosis subpopulations with EMSH reduced.
In contrast to WT M. tuberculosis, which preferentially resides in early endosomes, we observed that MtbΔwhiB3 mainly localized to acidified lysosomes and displayed a higher proportion of EMSH-oxidized bacilli. These findings support our earlier observations that lysosomes enrich EMSH-oxidized bacteria, whereas phagosomes with limited acidity (early endosomes) induce a reductive shift in EMSH of M. tuberculosis (15). Interestingly, whereas treatment with BafA1 prevented acidification of MtbΔwhiB3-containing phagosomes and reversed intramacrophage survival defect, the proportion of mutant bacilli with EMSH oxidized remained uninfluenced. One likely possibility is that the loss of WhiB3 compromised the ability of the mutant to respond to changes in the phagosomal environment via the mycothiol redox system. In line with this, several components of the mycothiol pathway, including MSH disulfide reductase involved in recycling MSSM to MSH, are down-regulated in MtbΔwhiB3 upon acid stress. Alternatively, MtbΔwhiB3 may exploit another antioxidant system, such as ergothionine (ERG), to respond to the intraphagosomal environment. A compensatory protective role of ERG has already been established in mycothiol-defective mycobacterial strains (38). Because Mrx1-roGFP2 does not respond to ERG (15), further work testing the role of ERG redox potential will allow more precise determination of relative contributions of ERG and MSH pathways in responding to the phagosomal milieu.
The acidic pH-induced reductive EMSH in WT M. tuberculosis most likely resulted from increased synthesis of MSH or a higher rate of MSSM reduction to MSH via the activity of NADPH-dependent MSH disulfide reductase. Expression data indicated a significant up-regulation of MSH-biosynthetic genes in response to acidic pH. Additionally, a decreased expression of respiratory genes involved in regenerating NAD+ cofactor (e.g. nuo operon), along with the up-regulation of fatty acid catabolic genes, which generate excessive NADH through β-oxidation, may further lead to accumulation of NADH/NADPH cofactors during acidity. Because excessive accumulation of NADH/NADPH is known to elevate endogenous ROS levels through autoxidation or via Fenton reaction (39, 40), a reductive shift in EMSH by NADPH-dependent conversion of MSSM to MSH via MSH disulfide reductase could be a mechanism to dispose of excess reductants. In this context, studies in M. tuberculosis have indicated only a marginal increase in the NADPH pool at acidic pH, whereas levels of cytoplasmic thiols, such as MSH and CoA-SH, were substantially elevated (33, 41), agreeing with our findings that the MSH pathway can function as a reductive sink to reduce toxicity associated with low pH. Further strengthening this connection is our finding showing the inability of MtbΔwhiB3 to maintain EMSH reduced, along with a previous report demonstrating massive accumulation of NADH/NADPH in MtbΔwhiB3 inside macrophages (17). The mycothiol redox system has recently been shown to interact with a major antioxidant enzyme; superoxide dismutase, to exert an efficient adaptive response during infection (42). In light of this, an early elevation in reductive capacity of mycothiol (EMSH reduced), in response to vacuolar pH, might be important to activate additional mechanisms to detoxify a range of hostile radicals that M. tuberculosis encounters later in the infection cycle (e.g. during immune activation). Altogether, these observations, along with the fact that external acidity does not increase H+ concentration inside M. tuberculosis (10), serve to implicate intrabacterial reductive EMSH as an internal physiological signal to which M. tuberculosis responds through WhiB3 to coordinate gene expression and survival. These findings were further strengthened by our data showing a complete loss of whiB3 induction along with other pH-responsive genes in an M. tuberculosis strain lacking mycothiol (MtbΔmshA) at acidic pH. Because WhiB3 is cytoplasmically located and implicated in responding to changes in intracellular redox conditions through redox-sensitive [4Fe-4S] cluster (13, 17), we hypothesize that the intramycobacterial reductive EMSH under acidic stress can promote accumulation and/or stabilization of the reduced form of the 4Fe-4S cluster ([4Fe-4S]1+-WhiB3), which directly or indirectly activates pH-responsive genes in M. tuberculosis. Alternatively, because the DNA binding activity of apo-WhiB3 (without an Fe-S cluster) is modulated by the redox state of its cysteine thiols (17), an interesting possibility is that the reversible S-mycothiolation of WhiB3 thiols in response to pH-mediated changes in EMSH may function as a redox-regulatory switch to modulate gene expression. The functional linkage between MSH and WhiB3, as revealed in this study, can now be exploited to understand the molecular mechanism(s) of how mycothiol exerts its influence on the redox behavior and gene regulatory properties of WhiB3.
We found that MtbΔwhiB3 has an impaired ability to arrest phagosomal maturation, which can be rescued by supplementation of surface-associated polyketide lipids from WT M. tuberculosis. These discoveries have significant implications in understanding mycobacterial pathogenesis, which suggests an intertwined association between immunomodulatory virulence factors and the core metabolic processes in M. tuberculosis. We propose that WhiB3-mediated synthesis of virulence factors, including secretory lipids and proteins (ESX-1 system), in response to intracellular redox changes associated with acidic pH will play a larger part, in both redox maintenance and in counteracting phagosomal acidification, to ensure long term persistence of M. tuberculosis (Fig. 10). Although our results indicate that WhiB3 is a major regulator of pH and redox homeostasis, other regulators, such as PhoP, that similarly modulate the expression of virulence factors to influence phagosomal maturation and intraphagosomal survival can provide overlapping control over pH and redox response in M. tuberculosis (33, 43). Finally, the physiological significance of our mechanistic findings comes from dramatic attenuation of MtbΔwhiB3 in the lungs of guinea pigs. Our results are in complete contrast with the previous findings on the MtbΔwhiB3 phenotype in vivo (11, 44). It is possible that hypoxic caseous TB granulomas formed in lungs of guinea pigs expose M. tuberculosis to a more acidic environment (45), which might have necessitated the need for a WhiB3-dependent redox sensing pathway for ensuring redox homeostasis, survival, and persistence of M. tuberculosis in vivo.
FIGURE 10.
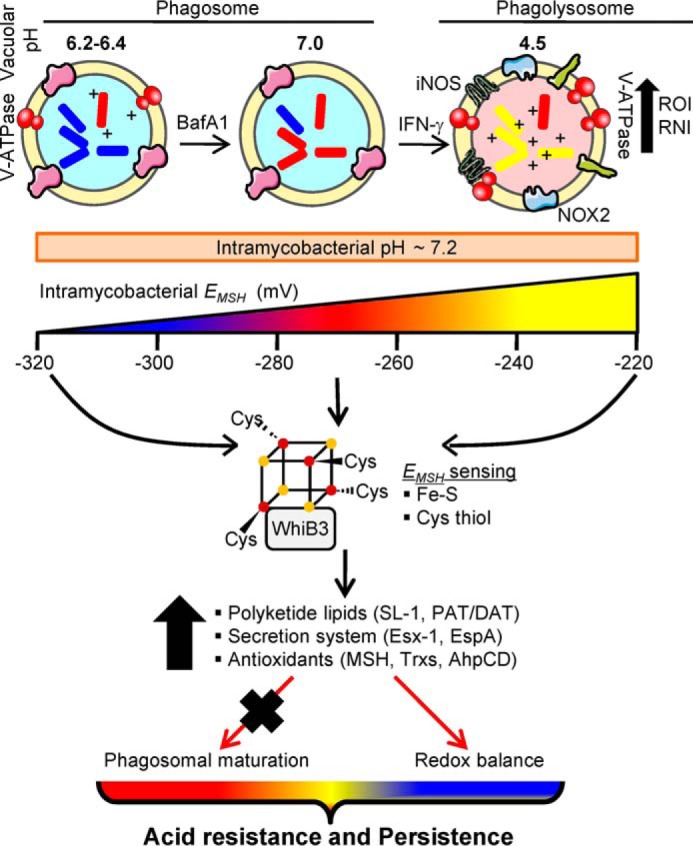
Model depicting the role of EMSH and WhiB3 in responding to acid stress during infection. In resting macrophages, M. tuberculosis impairs phagosome maturation and preferentially resides in a mildly acidic environment. Activation with IFN-γ induces phagosomal-lysosomal fusion to elevate the levels of proton (pH 4.5), reactive oxygen intermediates (ROI), and reactive nitrogen intermediates (RNI) in the microenvironment. Despite these changes in external pH, the internal pH of M. tuberculosis remains close to neutral (pH 7.2). Importantly, these variations in phagosomal pH induce dynamic changes in the EMSH of M. tuberculosis. Limited acidification inside resting macrophages induces a reductive shift in EMSH of M. tuberculosis (approximately −305 mV), whereas activation of macrophages induces oxidative shift (−240 mV). Pharmacological inhibition of phagosomal acidification by BafA1 neutralizes acidic pH to prevent reductive shift in EMSH of M. tuberculosis. M. tuberculosis responds to phagosomal acidification with the help of a putative redox-sensitive transcription factor, WhiB3. WhiB3 can sense changes in intrabacterial EMSH via its Fe-S cluster or cysteine thiols (S-mycothiolation) to modulate the expression of virulence genes involved in blocking phagosomal maturation (e.g. polyketides, secretory antigens) and redox homeostasis. Impaired ability of MtbΔwhiB3 to maintain mycothiol balance and block phagosomal maturation, along with the survival defect in vivo, suggests a central role of WhiB3 in regulating mycobacterial persistence in response to acid stress. The exact mechanisms by which WhiB3 senses pH-induced changes in internal EMSH via 4Fe-4S and/or cysteine thiols remain to be identified.
In summary, we have identified a new mechanism exploited by M. tuberculosis to respond to phagosomal pH. Our study provides a unique example of the tight connections forged between core redox machinery and virulence in mycobacterial pathogenesis. Furthermore, pH-induced redox signalling and its connection with gene expression and virulence may be relevant to other intracellular pathogens. For example, acidification is required for virulence expression in Salmonella (46), phagosomal escape of Listeria monocytogenes (47), and efficient replication of Legionella pneumophilla and Coxiella burnetti (48, 49). Thus, our findings have broad implications for several intracellular pathogens for which phagosomal pH plays a critical role in modulating virulence and long term persistence.
Author Contributions
M. M. and A. S. designed the research; M. M. and R. S. R. performed research; and M. M. and A. S. wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are thankful to the University of Delhi South Campus MicroArray Centre, New Delhi, for conducting microarray experiments. We also thank Department of Biotechnology, India, for providing the Tuberculosis Aerosol Challenge Facility at the International Centre for Genetic Engineering and Biotechnology. We thank Pankti Parikh for assistance in generating the MtbΔwhiB3 strain and MohamedHusen Munshi for lipid isolation from M. tuberculosis. We thank William R. Jacobs, Jr. (Albert Einstein College of Medicine) for the MtbΔmshA strain. We are also grateful to Manish Kumar and Dr. Sheetal Gandotra (Imaging Facility at CSIR-Institute of Genomics and Integrative Biology, New Delhi) for extensive technical help with all of the confocal microscopy experiments and analysis.
We thank the Department of Biotechnology (DBT), Ministry of Science and Technology, India BT/PR5020/MED/29/454/2012, BT/03/IYBA/2010 [AS]) and Wellcome-DBT India Alliance (500034/Z/09/Z [AS]) for financial support. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1 and S2.
- EMSH
- mycothiol redox potential
- MSH
- mycothiol
- BafA1
- bafilomycin A1
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- ERG
- ergothionine
- V-ATPase
- vacuolar H+-ATPase
- Mrx1
- mycoredoxin
- MOI
- multiplicity of infection
- PMA
- phorbol 12-myristate 13-acetate
- qRT-PCR
- quantitative RT-PCR
- roGFP
- redox-sensitive green fluorescent protein
- ROS
- reactive oxygen species
- RNS
- reactive nitrogen species.
References
- 1.Ehrt S., and Schnappinger D. (2009) Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schnappinger D., Ehrt S., Voskuil M. I., Liu Y., Mangan J. A., Monahan I. M., Dolganov G., Efron B., Butcher P. D., Nathan C., and Schoolnik G. K. (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohde K. H., Veiga D. F., Caldwell S., Balázsi G., and Russell D. G. (2012) Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 8, e1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong J. A., and Hart P. D. (1971) Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 134, 713–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hart P. D., and Armstrong J. A. (1974) Strain virulence and the lysosomal response in macrophages infected with Mycobacterium tuberculosis. Infect. Immun. 10, 742–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacMicking J. D., Taylor G. A., and McKinney J. D. (2003) Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science 302, 654–659 [DOI] [PubMed] [Google Scholar]
- 7.Schaible U. E., Sturgill-Koszycki S., Schlesinger P. H., and Russell D. G. (1998) Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160, 1290–1296 [PubMed] [Google Scholar]
- 8.Via L. E., Fratti R. A., McFalone M., Pagan-Ramos E., Deretic D., and Deretic V. (1998) Effects of cytokines on mycobacterial phagosome maturation. J. Cell Sci. 111, 897–905 [DOI] [PubMed] [Google Scholar]
- 9.Vandal O. H., Nathan C. F., and Ehrt S. (2009) Acid resistance in Mycobacterium tuberculosis. J. Bacteriol. 191, 4714–4721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandal O. H., Pierini L. M., Schnappinger D., Nathan C. F., and Ehrt S. (2008) A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 14, 849–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banaiee N., Jacobs W. R. Jr., and Ernst J. D. (2006) Regulation of Mycobacterium tuberculosis whiB3 in the mouse lung and macrophages. Infect. Immun. 74, 6449–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohde K. H., Abramovitch R. B., and Russell D. G. (2007) Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2, 352–364 [DOI] [PubMed] [Google Scholar]
- 13.Singh A., Guidry L., Narasimhulu K. V., Mai D., Trombley J., Redding K. E., Giles G. I., Lancaster J. R. Jr., and Steyn A. J. (2007) Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc. Natl. Acad. Sci. U.S.A. 104, 11562–11567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh A., Jain S., Gupta S., Das T., and Tyagi A. K. (2003) mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol. Lett. 227, 53–63 [DOI] [PubMed] [Google Scholar]
- 15.Bhaskar A., Chawla M., Mehta M., Parikh P., Chandra P., Bhave D., Kumar D., Carroll K. S., and Singh A. (2014) Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog. 10, e1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venkataraman B., Vasudevan M., and Gupta A. (2014) A new microarray platform for whole-genome expression profiling of Mycobacterium tuberculosis. J. Microbiol. Methods 97, 34–43 [DOI] [PubMed] [Google Scholar]
- 17.Singh A., Crossman D. K., Mai D., Guidry L., Voskuil M. I., Renfrow M. B., and Steyn A. J. (2009) Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 5, e1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hostetter J. M., Steadham E. M., Haynes J. S., Bailey T. B., and Cheville N. F. (2002) Cytokine effects on maturation of the phagosomes containing Mycobacteria avium subspecies paratuberculosis in J774 cells. FEMS Immunol. Med. Microbiol. 34, 127–134 [DOI] [PubMed] [Google Scholar]
- 19.Karim A. F., Chandra P., Chopra A., Siddiqui Z., Bhaskar A., Singh A., and Kumar D. (2011) Express path analysis identifies a tyrosine kinase Src-centric network regulating divergent host responses to Mycobacterium tuberculosis infection. J. Biol. Chem. 286, 40307–40319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao M., Streur T. L., Aldwell F. E., and Cook G. M. (2001) Intracellular pH regulation by Mycobacterium smegmatis and Mycobacterium bovis BCG. Microbiology 147, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 21.MacMicking J., Xie Q. W., and Nathan C. (1997) Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 [DOI] [PubMed] [Google Scholar]
- 22.Wong D., Bach H., Sun J., Hmama Z., and Av-Gay Y. (2011) Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc. Natl. Acad. Sci. U.S.A. 108, 19371–19376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan J. T., Young E. F., McCann J. R., and Braunstein M. (2012) The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect. Immun. 80, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturgill-Koszycki S., Schlesinger P. H., Chakraborty P., Haddix P. L., Collins H. L., Fok A. K., Allen R. D., Gluck S. L., Heuser J., and Russell D. G. (1994) Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681 [DOI] [PubMed] [Google Scholar]
- 25.Singh C. R., Moulton R. A., Armitige L. Y., Bidani A., Snuggs M., Dhandayuthapani S., Hunter R. L., and Jagannath C. (2006) Processing and presentation of a mycobacterial antigen 85B epitope by murine macrophages is dependent on the phagosomal acquisition of vacuolar proton ATPase and in situ activation of cathepsin D. J. Immunol. 177, 3250–3259 [DOI] [PubMed] [Google Scholar]
- 26.Clemens D. L., and Horwitz M. A. (1995) Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J. Exp. Med. 181, 257–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brodin P., Poquet Y., Levillain F., Peguillet I., Larrouy-Maumus G., Gilleron M., Ewann F., Christophe T., Fenistein D., Jang J., Jang M. S., Park S. J., Rauzier J., Carralot J. P., Shrimpton R., Genovesio A., Gonzalo-Asensio J. A., Puzo G., Martin C., Brosch R., Stewart G. R., Gicquel B., and Neyrolles O. (2010) High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. 6, e1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacGurn J. A., and Cox J. S. (2007) A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect. Immun. 75, 2668–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pethe K., Swenson D. L., Alonso S., Anderson J., Wang C., and Russell D. G. (2004) Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc. Natl. Acad. Sci. U.S.A. 101, 13642–13647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zullo A. J., and Lee S. (2012) Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J. Biol. Chem. 287, 12668–12678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Axelrod S., Oschkinat H., Enders J., Schlegel B., Brinkmann V., Kaufmann S. H., Haas A., and Schaible U. E. (2008) Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell. Microbiol. 10, 1530–1545 [DOI] [PubMed] [Google Scholar]
- 32.Metchnikoff E. (1905) Immunity to Infective Disease, pp. 182, Cambridge University Press, Cambridge, UK [Google Scholar]
- 33.Baker J. J., Johnson B. K., and Abramovitch R. B. (2014) Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol. Microbiol. 94, 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer A. J., and Dick T. P. (2010) Fluorescent protein-based redox probes. Antioxid. Redox Signal. 13, 621–650 [DOI] [PubMed] [Google Scholar]
- 35.Bhaskar A., Munshi M., Khan S. Z., Fatima S., Arya R., Jameel S., and Singh A. (2015) Measuring glutathione redox potential of HIV-1-infected macrophages. J. Biol. Chem. 290, 1020–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyagi P., Dharmaraja A. T., Bhaskar A., Chakrapani H., and Singh A. (2015) Mycobacterium tuberculosis has diminished capacity to counteract redox stress induced by elevated levels of endogenous superoxide. Free Radic. Biol. Med. 84, 344–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geiman D. E., Raghunand T. R., Agarwal N., and Bishai W. R. (2006) Differential gene expression in response to exposure to antimycobacterial agents and other stress conditions among seven Mycobacterium tuberculosis whiB-like genes. Antimicrob. Agents Chemother. 50, 2836–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ta P., Buchmeier N., Newton G. L., Rawat M., and Fahey R. C. (2011) Organic hydroperoxide resistance protein and ergothioneine compensate for loss of mycothiol in Mycobacterium smegmatis mutants. J. Bacteriol. 193, 1981–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan L. J., Levine R. L., and Sohal R. S. (1997) Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl. Acad. Sci. U.S.A. 94, 11168–11172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodmansee A. N., and Imlay J. A. (2003) A mechanism by which nitric oxide accelerates the rate of oxidative DNA damage in Escherichia coli. Mol. Microbiol. 49, 11–22 [DOI] [PubMed] [Google Scholar]
- 41.Buchmeier N. A., Newton G. L., and Fahey R. C. (2006) A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J. Bacteriol. 188, 6245–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nambi S., Long J. E., Mishra B. B., Baker R., Murphy K. C., Olive A. J., Nguyen H. P., Shaffer S. A., and Sassetti C. M. (2015) The oxidative stress network of Mycobacterium tuberculosis reveals coordination between radical detoxification systems. Cell Host Microbe 17, 829–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrer N. L., Gomez A. B., Neyrolles O., Gicquel B., and Martin C. (2010) Interactions of attenuated Mycobacterium tuberculosis phoP mutant with human macrophages. PLoS One 5, e12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steyn A. J., Collins D. M., Hondalus M. K., Jacobs W. R. Jr., Kawakami R. P., and Bloom B. R. (2002) Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proc. Natl. Acad. Sci. U.S.A. 99, 3147–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Via L. E., Lin P. L., Ray S. M., Carrillo J., Allen S. S., Eum S. Y., Taylor K., Klein E., Manjunatha U., Gonzales J., Lee E. G., Park S. K., Raleigh J. A., Cho S. N., McMurray D. N., Flynn J. L., and Barry C. E. 3rd (2008) Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 76, 2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helaine S., Cheverton A. M., Watson K. G., Faure L. M., Matthews S. A., and Holden D. W. (2014) Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beauregard K. E., Lee K. D., Collier R. J., and Swanson J. A. (1997) pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J. Exp. Med. 186, 1159–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maurin M., Benoliel A. M., Bongrand P., and Raoult D. (1992) Phagolysosomes of Coxiella burnetii-infected cell lines maintain an acidic pH during persistent infection. Infect. Immun. 60, 5013–5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sturgill-Koszycki S., and Swanson M. S. (2000) Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 192, 1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.