

Abstract
Peroxiredoxin 2 (Prx2) is a thiol protein that functions as an antioxidant, regulator of cellular peroxide concentrations, and sensor of redox signals. Its redox cycle is widely accepted to involve oxidation by a peroxide and reduction by thioredoxin/thioredoxin reductase. Interactions of Prx2 with other thiols are not well characterized. Here we show that the active site Cys residues of Prx2 form stable mixed disulfides with glutathione (GSH). Glutathionylation was reversed by glutaredoxin 1 (Grx1), and GSH plus Grx1 was able to support the peroxidase activity of Prx2. Prx2 became glutathionylated when its disulfide was incubated with GSH and when the reduced protein was treated with H2O2 and GSH. The latter reaction occurred via the sulfenic acid, which reacted sufficiently rapidly (k = 500 m−1 s−1) for physiological concentrations of GSH to inhibit Prx disulfide formation and protect against hyperoxidation to the sulfinic acid. Glutathionylated Prx2 was detected in erythrocytes from Grx1 knock-out mice after peroxide challenge. We conclude that Prx2 glutathionylation is a favorable reaction that can occur in cells under oxidative stress and may have a role in redox signaling. GSH/Grx1 provide an alternative mechanism to thioredoxin and thioredoxin reductase for Prx2 recycling.
Keywords: glutathionylation, hydrogen peroxide, mass spectrometry (MS), peroxiredoxin, redox regulation, glutaredoxin
Introduction
Thiol proteins regulate many cell functions, including metabolic pathways, gene transcription, phosphorylation cascades, and ion channel activity. Such redox regulation is achieved by a combination of (a) cysteine oxidation by hydrogen peroxide or other reactive oxygen species and (b) reduction of by cellular redoxin systems (1–3). The oxidation products are mostly disulfides, including mixed disulfides with GSH (4). The main reducing systems are thioredoxin/thioredoxin reductase (Trx/TrxR)2 and glutaredoxin (Grx)/GSH, both of which ultimately depend on NADPH (2, 5). Although these have different specificities and were initially regarded as acting in parallel, it is becoming apparent that the two systems interact (5–9).
Peroxiredoxins (Prxs) play a major role in redox regulation. These highly expressed thiol proteins react extremely rapidly with H2O2 and other peroxides and should be prime targets for these oxidants in cells (10, 11). They are best known as antioxidants that remove excess peroxides and control amounts available for other reactions, but there is increasing appreciation that specific Prxs are also involved in redox signaling as peroxide sensors that transmit oxidizing equivalents to other regulatory thiol proteins (12–16). However, mechanisms of redox regulation and the relevance of the contrasting roles of Prxs in specific aspects of cell metabolism and cell signaling are not well understood.
The Prxs all contain an active site Cys, the peroxidatic Cys (CysP), that is highly specific for peroxides. They are subdivided into subfamilies based on their active site architecture (17) with Prx2 belonging to the Prx1 (typical 2-Cys Prx) subfamily. These function as obligate homodimers that are non-covalently associated in their reduced form. The initial reaction in the peroxidase cycle of Prx2 (and other members of the subfamily) is between the CysP of each monomer and H2O2 to give a sulfenic acid (–SOH), which reacts with the resolving Cys (CysR) on the opposing chain of the homodimer to form a disulfide. The sulfenic acid can also be further oxidized by H2O2 (hyperoxidized) to the sulfinic acid, which will be present as the sulfinate (-SO2−) because of its low pKa. The rate constants for the H2O2 reactions are 1–2 × 107 m−1 s−1 for reduced Prx2 (18, 19) and ∼104 m−1 s−1 for the sulfenic acid (20). The peroxidase cycle is completed by regeneration of the reduced protein. Early work (21, 22) showed that the disulfides are efficiently reduced by Trx and TrxR, and this has been accepted as the major recycling mechanism (10, 11). However, redox interactions with other thiols have been reported (6, 14–16, 23, 24) and could provide an alternative recycling or signaling mechanism.
One potentially important reaction of 2-Cys Prxs is with GSH to form mixed disulfides as this could have a regulatory function or facilitate recycling of the protein. However, it is currently unknown whether their reactions with GSH are physiologically relevant. Glutathionylation of the isolated proteins has been observed but only after extended incubation with non-physiological GSSG concentrations (25, 26), and recycling of Prx1 and Prx2 by GSH and Grx1 was not apparent in initial studies (21). However, glutathione adducts have been detected in cell systems (25, 27, 28) and could therefore be physiologically relevant. We have characterized the interactions between GSH and Prx2 using mass spectrometry to detect products. We describe facile glutathionylation when physiological concentrations of GSH are added either to the disulfide or to reduced Prx2 during treatment with H2O2. Rapid reversal by Grx1 enabled GSH/Grx1 to complete the Prx2 peroxidase cycle, and we present evidence for Prx2 glutathionylation in erythrocytes from Grx1 knock-out mice.
Experimental Procedures
Expression of Recombinant Prxs
Recombinant His-tagged wild-type human Prx2 and mutant Prx2 with CysR mutated to Ser (C172S) were prepared as described (23) with a few modifications. Briefly, cDNA encoding human Prx2 with an amino-terminal Factor Xa cleavage site was cloned into a pET28a vector. Specific oligonucleotides were used to mutate this template by PCR to generate the cDNA constructs encoding the C172S mutant. The proteins were expressed in Escherichia coli and purified on His60 Ni Superflow Resin (Clontech) according to the manufacturer's instructions in the absence of reducing agents. His tags were removed by incubation with Factor Xa (Roche Applied Science) to give recombinant Prx2 proteins (beginning with Met) with no additional amino acids. The proteins were stored at −80 °C. Each isolated protein resolved as a single band at ∼22 kDa on reducing SDS-PAGE. MS analysis of the WT protein showed it was dimeric when isolated. Reduction gave a mass of 21,894 Da (theoretical, 21,892 Da) that increased to 22,269 Da on treatment with NEM (125 Da), consistent with derivatization of its three Cys residues. The isolated C172S mutant was monomeric but was 303 Da greater than the predicted mass of 21,876 Da (see “Results” for explanation).
Treatment with H2O2 and GSH
WT Prx2 was either used as prepared in its oxidized (disulfide) form or treated with 10 mm DTT for 1 h followed by removal of the reductant using a Micro Bio-Spin 6 column (Bio-Rad). The Prx2 concentration was determined using the Bio-Rad DC Protein Assay or using a DirectDetect spectrometer (Merck). Reactions with GSH and H2O2 were performed at 22 °C in 50 mm phosphate buffer, pH 7.4, containing 0.1 mm diethylenetriaminepentaacetic acid and 5 μm Prx2 unless otherwise stated. After incubation, samples were alkylated by treating for 15 min with 15 mm NEM. Samples for LC/MS were treated in 1 m guanidine hydrochloride to facilitate complete thiol alkylation (including the less reactive non-active site Cys, Cys-70).
Mass Spectrometry
For whole protein analysis, samples containing 0.5 μg of protein were injected onto an Accucore-150-C4 (50 × 2.1-mm, 2.6-μm) column (60 °C) using a Dionex Ultimate 3000 HPLC system coupled to a Velos Pro mass spectrometer (Thermo Scientific, San Jose, CA). Proteins were eluted with an acetonitrile gradient from 90% solvent A (0.1% formic acid in water) and 10% solvent B (0.1% formic acid in acetonitrile) to 80% solvent B over 4.6 min at a flow rate of 400 μl/min. Mass spectra for all charge states were acquired between m/z 400 and 2000 in positive mode, averaged over the full-length of each protein peak, and deconvoluted to yield the molecular masses and relative intensities using ProMass for Xcalibur (version 2.8; Novatia LLC, Monmouth Junction, NJ). The tune file was configured with a capillary temperature of 275 °C and a spray voltage of 4 kV. Three microscans were averaged, the maximum inject time was 10 ms, and automatic gain control target settings were 3 × 104 in full MS and 1 × 104 in MSn. The chromatographic conditions did not separate the different Prx species, and relative intensities were obtained from the deconvoluted spectral data. The accuracy of the deconvoluted masses was within 7 Da of theoretical masses. The masses used to identify glutathionylated species are given in Table 1. Quantification is based on peak intensity.
TABLE 1.
Theoretical masses used to identify the oxidized and glutathionylated forms of Prx2
| Species | Molecular massa |
|---|---|
| kDa | |
| Reduced monomer | 22.267 |
| Hyperoxidized monomer | 22.174 |
| Monomer with single glutathionylation | 22.447 |
| Monomer with double glutathionylation | 22.627 |
| Hyperoxidized monomer with single glutathionylation | 22.354 |
| Dimer | 44.030 |
| Dimer with single glutathionylation | 44.462 |
| Dimer with double glutathionylation | 44.642 |
| Hyperoxidized dimer | 44.189 |
a Based on complete alkylation of all cysteines, namely Cys-51 (CysP), Cys-70, and Cys-172 (CysR) in the reduced WT protein with NEM (+125 Da). Molecular mass of non-alkylated reduced monomer of Prx2 is 21,892 Da. Accuracy of MS analysis, 7 Da. In some samples, peaks corresponding to incomplete derivatization with NEM were detected. These were combined with the main peak for the particular species for quantification.
Peptide analyses were performed on chymotryptic digests (Promega; 1:60) using the same instrument as above and a C12 reverse-phase column (150 × 2.0 mm; Jupiter Proteo, 5-μm particle size). Elution was at 40 °C with a flow rate of 200 μl/min and a gradient of mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile). After 5 min at 5% B, the gradient increased linearly to 47% B over 30 min and then to 95% B for 4 min before equilibrating back to 5% B for 10 min. The capillary temperature and spray voltage were as above. There was no averaging of microscans. The eluted peptides were analyzed using a data-dependent nth order double play scan procedure. The five most abundant peptide peaks in a full scan (400–2000 m/z) were sequentially selected, and an MS/MS scan was performed using collisionally induced dissociation with a normalized collision energy of 40%. MS/MS spectra were recorded using dynamic exclusion with a repeat count of 5, a repeat duration of 30 s, and an exclusion duration of 60 s. A precursor mass list was generated of the masses expected from the Cys-containing peptides in both their modified and unmodified states.
Polyacrylamide Gel Electrophoresis and Western Blotting
Non-reducing SDS-PAGE was performed using 12% gels and a Bio-Rad Mini-Protean II apparatus. Proteins were transferred to polyvinylidene difluoride membranes for immunoblotting for Prx2 (Sigma; 1:10,000 dilution) or hyperoxidized Prx (Abfrontier, Seoul, Korea; 1:1000 dilution) as described (18). Bands were detected using the ECLPlus Detection System (GE Healthcare) and visualized with a ChemiDoc XRS gel documentation system (Bio-Rad). The relative intensities of the Prx monomer and dimer bands were quantified by densitometry using Quantity One software (Bio-Rad).
Rate Constants for the Reactions of Prx2 C172S with H2O2
The rate constant for reaction with the reduced protein was determined by competition with horseradish peroxidase (HRP) as described (18). The concentration of HRP was determined using ϵ403 = 1.02 × 105 m−1 cm−1, and the extent of conversion to compound I was monitored at 403 nm immediately after addition of H2O2. The rate constant for hyperoxidation of Prx2 C172S was determined by competition with catalase (20).
Human Erythrocytes
EDTA blood was obtained from healthy human volunteers with informed consent as approved by the New Zealand Southern Regional Ethics Committee. Erythrocytes were separated, washed, and diluted to 107 cells/ml in Hanks' buffer for treatment with H2O2.
Assessment of Prx2 Glutathionylation in Mouse Erythrocytes
All procedures for handling of mice were approved by the Institutional Animal Care and Use Committee at Boston University Medical Campus. Glrx1-deficient mice, generated as described (29), were backcrossed with the C57Bl/6J mouse (stock number 00664, The Jackson Laboratory, Bar Harbor, ME) until all progeny were homozygous for mutant nicotinamide nucleotide transhydrogenase. Female homozygous Glrx−/− mice (4–5 months old) and age-matched WT mice (Glrx+/+) were euthanized by exsanguination under isoflurane anesthesia. Blood samples were collected by cardiac puncture into EDTA, and 50 μl samples of pelleted erythrocytes were diluted 1:100 in PBS containing 5 mm glucose and 2 μm auranofin. After 10 min at 37 °C, the cells were mixed with H2O2 (final concentration, 0.5 mm), which was quenched with catalase (10 μg/ml) after 3 min. After further incubation for ∼10 min, the cells were spun down, mixed with 50 μl of 100 mm NEM for 15 min, and then lysed by freeze-thawing. Excess NEM was removed on microspin columns, and eluates were frozen at −80 °C before transporting on wet ice from Boston to Christchurch. Immunoprecipitations were carried out using rabbit anti-Prx2 (Sigma R8656) conjugated to Dynabeads (Dynal/Invitrogen). Eluates were diluted in PBS and incubated with the beads overnight at 4 °C. After four washes, Prx2 was eluted with 0.1 m glycine, pH 3; immediately neutralized with 67 mm Tris/HCl, pH 7.4; and subjected to LC/MS.
Materials
All biochemicals were from Sigma unless stated otherwise.
Results
Glutathionylation of Reduced Prx2
Reduced Prx2 was treated with H2O2 in the absence or presence of GSH, and the reaction mixtures were analyzed by LC/MS. Products were identified by deconvolution of spectra of the whole protein peak and relating to the theoretical masses in Table 1. In a previous study (20), we showed that low concentrations of H2O2 alone oxidize Prx2 to a disulfide-linked dimer with increasing concentrations progressively generating hyperoxidized dimer (with one disulfide bond) and then hyperoxidized monomer (structures shown in Fig. 1a). In this study, a 4-fold molar excess of H2O2 gave mainly disulfide plus a small amount of hyperoxidized dimer (Fig. 1b). GSH protected against hyperoxidation and inhibited disulfide formation with concomitant formation of glutathionylated products (Fig. 1c). Increasing concentrations of GSH gave a progressive increase in total glutathionylation (Fig. 1d). As shown in Fig. 1e for 8 mm GSH, the products were predominantly diglutathionylated monomers and dimers plus a small amount of monomer with one GSH attached (for structures, see Fig. 1a). Over time, the dimers gradually converted to glutathionylated monomers, but there was little evidence for regeneration of fully reduced Prx2 (not shown).
FIGURE 1.

Glutathionylation of reduced Prx2. a, structures of major Prx2 oxidation products observed in the absence or presence of GSH. For clarity, the CysP is positioned at the point of the symbol representing the monomer, and the CysR is at the wide end. G represents glutathionylation, DS represents disulfide, and other abbreviations are monomer (M), dimer (D), and hyperoxidized (H). The color coding and abbreviations are carried through into subsequent panels. Note that for the monoglutathionylated species either Cys could be glutathionylated. Although Prx2 functions as a non-covalent dimer, only covalently bonded species are shown as dimeric. b and c, typical examples of deconvoluted mass spectra of Prx2 (5 μm) after treating with 20 μm H2O2 at 22 °C in 50 mm phosphate buffer, pH 7.4, containing 0.1 mm diethylenetriaminepentaacetic acid in the absence and presence of 8 mm GSH, respectively. After 20 min, samples were treated for 15 min with 15 mm NEM in 1 m guanidine hydrochloride. Identified peaks are labeled. Minor peaks (shown in gray and each representing <5% of the total signal) were not assigned or analyzed. d, concentration-dependent glutathionylation with 20 μm H2O2. All glutathionylated species are combined into a single plot. Quantification from MS data is based on signal intensities. e, proportions of individual glutathionylated and non-glutathionylated species after treatment of reduced Prx2 and 8 mm GSH for 30 min with 20 or 240 μm H2O2. f, Western blot of non-reducing SDS-PAGE with antibody against hyperoxidized Prxs showing protection of purified Prx2 (5 μm) against hyperoxidation by a low concentration and less protection at higher concentrations of H2O2.
Inhibition of hyperoxidation by GSH was also apparent when non-reducing gels were blotted with an antibody against the hyperoxidized protein (Fig. 1f). There was strong protection at lower concentrations of H2O2, but this lessened as the extent of hyperoxidation increased. With an H2O2 concentration that gave predominantly hyperoxidized products, MS analysis showed that GSH inhibited the formation of Prx2 disulfide with a corresponding increase in glutathionylation but had little effect on the extent of hyperoxidation (Fig. 1e).
Glutathionylation of Prx2 Sulfenic Acid
Glutathionylation of Prx2 could occur by disulfide exchange with oxidized Prx2 and/or by reaction of the sulfenic acid with GSH. The latter reaction would have to compete with other reactions of the sulfenic acid, namely condensation with CysR and hyperoxidation by H2O2 to the sulfinate (20). To establish whether GSH reacts with the sulfenic acid, we generated recombinant Prx2 lacking CysR (C172S). Interestingly, when the mutant protein was purified without adding a reducing agent, MS analysis (Fig. 2a) showed that its mass was higher than predicted by an amount equivalent to the addition of GSH (305 Da). Reduction with DTT removed the extra mass (Fig. 2b), and subsequent alkylation gave a mass of 22,133 Da, which corresponds to the increase of 250 Da expected for addition of NEM to two Cys residues. When the protein was precipitated following reduction and NEM treatment, the NEM adduct of GSH was detected by LC/MS in the supernatant. Taken together, these data indicate that the mutant protein becomes stably glutathionylated when expressed in E. coli.
FIGURE 2.

Reactions of the C172S mutant of Prx2 with H2O2 and GSH. Deconvoluted mass spectra of the C172S mutant as isolated from E. coli under non-reducing conditions (a), following reduction with DTT and showing loss of 303 Da (b), following treatment of reduced C172S (5 μm) with a 2-fold excess of H2O2 (c), and showing trapping of the sulfenic acid with dimedone (50 mm) present during addition of equimolar H2O2 (d) are shown. e, concentration-dependent glutathionylation of CysP of C172S mutant of Prx2 and corresponding inhibition of hyperoxidation. Purified recombinant C172S Prx2 (5 μm) was reduced, mixed with varying concentrations of GSH, and subjected to reaction with 10 μm H2O2. Data represent means ± S.D. (error bars) from four individual experiments. Kinetic analysis of the data was performed using Berkeley Madonna (as in Ref. 20) and rate constants for Reactions 1 and 2 in Table 2. kGSH was varied to give the best fit (plotted), which was obtained with a value of 500 m−1 s−1.
When the C172S mutant was treated with H2O2, it was readily hyperoxidized with a 2-fold molar excess giving almost complete hyperoxidation (Fig. 2c). Low concentrations of GSH inhibited hyperoxidation, and the major product was a monoglutathione adduct (Fig. 2e).
Before examining the kinetics and mechanism of glutathionylation of the C172S mutant, we first characterized its reactions with H2O2. Dimedone trapping (Fig. 2d) showed that the initial oxidation product is the sulfenic acid. Not all the Prx2 was trapped, which is consistent with the reaction of dimedone with the sulfenic acid being slow (30). The other masses detected by MS correspond to the sulfinate (with one NEM) and the reduced protein. Theoretically, with equimolar H2O2, all the Prx2 C172 should be converted to the sulfenic acid without any hyperoxidation and with no reduced protein remaining. However, sulfenic acids are unstable, and it is likely that the other products arose through dismutation (31). Consistent with this explanation, when Prx2 C172S was treated with equimolar H2O2 (in the absence of dimedone) and analyzed by LC/MS without adding NEM, half of the protein was hyperoxidized, and the other half was reduced (not shown). It was necessary to prevent this dismutation when measuring the rate constant for hyperoxidation.
The rate constant for reaction of reduced C172S Prx2 with H2O2 was measured in a competition assay with HRP. The observed concentration-dependent inhibition of HRP oxidation (Fig. 3a) gave a second order rate constant (k1 in Table 2) of 2.7 × 107 m−1 s−1, which is similar to values of 1.3 × 107 and 1.7 m−1 s−1 measured for wild-type Prx2 (18, 19). The rate constant for Reaction 2 between the sulfenic acid and H2O2 (hyperoxidation) was measured by adding concentrations of catalase that were sufficient to give progressive inhibition of Reaction 2 but not enough to inhibit Reaction 1 and modeling with varying k2 to give the best fit of the data in Fig. 3b. The k2 value obtained of 1.2 × 104 m−1 s−1 is no different from that measured for wild-type protein (20).
FIGURE 3.

Determination of rate constants for reactions of H2O2 with the reduced thiol (a) and sulfenic acid (b) forms of Prx2 C172 S mutant. a, HRP (10 μm) was mixed with various concentrations of Prx2 C172S and then treated with 8 μm H2O2 in two independent experiments (error bars show range). The fractional inhibition of HRP oxidation (F) was determined at each concentration, and the rate constant was determined from the slope of the plot of (F/1 − F)kHRP[HRP] against Prx2 concentration. b, reduced protein (5 μm) was mixed with bovine catalase at the stated concentration and then treated with 10 μm H2O2. Within 1 min, additional catalase was added to all samples to quench any residual H2O2, and GSH (1 mm) was added to react with the sulfenic acid and prevent formation of the sulfinic acid through dismutation. The proportions of hyperoxidized protein and glutathionylated protein (which corresponds to the initial sulfenic acid) were measured by MS. The data points are from two independent experiments. The second order rate constant was calculated by determining the percentage of hyperoxidation as a function of catalase concentration and fitting the data to a kinetic model using Berkeley Madonna software and rate constants for Reactions 1, 2, 6, and 7 in Table 2. Simulations were performed as described previously (20) varying k4 to obtain the best fit. The solid line was obtained with k2 set at 1.2 × 104 m−1 s−1; the dashed lines were obtained with 5 × 103 (bottom) and 2 × 104 m−1 s−1 (top). The curve fitted well with k2 = 1.2 × 104 m−1 s−1, which is the same value as determined for wild-type Prx2 (20).
TABLE 2.
Rate equations used for kinetic analysis
Rate constants for Reactions 1 and 2 are for the C172S mutant and were determined from the data in Fig. 3. They are close to values for the wild-type protein (18–20); k4 for the C172S mutant was determined from the data in Fig. 2. Other values and the methods used for kinetic analysis have been described previously (18, 20).
| Equation | Rate constant (k) | Reaction |
|---|---|---|
| PrxSH + H2O2 → PrxSOH | 2.7 × 107 m−1 s−1 | 1 |
| PrxSOH + H2O2 → PrxSO2H | 12,000 m−1 s−1 | 2 |
| PrxSOH → Prx(SS) | 2 s−1 | 3 |
| PrxSOH + GSH → PrxSSG | 500 m−1 s−1 | 4 |
| HRP(reduced) + H2O2 → HRP-compound I | 1.7 × 107 m−1 s−1 | 5 |
| Catalase (reduced) + H2O2 → Catalase (oxidized) + H2O | 5 × 106 m−1 s−1 | 6 |
| Catalase (oxidized) + H2O2→ Catalase (reduced) + O2 | 1 × 107 m−1 s−1 | 7 |
To determine the rate constant for reaction of the sulfenic acid with GSH, the data in Fig. 2e were fitted to a kinetic model in which GSH (Reaction 4; Table 2) competes with H2O2 (Reaction 2) for the sulfenic acid. Using the values determined for k1 and k2, the data fit well with a k4 of 500 m−1 s−1. This value is 25-fold less than the rate constant for the sulfenic acid reacting with H2O2. With the proviso that the WT protein reacts at the same rate, millimolar concentrations of GSH should inhibit its hyperoxidation by 20 μm H2O2 but have limited effect with 240 μm as was observed in Fig. 1e. Also, with a rate constant (k3) of 2 s−1 for internal disulfide formation (20), we can calculate that disulfide formation and glutathionylation should be equally effective with ∼4 mm GSH. Therefore, WT Prx2 should be glutathionylated via the sulfenic acid under physiological conditions.
Glutathionylation by Disulfide Exchange with Oxidized Prx2
Incubation of oxidized WT Prx2 with GSH resulted in time-dependent formation of both dimer and monomer with one bound glutathione (Fig. 4a). Over time, these species gradually converted to the fully reduced protein. Gradual reduction to monomeric species was also apparent on SDS-PAGE analysis (Fig. 4b). Thus Prx2 can be glutathionylated by disulfide exchange, but in contrast to treating the reduced protein with H2O2, species with two bound GSH molecules are not significant products.
FIGURE 4.

a, time course of glutathionylation of oxidized Prx2 by exchange with GSH. Oxidized Prx2 was incubated with GSH (8 mm), and conversion to glutathionylated species over time was followed by MS analysis as in Fig. 1. ●, disulfide (DS); ▴, dimer + one GSH (D+G); □, monomer + one GSH (M+G); ○, reduced monomer (M). Results are means and S.E. (error bars) from six to 10 independent experiments. b, Coomassie-stained non-reducing gel showing the time course of reduction of oxidized Prx2 with 8 mm GSH and where indicated 0.3 μm Grx1. c, Prx2 species distribution 20 min after treating oxidized Prx2 with 8 mm GSH and 250 μm H2O2. Samples were analyzed by LC/MS, and results are means and S.E. (error bars) from five independent experiments.
When H2O2 (20 or 250 μm) was present with oxidized Prx2 and 8 mm GSH, a different product profile was observed (Fig. 4c). By 30 min, almost all the protein was converted to glutathionylated monomers with the majority having two GSH molecules bound. Even at the higher H2O2 concentration, little of the Prx2 became hyperoxidized.
Location of Glutathione Adducts on CysP and CysR
We first examined which thiols became glutathionylated when oxidized Prx2 was treated with GSH and H2O2 as in Fig. 4c. MS analysis confirmed that under these conditions the treated protein was all monomeric with predominantly two GSH molecules attached. Reduced thiols were blocked with NEM, and then disulfides were reduced and blocked with iodoacetamide (IAM). Therefore, IAM adducts should have arisen from mixed disulfides with GSH. MS analysis of the blocked protein showed that the majority had two IAM molecules and one NEM attached with a lesser fraction with one IAM and two NEM molecules. Peptide analysis showed good signals for CysP and CysR as IAM adducts with no good match for NEM derivatives (Table 3). Cys-70 was detectable only as an NEM adduct. These results indicate that both CysP and CysR were glutathionylated.
TABLE 3.
Cys peptides detected after treating Prx2 with GSH and H2O2
Oxidized Prx2 (5 μm) was treated with 8 mm GSH and 240 μm H2O2 for 30 min, then blocked with NEM (15 mm). After 10 min, DTT (20 mm) was added to block residual NEM and reduce disulfides in the Prx2, and then iodoacetamide (50 mm) was added to derivatize the freed thiols. LC/MS/MS was performed on a chymotryptic digest. * represents position of IAM or NEM.

The site of the single GSH on the glutathionylated monomer prepared by disulfide exchange was also examined. Oxidized Prx2 was incubated for 15 min with 8 mm GSH and analyzed as above except that the monomeric species were separated from dimers by PAGE before reduction and treatment with IAM. This was necessary to avoid labeling Cys residues from internal disulfides. Peptide analysis showed IAM adducts on both CysP and CysR, indicating a mixture of species with one or the other glutathionylated.
Recycling of Glutathionylated Prx2 by Glutaredoxin
When Prx2 disulfide was incubated with 8 mm GSH in the presence of Grx1, MS analysis showed that it was reduced much more rapidly than with GSH alone and gave no detectable glutathionylated species (Fig. 5a). Corresponding gel electrophoresis showed almost complete reduction to the monomer within 15 min (Fig. 4b). These results show that oxidized Prx2 can be fully reduced by the GSH/Grx1 system and that glutathionylation rather than deglutathionylation is rate-limiting.
FIGURE 5.

Reduction of Prx2 disulfide and support of Prx2 peroxidase activity by GSH plus Grx1. a, MS analysis of products observed after incubation of 5 μm Prx2 disulfide with 8 mm GSH in the presence or absence of Grx1 (0.3 μm). Results are from a representative experiment. b, peroxidase activity of Prx2/GSH/Grx1 measured as consumption of H2O2 (20 μm) by 3 mm GSH in the absence or presence of Prx2 (3 μm) and Grx1 (0.2 μm). Reactions were at 37 °C in 50 mm phosphate buffer, pH 7.4, and samples were removed at intervals after H2O2 addition to measure residual H2O2 using the ferrous oxidation-xylenol orange assay (40). Results are from two independent experiments with data points for each either overlapping or shown separately. c, H2O2 consumption by Prx2 (3 μm) in the presence of 1 mm DTT (■) or by DTT alone (○). M, monomer; D, dimer; G, GSH; DS, disulfide.
Peroxidase Activity of Prx2/GSH/Grx1
GSH plus Grx1 supported the peroxidase activity of Prx2 (Fig. 5b). As H2O2 reacts directly with GSH (32), H2O2 was slowly consumed when the two were incubated together. Prx2 alone had little effect on the rate of consumption, but this was substantially increased when both Prx2 and Grx1 were present. The rate of H2O2 consumption increased with increasing Prx2 concentration, and the Grx1 concentration used in Fig. 4b was optimal (not shown). Grx1 alone did not catalyze removal of H2O2 by GSH: at a time when 44% of the H2O2 was consumed by GSH alone, consumption by GSH plus Grx1 was unchanged at 42%. The peroxidase activity of Prxs is frequently measured using DTT as reductant. The rate of Prx2-dependent H2O2 consumption with Grx1 and 3 mm GSH was approximately half that observed with 1 mm DTT (Fig. 5c).
Glutathionylation and Involvement of GSH/Grx1 in Prx2 Reduction in Erythrocytes
To examine cellular mechanisms for Prx2 reduction, human red cells were treated with H2O2 in the presence of glucose, and Prx2 was monitored by non-reducing SDS-PAGE. Under these conditions, Prx2 is converted initially to a dimer (Fig. 6, lane 2), and then as the H2O2 is consumed, it is re-reduced by cellular recycling mechanisms (lanes 3–5). When the cells were treated in the presence of dinitrochlorobenzene (DNCB), which inhibits TrxR and reacts with other thiols, recycling was inhibited (lanes 9–11 and Ref. 33). However, the potent and more specific TrxR inhibitor auranofin (34) did not prevent Prx2 reduction (lanes 6–8). Cellular GSH was also measured. It was not affected by auranofin, but DNCB caused a loss of ∼40%. These results imply that Prx2 recycling in erythrocytes can occur independently of the Trx/TrxR system.
FIGURE 6.

Inhibition of Prx2 reduction in erythrocytes by DNCB but not by the TrxR inhibitor auranofin. Human erythrocytes (107 cells/ml in Hanks' buffer) were preincubated at 37 °C for 15 min with either 2 μm auranofin, 0.1 mm DNCB, or no addition and then treated with H2O2 (20 μm). After 5 min, 2 μg/ml catalase was added, and then samples were taken at intervals, blocked with 30 mm NEM, separated by SDS-PAGE, and immunoblotted for Prx2. Results are from a representative experiment. The middle band represents a dimer of hemoglobin that reacts with the ECL reagent.
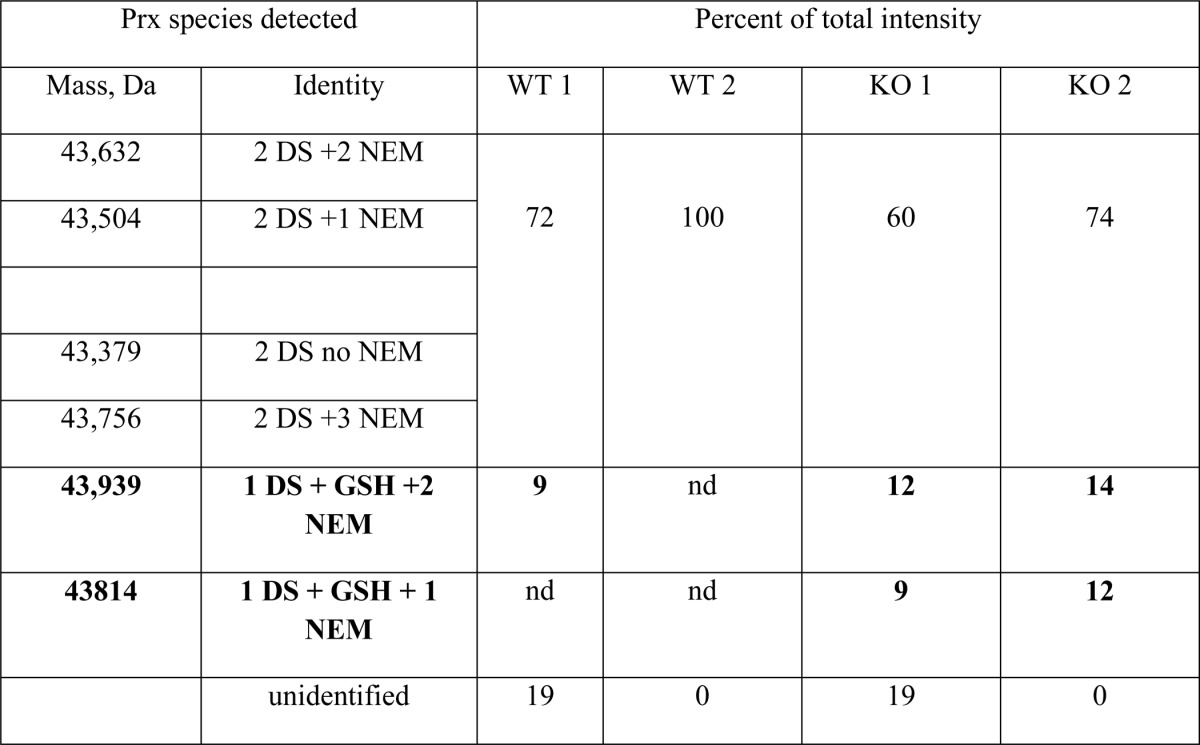
As Prx2 recycling by Grx1/GSH proceeds via a mixed disulfide intermediate and deglutathionylation is faster than glutathionylation (Fig. 5a), we reasoned that detection of mixed disulfides would be more likely in cells lacking Grx1. Therefore, we isolated erythrocytes from Glrx−/− and WT mice, exposed them to H2O2, and analyzed immunoprecipitated Prx2 by LC/MS for glutathionylated adducts. In all samples, the majority of the protein was dimeric Prx2. In an initial experiment, a small amount of glutathionylated Prx2 was detected in erythrocytes from two knock-out mice but not paired WT controls. Although this result was promising, blocking with NEM was not efficient, and GSH may have been lost by exchange. Another experiment was performed with more efficient blocking. In this case (Table 4), 20–26% of the Prx2 from the knock-out cells had a mass corresponding to a GSH adduct compared with 0–8% for the WT. Furthermore, reduction of the immunoprecipitated Prx2 gave detectable release of GSH that was ∼4-fold higher for the Grx1-deficient cells.
TABLE 4.
Detection of Glutathionylated Prx2 in Erythrocytes from Glrx-deficient mice
Isolated erythrocytes from two knockout (KO) and two wild-type (WT) mice were treated with H2O2 and processed as described under “Experimental Procedures.” Proteins were immunoprecipitated with anti-Prx2 and detected by LC/MS. Species representing < 5% of total signal have been excluded. Bold masses correspond to glutathionylated species; nd, not detected; DS, disulfide. The detected mass of 43,379 Da with no NEM or GSH is as predicted for mouse Prx2 (Swiss-Prot) with the same N-terminal acetylation observed in human erythrocytes (38). Eluates containing immunoprecipitated Prx2 were further analyzed for released GSH following reduction with DTT. Analysis of equal volumes by isotope dilution mass spectrometry (39) gave 3 or 5 times more GSH for the KO compared with the WT samples.
Discussion
We have shown that Prx2, a typical 2-Cys Prx, readily forms glutathionylated products on one or both of its active site CysP and CysR residues when oxidized in the presence of physiological concentrations of GSH. GSH inhibited intersubunit disulfide formation and, when the Prx2 was exposed to modest excesses of H2O2, protected against hyperoxidation. The GSH adducts were deglutathionylated efficiently by Grx1, and GSH/Grx1 was able to support peroxidase activity. When 2-Cys Prxs were first recognized as peroxidases, they were called thioredoxin peroxidases because they could be reduced by Trx/TrxR and NADPH (35). This system is widely accepted as the physiological recycling mechanism. Based on our results, the GSH/Grx system needs to be considered as an alternative. It is likely that recycling by GSH/Grx1 is slower, and this may be why it was not recognized previously (21). However, it may be more prominent in situations such as in the erythrocyte where TrxR activity is extremely low (33). Indeed the ability of DNCB but not the more specific TrxR inhibitor auranofin to inhibit recycling of erythrocyte Prx2 implies that TrxR activity is not required and is consistent with a GSH/Grx1 reduction mechanism. There may, however, be more than one way of bypassing TrxR as Grx1 is also able to reduce Trx (7, 8).
Our kinetic data with isolated Prx2 imply that glutathionylation should be fast enough to occur in cells. As deglutathionylation by Grx1 was faster, we reasoned that glutathione adducts may not accumulate in cells containing active Grx1. Therefore, we examined erythrocytes from Glrx−/− mice. Glutathionylated Prx2 was detected in these cells following treatment with H2O2 and auranofin with levels being much lower or absent in WT cells. There was some experimental variation in the amounts detected, possibly because of slow deglutathionylation by GSH itself or disulfide exchange during processing. However, it is hard to envisage how GSH adducts could form as an artifact, and our findings are good evidence that glutathionylation and Grx1-dependent deglutathionylation occur physiologically.
We identified two mechanisms of glutathionylation, one involving disulfide exchange with oxidized Prx2 and the other via the sulfenic acid. Both occurred within minutes and resulted in substantial adduct formation. Intersubunit disulfide formation was either reversed or inhibited, and hyperoxidation caused by modest excesses of H2O2 was suppressed. This indicates that GSH could play a physiological role in protecting Prx2 from hyperoxidation during oxidative stress.
Proposed mechanisms for Prx2 glutathionylation are represented by the schemes in Fig. 7. In the exchange mechanism (Fig. 7A) with oxidized Prx2 where only species with one GSH bound were detected in significant amounts, our results fit with the active site disulfides reacting sequentially to give first dimeric (a), then monomeric single GSH adducts (b), and then eventually complete reduction (c). Our finding of GSH adducts on both CysP and CysR supports the exchange reactions shown in Fig. 7A, although it is possible that the GSH initially reacts with one of the Cys residues and then redistributes. For glutathionylation of reduced Prx2 during treatment with H2O2 (Fig. 7B), the initial product would be the CysP sulfenic acid (d), which in the typical Prx redox cycle condenses with the opposing CysR to form the disulfide (e). Reactions of the sulfenic acid with GSH (f and g) compete with condensation. Based on the rate constant of 500 m−1 s−1 measured with the resolving mutant, it can be calculated that millimolar concentrations of GSH would be sufficiently competitive to give substantial glutathionylation. The expected products would be monoglutathionylated at each CysP and either monomeric or dimeric. However, most of the observed products were diglutathionylated on CysP and CysR, indicating a more complex mechanism. CysR reacts slowly with H2O2, so it is unlikely to become glutathionylated via an oxidative mechanism. However, exchange between glutathionylated CysP and CysR (h and i) would generate reduced CysP, which would be rapidly oxidized to the sulfenic acid (j and k) and then form a second GSH adduct (l and m). Thus glutathionylation of CysP and CysR is well explained by a combination of oxidative and exchange mechanisms.
FIGURE 7.

Proposed mechanism for glutathionylation of Prx2 by exchange with the oxidized protein and by oxidation of the reduced protein. Boxes indicate the major glutathionylated products observed under each condition. As in Fig. 1a, the CysP is positioned at the point of the symbol representing the monomer, and the CysR is at the wide end. Glutathione adducts are represented as G in yellow. a–m represent proposed steps in the reaction mechanism and are explained in the text.
Treatment of oxidized Prx2 with H2O2 and GSH gave a somewhat different picture. Glutathionylation was more extensive than with exchange alone, and there was more reduction and diglutathionylation. Also there was much less hyperoxidation than when reduced Prx2 was treated with the same concentration of H2O2. The extensive glutathionylation must have required considerable redox cycling, including sulfenic acid generation; however, the sulfenic acid apparently resisted hyperoxidation. Further investigation is needed to explain this result.
Formation of a stable mixed disulfide at the active site of Prx2 is perhaps surprising as proteins that form internal disulfides generally displace an external thiol such as GSH with their resolving Cys. A feature of mammalian 2-Cys Prxs is that CysP and CysR are well spaced. Disulfide formation requires transformation from a fully folded to locally unfolded conformation, and this reaction is relatively slow (10, 20, 36). Although the structures of the glutathionylated species are not known, their stability suggests that the conformational change required to displace the GSH is not favored.
There are several other reports of glutathionylation of mammalian 2-Cys Prxs. Park et al. (25, 26), who generated adducts by overnight incubation of reduced Prxs with 10 mm GSSG, focused mainly on glutathionylation of Cys-83 in Prx1 (which is not present in Prx2) and reversal by sulfiredoxin. Others have detected glutathionylated Prxs in proteomic screens and observed secretion from cells under oxidative and inflammatory stress (27, 28). Reduction of Prx3 by Grx2 has also been observed, but in this case, the disulfide was reduced directly without involving a glutathionylated intermediate (6). The ease of active site glutathionylation, as shown here for Prx2 and which may apply to other 2-Cys Prxs, has not previously been recognized.
Glutathionylation of proteins is increasingly being recognized as an important mechanism for redox regulation. Therefore, it is possible that glutathionylated Prxs have a function in their own right. The high reactivity of Prxs makes them well suited as peroxide sensors that transmit intracellular redox signals. One mechanism of signal transmission could involve oxidation of the Prx to a GSH adduct and then transfer of the GSH to a target protein. Alternatively glutathionylation could cause a conformational change that alters binding interactions. From a broader perspective, a similar mechanism of mixed disulfide formation as described here for GSH could operate for Prx2 and other thiol proteins. Formation and resolution of a mixed disulfide would transfer oxidizing equivalents from the Prx to the target protein and provide a thiol relay mechanism for signal transmission. Our findings reinforce studies where this mechanism has been invoked, for example with the yeast and mammalian transcription factors YAP1 and STAT3 (12, 14–16). Mammalian 2-Cys Prxs may be particularly well suited to this sensor role not only because of their high peroxide reactivity but also the slow rate of dimerization of their sulfenic acid. Theories as to why this reaction is slow have mostly focused on it enabling time for hyperoxidation (36, 37). Our observations raise an alternative view that it provides a window of opportunity for the sulfenic acid to react with GSH or other protein thiols.
Author Contributions
C. C. W and A. V. P. conceived the study, designed the experiments, and wrote the manuscript. A. V. P. carried out most of the experiments, P. E. P. provided recombinant proteins, L. N. P. performed mass spectrometry, J. B. B. performed the mouse studies in consultation with M. M. B., and M. S. contributed data on human erythrocytes.
Acknowledgments
We thank Dr. Guy Salveson (Burnham Institute) for suggesting that the isolated resolving mutant might be glutathionylated, Dr. Nina Dickerhof for performing the LC/MS analyses for GSH, Drs. R. Matsui and Y. Janssen-Heininger for providing the Glrx−/− knock-out mice, and Andrew Das for assistance with the figures.
This work was supported by grants from the Health Research Council of New Zealand; the NHLBI, National Institutes of Health, Department of Health and Human Services, under Contract HHSN268201000031C; National Institutes of Health Grants PO1 HL068758, R01 DK103750, and R37 HL104017; American Heart Association Grant 16GRNT27660006; and National Institutes of Health Training Grant T32 HL007501 (to J. B.). The authors have no conflicts of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- DNCB
- dinitrochlorobenzene
- Grx
- glutaredoxin
- IAM
- iodoacetamide
- NEM
- N-ethylmaleimide
- Prx
- peroxiredoxin
- CysP
- peroxidatic Cys
- CysR
- resolving Cys
- GSSG
- oxidized glutathione.
References
- 1.Winterbourn C. C., and Hampton M. B. (2008) Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 45, 549–561 [DOI] [PubMed] [Google Scholar]
- 2.Hanschmann E. M., Godoy J. R., Berndt C., Hudemann C., and Lillig C. H. (2013) Thioredoxins, glutaredoxins, and peroxiredoxins—molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 19, 1539–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marinho H. S., Real C., Cyrne L., Soares H., and Antunes F. (2014) Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2, 535–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mieyal J. J., and Chock P. B. (2012) Posttranslational modification of cysteine in redox signaling and oxidative stress: focus on s-glutathionylation. Antioxid. Redox Signal. 16, 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J., and Holmgren A. (2014) The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 [DOI] [PubMed] [Google Scholar]
- 6.Hanschmann E. M., Lönn M. E., Schütte L. D., Funke M., Godoy J. R., Eitner S., Hudemann C., and Lillig C. H. (2010) Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J. Biol. Chem. 285, 40699–40705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Y., Zhang H., Lu J., and Holmgren A. (2012) Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 287, 38210–38219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H., Du Y., Zhang X., Lu J., and Holmgren A. (2014) Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 21, 669–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casagrande S., Bonetto V., Fratelli M., Gianazza E., Eberini I., Massignan T., Salmona M., Chang G., Holmgren A., and Ghezzi P. (2002) Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc. Natl. Acad. Sci. U.S.A. 99, 9745–9749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall A., Karplus P. A., and Poole L. B. (2009) Typical 2-Cys peroxiredoxins—structures, mechanisms and functions. FEBS J. 276, 2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhee S. G., and Woo H. A. (2011) Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger HO, and protein chaperones. Antioxid. Redox. Signal. 15, 781–794 [DOI] [PubMed] [Google Scholar]
- 12.Winterbourn C. C., and Hampton M. B. (2015) Redox biology: signaling via a peroxiredoxin sensor. Nat. Chem. Biol. 11, 5–6 [DOI] [PubMed] [Google Scholar]
- 13.Randall L. M., Ferrer-Sueta G., and Denicola A. (2013) Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzymol. 527, 41–63 [DOI] [PubMed] [Google Scholar]
- 14.Delaunay A., Pflieger D., Barrault M. B., Vinh J., and Toledano M. B. (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481 [DOI] [PubMed] [Google Scholar]
- 15.Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 [DOI] [PubMed] [Google Scholar]
- 16.Jarvis R. M., Hughes S. M., and Ledgerwood E. C. (2012) Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 53, 1522–1530 [DOI] [PubMed] [Google Scholar]
- 17.Nelson K. J., Knutson S. T., Soito L., Klomsiri C., Poole L. B., and Fetrow J. S. (2011) Analysis of the peroxiredoxin family: using active-site structure and sequence information for global classification and residue analysis. Proteins 79, 947–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peskin A. V., Low F. M., Paton L. N., Maghzal G. J., Hampton M. B., and Winterbourn C. C. (2007) The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885–11892 [DOI] [PubMed] [Google Scholar]
- 19.Manta B., Hugo M., Ortiz C., Ferrer-Sueta G., Trujillo M., and Denicola A. (2009) The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 484, 146–154 [DOI] [PubMed] [Google Scholar]
- 20.Peskin A. V., Dickerhof N., Poynton R. A., Paton L. N., Pace P. E., Hampton M. B., and Winterbourn C. C. (2013) Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidative cysteine. J. Biol. Chem. 288, 14170–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chae H. Z., Kim H. J., Kang S. W., and Rhee S. G. (1999) Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 45, 101–112 [DOI] [PubMed] [Google Scholar]
- 22.Seo M. S., Kang S. W., Kim K., Baines I. C., Lee T. H., and Rhee S. G. (2000) Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 275, 20346–20354 [DOI] [PubMed] [Google Scholar]
- 23.Pace P. E., Peskin A. V., Han M. H., Hampton M. B., and Winterbourn C. C. (2013) Hyperoxidized peroxiredoxin 2 interacts with the protein disulfide-isomerase ERp46. Biochem. J. 453, 475–485 [DOI] [PubMed] [Google Scholar]
- 24.Tavender T. J., Springate J. J., and Bulleid N. J. (2010) Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park J. W., Mieyal J. J., Rhee S. G., and Chock P. B. (2009) Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J. Biol. Chem. 284, 23364–23374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J. W., Piszczek G., Rhee S. G., and Chock P. B. (2011) Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 50, 3204–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salzano S., Checconi P., Hanschmann E. M., Lillig C. H., Bowler L. D., Chan P., Vaudry D., Mengozzi M., Coppo L., Sacre S., Atkuri K. R., Sahaf B., Herzenberg L. A., Herzenberg L. A., Mullen L., and Ghezzi P. (2014) Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. U.S.A. 111, 12157–12162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Checconi P., Salzano S., Bowler L., Mullen L., Mengozzi M., Hanschmann E. M., Lillig C. H., Sgarbanti R., Panella S., Nencioni L., Palamara A. T., and Ghezzi P. (2015) Redox proteomics of the inflammatory secretome identifies a common set of redoxins and other glutathionylated proteins released in inflammation, influenza virus infection and oxidative stress. PLoS One 10, e0127086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho Y. S., Xiong Y., Ho D. S., Gao J., Chua B. H., Pai H., and Mieyal J. J. (2007) Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 43, 1299–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klomsiri C., Nelson K. J., Bechtold E., Soito L., Johnson L. C., Lowther W. T., Ryu S. E., King S. B., Furdui C. M., and Poole L. B. (2010) Use of dimedone-based chemical probes for sulfenic acid detection evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol. 473, 77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagy P., and Winterbourn C. C. (2010) in Advances in Molecular Toxicology (Fishbein J. C., ed) pp. 183–222, Elsevier, New York [Google Scholar]
- 32.Winterbourn C. C., and Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 33.Low F. M., Hampton M. B., Peskin A. V., and Winterbourn C. C. (2007) Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 109, 2611–2617 [DOI] [PubMed] [Google Scholar]
- 34.Gromer S., Arscott L. D., Williams C. H. Jr., Schirmer R. H., and Becker K. (1998) Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 273, 20096–20101 [DOI] [PubMed] [Google Scholar]
- 35.Rhee S. G., Chae H. Z., and Kim K. (2005) Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 38, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 36.Karplus P. A. (2015) A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 80, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood Z. A., Poole L. B., and Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 [DOI] [PubMed] [Google Scholar]
- 38.Seo J. H., Lim J. C., Lee D. Y., Kim K. S., Piszczek G., Nam H. W., Kim Y. S., Ahn T., Yun C. H., Kim K., Chock P. B., and Chae H. Z. (2009) Novel protective mechanism against irreversible hyperoxidation of peroxiredoxin: Nα-terminal acetylation of human peroxiredoxin II. J. Biol. Chem. 284, 13455–13465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harwood D. T., Kettle A. J., Brennan S., and Winterbourn C. C. (2009) Simultaneous determination of reduced glutathione, glutathione disulphide and glutathione sulphonamide in cells and physiological fluids by isotope dilution liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 3393–3399 [DOI] [PubMed] [Google Scholar]
- 40.Wolff S. P. (1994) Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 233, 182–189 [Google Scholar]