

Abstract
The differentiation and fusion of myoblasts into mature myotubes are complex processes responding to multiple signaling pathways. The function of Akt/PKB is critical for myogenesis, but less is clear as to the regulation of its isoform-specific expression. Bexarotene is a drug already used clinically to treat cancer, and it has the ability to enhance the commitment of embryonic stem cells into skeletal muscle lineage. Whereas bexarotene regulates fundamental biological processes through retinoid X receptor (RXR)-mediated gene expression, molecular pathways underlying its positive effects on myogenesis remain unclear. In this study, we have examined the signaling pathways that transmit bexarotene action in the context of myoblast differentiation. We show that bexarotene promotes myoblast differentiation and fusion through the activation of RXR and the regulation of Akt/PKB isoform-specific expression. Interestingly, bexarotene signaling appears to correlate with residue-specific histone acetylation and is able to counteract the detrimental effects of cachectic factors on myogenic differentiation. We also signify an isoform-specific role for Akt/PKB in RXR-selective signaling to promote and to retain myoblast differentiation. Taken together, our findings establish the viability of applying bexarotene in the prevention and treatment of muscle-wasting disorders, particularly given the lack of drugs that promote myogenic differentiation available for potential clinical applications. Furthermore, the model of bexarotene-enhanced myogenic differentiation will provide an important avenue to identify additional genetic targets and specific molecular interactions that we can study and apply for the development of potential therapeutics in muscle regeneration and repair.
Keywords: Akt PKB, cancer, gene regulation, myogenesis, nuclear receptor, cachexia
Introduction
Many diseases and underlying conditions, such as cancer, aging, AIDS, inflammation, congestive heart failure, and chronic obstructive pulmonary diseases often present with muscle-wasting disorders characterized by a progressive loss of skeletal muscle mass (1). They can be extremely debilitating and correlate with a poor quality of life and high mortality rate. Pharmacotherapy that can prevent the muscle loss would be a solution, but currently no such drugs are approved for clinical application. Understanding the molecular mechanisms governing the differentiation and fusion of skeletal myoblasts will be critical to the finding of a treatment regime for muscle wasting disorders.
The formation of myoblasts from myogenic progenitors and their subsequent differentiation into mature skeletal myocytes are highly ordered processes coordinated by multiple myogenic regulatory factors, including Myf5, MyoD, and myogenin (2). Myf5 and MyoD are expressed in proliferating myoblasts and are responsible for the commitment of skeletal muscle lineage (3), whereas myogenin is expressed in differentiating myoblasts and controls the differentiation and fusion of myoblasts into myotubes (3).
Akt/PKB is a serine/threonine kinase important for the regulatory events of many cellular activities (4–6). There are three isoforms of Akt in mammals, namely Akt1, Akt2, and Akt3. Although Akt1 is the predominant isoform found in most tissues, Akt2 is highly expressed in skeletal muscle and liver (7, 8). Akt3 expression is more restricted and most abundant in the brain (8). Because of sequence and structure similarities, the Akt isoforms share some overlapping functions and can compensate for the loss of one another (9). Nonetheless, it is increasingly recognized that Akt1 is mainly involved in cellular survival pathways and Akt2 in glucose homeostasis. Although the function of Akt3 is less clear, it has been implicated in brain development (9–14).
The RXRs3 belong to the nuclear receptor superfamily. They function as transcription factors and are amenable to ligand activation (15). There are three subtypes of RXR, explicitly RXRα, RXRβ, and RXRγ. Notably, the RXRs are able to constitutively bind to DNA motifs as homodimers or as dimerization partners for other nuclear receptors (16, 17). The DNA-binding specificity of the RXRs is determined by the type of dimerization and the number of spacer nucleotides between the two direct repeats of the canonical binding sequence, and ligand activation of RXR occurs in homodimers or permissive heterodimers (18, 19).
Bexarotene is an RXR-selective ligand that has been approved by the Food and Drug Administration for use in the treatment of refractory or persistent cutaneous T-cell lymphoma, and it has been shown to reduce tumor growth in several additional cancers as well (20, 21). Moreover, bexarotene is an efficient enhancer for the specification of skeletal muscle lineage (22). Although bexarotene regulates fundamental biological processes through RXR-mediated gene expression (23), molecular pathways underlying its antitumor effects and its positive effects on myogenic differentiation are less clear. Thus, it is fundamental to understand on a molecular level how different molecular pathways converge to mediate bexarotene action in specific cellular settings.
In this study, we have examined the signaling pathways that transmit bexarotene action in the context of myoblast differentiation. Our studies have determined a role for Akt2 in RXR-selective signaling to promote and retain myogenic differentiation. Our data also suggest a potential application for bexarotene in muscle regeneration and repair.
Experimental Procedures
Cell Culture and Reagents
Mouse primary myoblasts were isolated as described previously (24). Briefly, lower hind limb muscles from 6- to 8-week-old C57BL/6 female mice (Charles River laboratories, gift from Dr. Wiper-Bergeron) were dissected and digested with dispase and collagenase. Isolated cells were grown on Matrigel-coated dishes in DMEM supplemented with 20% FBS, 10% horse serum in the presence of 10 ng/ml basic FGF and 2 ng/ml HGF, at 37 °C with 5% CO2. To induce differentiation, the medium of 70% confluent cell cultures was changed to DMEM containing 2% FBS and 10% horse serum. The C2C12 myoblasts (ATCC) were maintained in GM, DMEM supplemented with 10% FBS, at 37 °C with 5% CO2. To induce differentiation, the medium of 80% confluent cell cultures was changed to differentiation medium, DMEM supplemented with 2% horse serum, for 1–4 days (25). Human prostate cancer cells (PC3, ATCC) were maintained in RPMI 1640 medium (Gibco) with 10% FBS. PC3-conditioned media were collected 48 h after the medium change for 90% confluent PC3 cultures. Prior to differentiation, C2C12 myoblasts were grown in GM supplemented with PC3-conditioned medium (1:1 ratio) for 48 h and then the medium was changed to fresh differentiation medium for differentiation. Mock-conditioned medium was obtained from proliferating C2C12 cultures. Bexarotene was from LC Laboratories, and UVI3003 was from Tocris.
Immunofluorescence Microscopy
Following differentiation, the cells were first fixed with cold methanol, rehydrated in PBS, and incubated with myosin heavy chain antibody (hybridoma MF20) overnight, washed with PBS, and incubated with Alexa Fluor® 594 secondary antibody (Invitrogen). The cells were also incubated with 0.1 μg/ml Hoechst to stain the DNA. Axiovert 200 M microscope, AxioCam HRM camera, and AxioVision Rel 4.6 software (Zeiss) were used to capture the images through a ×10 objective. For each coverslip, five random images were analyzed, and the percentage of skeletal myocytes was determined as the fraction of myosin heavy chain-positive cells relative to the total number of nuclei. ImageJ software was used for cell counting. Student's t tests were used for the statistical analysis.
Western Analysis
Whole cell extracts were prepared as described previously (26). Protein concentrations were determined using a protein assay dye reagent (Bio-Rad) and multiscan spectrum photospectrometer (Thermo). Equal amounts of protein were resolved on SDS-polyacrylamide gel and transferred onto an Immuno-Blot PVDF membrane (Bio-Rad). Scion Image software (Scion Corp.) was used for quantifications. Antibodies against Akt isoforms were from Cell Signaling; RXRα was from Santa Cruz Biotechnology and cyclophilin B was from Abcam. Myogenin antibody was from hybridoma F5D and β-tubulin was from E7 (22).
Reverse Transcription PCR Analysis
RNA was isolated using total RNA kit I and on-column DNase I digestion was performed using the DNase I set (Omega). Total RNA was quantified by Nanodrop (ND-1000). Reverse transcription was performed using a High Capacity cDNA reverse transcription kit (Applied Biosystems). qPCR was conducted using a SYBR® Green and ROX PCR Master Mix and HotStarTaq DNA polymerase (Qiagen) on an Mx3000P platform (Stratagene). Quantification of the targets, normalized to the Gapdh endogenous reference and relative to a calibrator control, was calculated using the formula 2−ΔΔCT. MyoD and Abca1 primers have been described previously (25, 27). The following primers were used: myogenin primers forward, ATCCAGTACATTGAGCGCCTAC, and reverse, AGCAAATGATCTCCTGGGTTGG; Akt1 primers forward, CCTGAAGCTGGAGAACCTCA, and reverse, TTCATAGTGGCACCGTCCTT; Akt2 primers forward, GCGCAAGGAGGTCATCATT, and reverse, GCATACTTGAGGGCTGTAAGG; Akt3 primers forward, AGTATGACGACGACGGCAT, and reverse, GTAGAGATGTCCAGGAATCAGTC.
shRNA Knockdown
C2C12 myoblasts were grown in GM to about 30% confluence and transduced at a multiplicity of infection of 40 with lentiviral particles targeting Rxrα or Akt isoforms in the presence of Polybrene (5 μg/ml) according to the manufacturer's protocol (Santa Cruz Biotechnology). A nonsilencing shRNA was used as a negative control. Puromycin (2 μg/ml) was used to select pooled stable clones.
ChIP Analysis
Cells were cross-linked and sonicated with a Bioruptor®. Chromatin was immunoprecipitated as described previously (25). Normal IgG antiserum was used as a negative control. Purified DNA was quantified using a NanoDrop spectrophotometer (ND-1000). qPCR was performed on the Mx3000P platform (Stratagene). Input DNA was used to generate a standard curve for each immunoprecipitate in the amplification. Quantification was determined as the abundance of target DNA as a percentage of input DNA (25). Each ChIP was repeated at least three times. Antibodies against H3K18ac and H3K27ac were from Abcam. Primer pairs used for the qPCR amplification were as follows: Abca1 promoter forward, TGCCGCGACTAGTTCCTT, and reverse, TCTCCACGTGCTTTCTGCT; Akt2 locus forward, CTTACTGTGGTCCCTAAGCAGG, and reverse, GGCAAGCCAAGATCACAAGC; Ctl locus forward, CCTGAGTATCTGGTAGGGTGTC, and reverse, GCATTTAAGAGGGCCCAGAGT.
Bioinformatic Analysis
ChIP-seq data were exported from the European Nucleotide Archive, and the sequencing reads were aligned to the mouse genome version mm9 using the short read aligner Bowtie (28). The genome coverage of reads from each sample was computed and subsequently visualized in a genome browser. Local peaks in read density identified using MACS (29) were used to select regions of interest at the Akt2 locus and guide the design of primers for ChIP-qPCR. To identify consensus RXR-binding motifs within the region of interest, the sequence was scanned with the position weight matrix for RXRα available from the JASPAR CORE database of transcription factor binding preferences.
Results
Bexarotene Enhances Myoblast Differentiation
RXR is very important for early embryonic development (30–32). Therefore, we examined the effects of bexarotene, an RXR-selective ligand, on myoblast differentiation. First, we employed a primary myoblast model in which mouse primary myoblasts were isolated from lower hind limb muscles (24), and differentiation was induced in the presence or absence of bexarotene. As shown in Fig. 1, A–C, the addition of bexarotene at a concentration close to Kd values (30 nm) significantly enhanced not only the differentiation but also the fusion of primary myoblasts by day 1 of differentiation as determined by quantitative microscopy. The ability of bexarotene to enhance myogenic differentiation was also illustrated by a significant increase in MyoD gene expression, about 1.5-fold compared with untreated myoblasts, by 12 h of differentiation as assessed by RT-qPCR analysis (Fig. 1D). More importantly, the expression of myogenin, a terminal differentiation factor and muscle identity marker, was further increased significantly by 2.5-fold following 24 h of differentiation in the presence of bexarotene (Fig. 1E).
FIGURE 1.

Effects of bexarotene on primary myoblast differentiation. A, primary myoblasts were differentiated in the presence or absence of bexarotene (Bex, 30 nm) and stained for quantitative microscopy. Differentiation was defined as the percentage of myogenic nuclei relative to the total number of nuclei. Error bars represent the standard deviations of three independent experiments (*, p < 0.05). B, fusion rate was defined as the number of nuclei per myocyte (**, p < 0.01). C, representative microscopy images stained for myosin heavy chain (red) and nuclei (blue) on day 2 of differentiation. D, levels of MyoD mRNA were determined by RT-qPCR and presented as fold changes relative to proliferating primary myoblasts (0h), normalized to Gapdh (n = 3). E, myogenin (Myog) mRNA was analyzed in parallel.
Next, we employed a very well characterized C2C12 myoblast model (33), because C2C12 cells are amenable to genetic manipulation and stable clones retain their capacity to differentiate. Similar to primary myoblasts, the differentiation and fusion of C2C12 myoblasts were also significantly enhanced by the presence of bexarotene (30–50 nm), as determined by quantitative microscopy (Fig. 2, A–C). Moreover, the levels of myogenin mRNA were significantly augmented by about 3-fold following the addition of bexarotene (Fig. 2D). The augmentation of myogenin gene expression was further corroborated by an increase in the levels of myogenin protein compared with untreated cells (Fig. 2E). Taken together, our data demonstrate that bexarotene acts as a molecular enhancer of myoblast differentiation, possibly through RXR-selective signaling.
FIGURE 2.
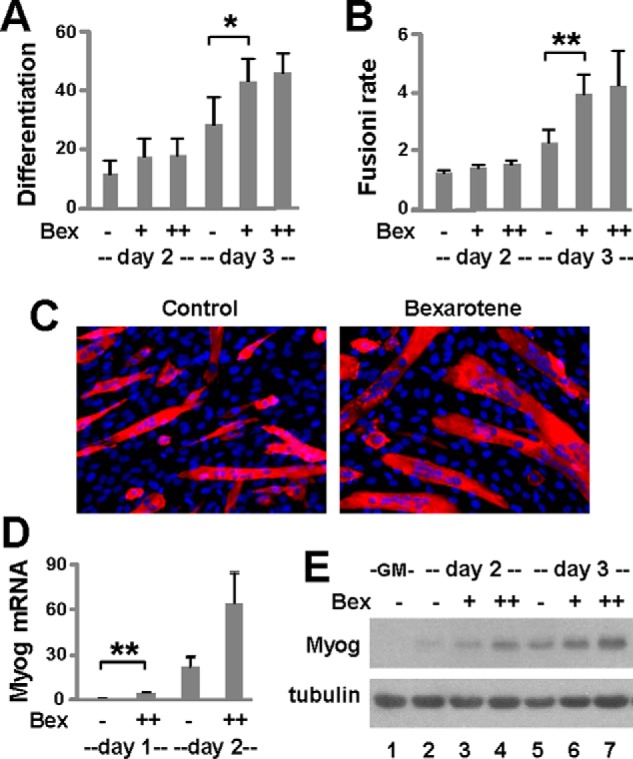
Bexarotene signaling in myoblast differentiation. A, C2C12 myoblasts were differentiated with bexarotene (Bex, 30 and 50 nm) and stained for quantitative microscopy. Differentiation was defined as the percentage of myogenic nuclei relative to the total number of nuclei. Error bars represent the standard deviations of five independent experiments (*, p < 0.05). B, fusion rate was defined as the average number of nuclei per myocyte (**, p < 0.01). C, shown are representative images stained for myosin heavy chain (red) and nuclei (blue). D, levels of myogenin (Myog) mRNA were analyzed by RT-qPCR and presented as a fold change relative to day 1 untreated differentiating myoblasts, normalized to Gapdh (n = 3). E, myogenin protein was examined by Western blotting. Proliferating myoblasts (GM) were included as controls and β-tubulin as a loading control.
Role of RXR in Bexarotene-enhanced Myoblast Differentiation
To assess whether bexarotene-enhanced myoblast differentiation is mediated through RXR activation, we first employed a potent RXR antagonist UVI3003 (34). C2C12 myoblasts were differentiated with bexarotene in the presence of high concentrations of UVI3003, about 30- and 150-fold of Kd values. As shown in Fig. 3, A–C, cotreatment with the RXR antagonist attenuated the positive effects of bexarotene on the differentiation and fusion of C2C12 myoblasts as determined by quantitative microscopy, whereas treatment with UVI3003 in the absence of bexarotene did not affect normal myoblast differentiation. Furthermore, although the levels of myogenin protein were not reduced by UVI3003 in normal myoblast differentiation, bexarotene-enhanced myogenin expression was blocked by the addition of this RXR antagonist, as revealed by quantitative Western analysis (Fig. 3, D and E), indicating that bexarotene enhances myogenic differentiation through RXR-selective signaling.
FIGURE 3.

Effects of RXR antagonist on myoblast differentiation. A, C2C12 myoblasts were differentiated with bexarotene (Bex, 50 nm) in the presence of RXR antagonist UVI 3003 (UVI, 1 and 5 μm) for 4 days. Differentiation was defined as the percentage of myogenic nuclei relative to total number of nuclei (**, p < 0.01; n = 4). B, fusion rate was defined as the average number of nuclei per myocyte. C, representative images stained for myosin heavy chain (red) and nuclei (blue). Ctl, control. D, levels of myogenin (Myog) protein were examined by Western blotting. Proliferating myoblasts were used as controls (lane 1) and β-tubulin as a loading control. E, quantification of myogenin protein is presented as a fold change relative to untreated differentiating myoblasts (n = 4).
RXRα is the main subtype of RXR involved in embryonic development (30, 31) and the predominant subtype expressed in skeletal muscle (GSE41338 (35)). Therefore, we targeted RXRα to delineate the role of RXR in myogenic differentiation. A subtype-specific shRNA was used to knock down RXRα, and a nonsilencing shRNA was used as a negative control (Fig. 4A). Validation experimentation showed that induction of Abca1, a direct target gene, by bexarotene was blunted in the pooled shRXRα stable cells compared with the control cells as determined by RT-qPCR analysis (Fig. 4B). Differentiation of the RXRα knockdown cells was then induced in the presence or absence of bexarotene. As shown in Fig. 4, C–E, knockdown of the RXRα subtype prevented bexarotene-enhanced myoblast differentiation and fusion events significantly. In addition, the RXRα knockdown encumbered bexarotene-enhanced myogenin gene expression as determined by RT-qPCR analysis (Fig. 4F). The impaired myogenin gene expression following RXRα knockdown was also substantiated by a decrease in the levels of myogenin protein (Fig. 4G). Thus, bexarotene enhances myogenic differentiation through the function of RXR as a transcription factor. Interestingly, the baseline of myogenin expression appears to be also affected by the knockdown of RXRα (Fig. 4, F and G), suggesting a role for unliganded RXR in myogenin expression.
FIGURE 4.

Role of RXR in bexarotene-enhanced myoblast differentiation. A, levels of RXRα protein were examined by Western blotting following RXRα knockdown (shRXRα), and a non-silencing shRNA (shCtl) was used as a control. B, mRNA levels of Abca1 were assessed by RT-qPCR and presented as the fold change in relation to proliferating myoblasts (GM, n = 3). C, RXRα knockdown cells were differentiated with bexarotene (Bex, 50 nm) for 4 days and processed for microscopy. Differentiation was defined as the percentage of myogenic nuclei relative to the total number of nuclei (**, p < 0.01; n = 4). D, fusion rate was defined as the average number of nuclei per myocyte. E, shown are representative images stained for myosin heavy chain (red) and nuclei (blue). F, mRNA levels of myogenin (Myog) were determined by RT-qPCR and presented as the fold change in relation to proliferating myoblasts (n = 3). G, myogenin protein was examined by Western blotting. Proliferating myoblasts were used as controls (lanes 1 and 4). DM, differentiation medium.
Bexarotene Augments Akt2 Isoform Expression
Akt is activated and stabilized during myoblast differentiation (36). Therefore, we examined the effects of bexarotene on the expression of Akt isoforms to probe for molecular pathways that mediate RXR function during myogenic differentiation. We detected a significant increase in the expression of Akt2, but not Akt1 or Akt3, during the early stages of myoblast differentiation as revealed by quantitative Western analysis (Fig. 5, A and B). More importantly, bexarotene enhanced Akt2 expression during myoblast differentiation significantly but had no such effects on Akt1 or Akt3 expression (Fig. 5, A and B). This further increase in Akt2 expression by bexarotene was mirrored in parallel by significant increases in the levels of myogenin and myosin heavy chain protein (Fig. 5, A, C, and D).
FIGURE 5.

Effects of RXR signaling on Akt isoform expression. A, C2C12 myoblasts were differentiated with bexarotene (Bex, 50 nm). Protein levels of Akt1, Akt2, Akt3, myogenin (Myog), and myosin heavy chain (MyHC) were examined using Western blotting. Proliferating myoblasts (GM) were included as controls and β-tubulin as a loading control. B, quantification of Akt isoform expression is presented as fold changes relative to proliferating myoblasts (**, p < 0.01; n = 5). C, quantification of myogenin protein is presented as a fold change in relation to day 1 untreated differentiating myoblasts (n = 5). D, quantification of myosin heavy chain protein is plotted as the fold change relative to day 2 untreated differentiating myoblasts (n = 5). E, Akt1, Akt2, Akt3, and myogenin expression were examined using Western blotting following RXRα knockdown (shRXRα). A non-silencing shRNA (shCtl) was used as a control. DM, differentiation medium. F, primary myoblasts were differentiated with bexarotene. Protein levels of Akt2 and myogenin were examined using Western blotting. Proliferating primary myoblasts were included as controls (0h) and cyclophilin B (CypB) as a loading control. G, quantification of Akt2 protein is presented as a fold change relative to proliferating primary myoblasts. Error bars represent the standard deviations of four independent experiments. H, quantification of myogenin protein is presented as a fold change relative to day 1 untreated differentiating primary myoblasts (*, p < 0.05).
We next examined the role of RXR on Akt isoform-specific expression using our established RXRα knockdown cells. As shown in Fig. 5E, knockdown of RXRα attenuated the positive effects of bexarotene on Akt2 expression, while having no impact on Akt1 and Akt3 expression regardless of treatment as determined by Western analysis.
In addition, during primary myoblast differentiation, the level of Akt2 protein increased significantly by about 2-fold and was further enhanced by another 2-fold following bexarotene treatment (Fig. 5, F and G). The positive effect of bexarotene on primary myoblast differentiation was corroborated by a significant increase in myogenin expression (Fig. 5, F and H). Thus, bexarotene enhances myogenic differentiation possibly through the regulation of Akt2 gene expression.
Akt2 Is Important for Bexarotene-enhanced Myoblast Differentiation
To discern the contribution of Akt isoforms to bexarotene-enhanced myoblast differentiation, we knocked down each individual Akt isoform using isoform-specific shRNA while using a nonsilencing shRNA as the negative control (Fig. 6, A–C). Differentiation of the pooled stable cells was induced in the presence of bexarotene. As shown in Fig. 6, D–F, knockdown of Akt1 decreased the frequency of myoblast differentiation and fusion, but the cells were still able to respond to bexarotene resulting in significantly higher rates of differentiation and fusion events as determined by quantitative microscopy. Similarly, knockdown of Akt3 did not prevent the enhancement effect of bexarotene on myogenic differentiation (Fig. 6, D–F). In contrast, knockdown of Akt2 attenuated bexarotene-enhanced myoblast differentiation and fusion events, while having little impact on normal myoblast differentiation (Fig. 6, D–F). The differential capacities of Akt isoforms to mediate bexarotene action were also mirrored in the protein levels of myogenin and myosin heavy chain, identity markers of skeletal myocytes (Fig. 6G). Thus, our data indicate that Akt2 is the major isoform involved in mediating bexarotene action to promote myogenic differentiation.
FIGURE 6.

Roles of Akt isoform signaling in myoblast differentiation. A–C, individual Akt isoforms were examined using Western blotting following isoform-specific knockdowns (shAkt1, shAkt2, and shAkt3). A non-silencing shRNA (shCtl) was used as a control and β-tubulin as a loading control. D, cells were differentiated with bexarotene (Bex, 50 nm) for 3 days and stained for microscopy. Differentiation was defined as the percentage of myogenic nuclei relative to the total number of nuclei (*, p < 0.05; **, p < 0.01; n = 4). E, fusion rate was the average number of nuclei per myocyte. F, representative images stained for myosin heavy chain (red) and nuclei (blue). Ctl, control. G, protein levels of myogenin (Myog) and myosin heavy chain (MyHC) on day 1 of differentiation were determined using Western blotting following Akt isoform-specific knockdowns. Proliferating primary myoblasts (GM) were included as controls and cyclophilin B (CypB) as a loading control. H, relative abundance of Akt1, Akt2, and Akt3 mRNA in differentiating myoblasts is presented as the fold change relative to that of Akt1 in proliferating myoblasts (n = 5).
In addition, RT-qPCR analysis revealed that the relative abundance of Akt2 and Akt3 isoforms was significantly lower than that of Akt1, about 40 and 20% of Akt1, respectively (Fig. 6H). More importantly, although the levels of Akt1 and Akt3 mRNA remained relatively constant during myoblast differentiation, the levels of Akt2 mRNA increased significantly to a similar level as Akt1 by day 2 of differentiation (Fig. 6H). Taken together, our data suggest that bexarotene exerts its enhancement effects on myogenic differentiation through RXR-mediated Akt2 gene regulation, and thus Akt2 can be specifically targeted to achieve more efficient myogenic differentiation.
Bexarotene-responsive Histone Acetylation at the Akt2 Locus
To determine whether bexarotene affects Akt2 gene expression at the level of transcription, we examined Akt2 mRNA levels during bexarotene-enhanced myogenic differentiation. As shown in Fig. 7A, the mRNA levels of Akt1 and Akt3 in differentiating myoblasts were similar to those in proliferating myoblasts and were not affected by the presence of bexarotene as determined by RT-qPCR analysis. However, Akt2 mRNA levels increased significantly by about 1.5-fold in differentiating myoblasts compared with proliferating myoblasts (Fig. 7A). Moreover, treatment with bexarotene increased Akt2 mRNA significantly by another 1.5-fold in differentiating myoblasts (Fig. 7A), suggesting that RXR-selective signaling is involved in Akt2 gene regulation.
FIGURE 7.

Histone acetylation at the Akt2 locus. A, transcript level of each Akt isoform on day 1 of differentiation was determined by RT-qPCR analysis. Quantification is presented as the fold change relative to respective isoforms in proliferating myoblasts (GM) normalized to Gapdh (*, p < 0.05; n = 5). DM, differentiation medium. B, binding of RXR to the Akt2 locus was determined in parallel by quantitative ChIP analysis using antibodies against RXRα. Normal IgG antiserum was used as a negative control. Quantification is presented as the percentage of enrichment in relation to the input chromatin DNA. The Abca1 promoter was used as a positive control. C, ChIP analysis was performed using antibodies against H3K18ac or H3K27ac with the same batch of chromatin (**, p < 0.01; n = 3). Proliferating myoblasts were used as controls. Error bars represent the standard deviation of three independent experiments. An intergenic region (Ctl) was included in the qPCR analysis as a negative control. D, Western analysis of global levels of H3K18ac and H3K27ac. Protein levels of H3, RXRα, and myogenin (Myog) were examined in parallel with β-tubulin as a loading control.
Both RXRα and Akt2 are highly expressed in liver (GSE41338 (35)). We therefore used publicly available RXRα ChIP-seq reads (23) from mouse liver tissue for visualization of RXR occupancy at the Akt2 locus. An enrichment of RXR and a putative bexarotene-responsive region was identified ∼12 kb upstream of the transcription start site. A consensus RXRα-binding motif (DR1) was also found within this region using the position weight matrix given in the JASPAR CORE database of transcription factor profiles. Using an antibody against RXRα, we subsequently validated the binding of RXRα to this region during bexarotene-enhanced myoblast differentiation by ChIP-qPCR analysis. Normal IgG antiserum was used as a negative control in the ChIP analysis. As shown in Fig. 7B, RXRα binding was detected at the Abca1 promoter and the Akt2 region in proliferating myoblasts and differentiating myoblasts in the presence or absence of bexarotene. Thus the occupancy of RXRα at the Akt2 locus is constitutive. We next wished to determine whether the binding of RXR to this region couples with bexarotene-responsive and residue-specific histone acetylation.
H3K18ac is often associated with enhancer activity and has been linked to the function of nuclear receptors (37). This putative bexarotene-responsive region is also marked by peaks in the H3K18ac and H3K27ac signal according to ChIP-seq reads from C2C12-proliferating myoblasts and myotubes (38, 39). We therefore examined the status of H3K18ac and H3K27ac in parallel at this region during bexarotene-enhanced myoblast differentiation using ChIP-qPCR analysis. Consistent with published ChIP-seq data, enrichment in H3K18ac signals was readily detectable at this region in proliferating myoblasts compared with a control locus (Fig. 7C). Most importantly, H3K18ac signals increased significantly on day 1 of differentiation by about 2-fold and were further significantly increased by another 1.5-fold following the addition of bexarotene (Fig. 7C). The increase in H3K18ac signals was not only correlated with the up-regulation of Akt2 mRNA (Fig. 7A) but was also mirrored by an increase in myogenin expression (Fig. 7D).
Similar to H3K18ac, enrichment of H3K27ac was also readily detectable at this RXR-bound region in proliferating myoblasts (Fig. 7C). However, increase in H3K27ac on day 1 of differentiation and its further augmentation in the presence of bexarotene were moderate (Fig. 7C) in line with a previous report (40). Nonetheless, global levels of H3K18ac, H3K27ac, and H3 were relatively constant during the early stage of differentiation and not affected by the addition of bexarotene (Fig. 7D). Taken together, our study identifies an RXR-bound region that is marked by bexarotene-responsive and residue-specific histone acetylation within the Akt2 locus and suggests that this region may confer RXR function to regulate Akt2 isoform-specific expression.
Capacity of Bexarotene to Retain Myogenic Differentiation Following Cachectic Insult
To explore the potential application of bexarotene-enhanced myogenic differentiation, we employed a well established human prostate cancer cell (PC3)-conditioned muscle-wasting model relevant to cancer cachexia (41). C2C12 myoblasts were grown in PC3- or mock-conditioned media for 2 days, and then differentiation was induced in fresh media in the presence of bexarotene. Consistent with previous reports, PC3-conditioned media inhibited significantly the differentiation and fusion of myoblasts as determined by quantitative microscopy (Fig. 8, A–C). Remarkably, bexarotene was able to counter the detrimental effects of tumor-derived factors and retained significantly the differentiation and fusion of myoblasts following cachectic insult (Fig. 8, A–C). The ability of bexarotene to partially rescue myoblast differentiation following cachectic insult was also reflected in partial recovery of myogenin expression as assessed by quantitative Western analysis (Fig. 8, D and E).
FIGURE 8.

Capacity of bexarotene to retain myogenic differentiation following cachectic insult. A, C2C12 myoblasts were cultured with mock- or PC3-conditioned media for 2 days and then differentiated in fresh media in the presence of bexarotene (Bex, 50 nm) for 3 days and stained for microscopy. Differentiation was defined as the percentage of myogenic nuclei in relation to the total number of nuclei (*, p < 0.05; **, p < 0.01; n = 4). B, fusion rate was defined as the average number of nuclei per myocyte. C, representative images stained for myosin heavy chain (red) and nuclei (blue). Ctl, control. D, protein levels of Akt1, Akt2, and myogenin (Myog) on day 1 of differentiation were analyzed using Western blotting. Mock-conditioned (lane 1) and PC3-conditioned (lane 2) proliferating myoblasts (GM) were used as controls and β-tubulin as a loading control. E, quantification of myogenin protein is presented as a fold change relative to mock-conditioned untreated differentiating myoblasts (n = 5). F, quantification of Akt2 on day 2 of differentiation is presented as a fold change fold change relative to mock-conditioned proliferating myoblasts (n = 5).
Most intriguingly, although PC3-conditioned media did not have much impact on the levels of Akt1, the most abundant isoform, it significantly prevented the up-regulation of Akt2 during the early stages of myoblast differentiation (Fig. 8, D and F). In addition, treatment with bexarotene significantly alleviated the impairment of Akt2 expression caused by tumor-derived factors (Fig. 8, D and F). Therefore, PC3-conditioned media inhibit myoblast differentiation at least in part through the repression of Akt2 gene expression, and bexarotene partially rescues the differentiation and fusion of myoblasts through the regulation of Akt2 isoform-specific expression. Taken together, our studies suggest a potential use of bexarotene in the prevention or treatment of cancer-related muscle atrophy.
Discussion
We have examined the effects of an RXR-selective ligand on myogenic differentiation. We show that bexarotene promotes myoblast differentiation and fusion through the regulation of Akt2 isoform-specific expression. We also show that bexarotene is able to counteract the detrimental effects of cachectic factors on myogenic differentiation. Our findings establish the feasibility of applying this RXR-selective ligand to prevent and treat muscle-wasting disorders. In addition, the model of bexarotene-enhanced or -retained myogenic differentiation will provide an important avenue to identify additional bexarotene target genes and specific interactions that we can study and apply to the development of potential therapeutics in muscle regeneration and repair.
Our finding that bexarotene, a Food and Drug Administration-approved drug, enhances the differentiation and fusion of both primary and C2C12 myoblasts (Figs. 1 and 2) is novel and significant, given the lack of small molecules that enhance myogenic differentiation available for potential clinical applications. Most importantly, our observation that bexarotene can partially rescue tumor factor-induced muscle wasting (Fig. 8) presents a potential solution, as it is a drug that is already used clinically and there is currently no efficient pharmacotherapy that can treat or prevent muscle atrophy.
The function of RXR is essential for early embryonic development. Although RXRα null mice die in utero and have myocardial and ocular malformations, RXRβ and RXRγ null mutants are viable and appear to be normal (32, 42). Although the role of RXR in myogenic differentiation is largely unclear, advances in next-generation sequencing have allowed the mapping of genome-wide RXRα-binding sites in other experimental systems (23, 40, 43). Here, we demonstrate that bexarotene enhances myogenic differentiation through the activation of RXR (Figs. 3 and 4). Moreover, Akt2 gene expression appears to be under the control of RXR (Fig. 5). Interestingly, although Akt1 contributes to normal myoblast differentiation, Akt2 is essential for bexarotene-enhanced myoblast differentiation, particularly for the fusion events (Fig. 6). Thus, the Akt2 isoform is an important mediator of bexarotene action in the context of myogenic differentiation.
Histone acetylation can offer a useful readout for enhancer activity, but it is less clear whether it is a cause or a consequence of enhancer activation. We recently used the C2C12 model of myogenesis to profile the pattern of histone acetylation in MyoD gene regulation, because these cells provide a more homogeneous population (compared with primary myoblasts) that can be differentiated in synchronicity to provide a better gauge of chromatin dynamics during differentiation (25). In addition, studies of gene expression in this widely used model consistently provide results that are confirmed in primary tissue cells (38, 39). In this study, we identified a potential RXR binding region within the Akt2 locus (Fig. 7). Intriguingly, RXR-selective signaling is coupled significantly with H3K18 acetylation but not H3K27 acetylation (Fig. 7). Recent genome-wide studies have identified H3K27 acetylation as a transcription start site-preferred mark (44). In addition, it has been shown that H4K5/8 acetylation, but not H3K27 acetylation, increases on putative RXR-bound enhancers upon RXR ligand activation (40). Thus our study provides additional molecular insights into how mark-specific histone acetylation may be related to bexarotene-responsive locus activation and consequently gene transcription.
It is worth noting that although baseline myogenin expression, in the absence of ligand, is not affected by RXR antagonist (Fig. 3), it is reduced by the knockdown of RXR (Fig. 4). Apart from activating gene transcription upon ligand induction, the function of RXR in the absence of ligand is also important for establishing chromatin signatures at genomic loci (40, 45, 46). Thus, our data suggest that RXR knockdown per se would perturb RXR-mediated chromatin modifications at regulatory loci and subsequently affect gene expression, i.e. that an inactivated RXR also plays an active role in gene regulation.
The kinase activity of Akt is regulated by phosphorylation of serine 473 and threonine 308 that is conserved in all Akt isoforms (47). Consequently, the level of Akt proteins in general determines the rate and extent of skeletal muscle development (48). As such, Akt isoform-specific function often stems from tissue-restrictive expression and cellular localization rather than regulation of enzymatic activity. Nonetheless, little is known as to how Akt isoform-specific expression per se is regulated. Our data establish that not only does each Akt isoform have a distinct role in bexarotene-enhanced myogenic differentiation but also how Akt2 can be specifically targeted at the level of transcription to achieve a higher efficiency of differentiation, specifically in view of muscle regeneration and repair.
Although cancer cachexia is often considered as a condition associated with advanced malignancy, many patients suffer from weight loss caused by muscle wasting at the early stage of cancer. Cytokines and tumor-derived factors have been linked to the down-regulation of myogenic regulatory factors and muscle-related proteins (49–55), but the molecular basis for the pathophysiology of cachexia remains unclear. It is known that RXR is involved in inflammation and immune processes and forms permissive heterodimers with metabolic sensor receptors (56). If bexarotene is able to counteract cachectic insult and to retain myogenic differentiation (Fig. 8), dissecting the underlying mechanisms of bexarotene's capacity to promote myoblast differentiation or to protect against cachectic insult will allow us to uncover the molecular basis of cancer-associated muscle atrophy and consequently develop new strategies to prevent and treat cachexia.
Author Contributions
J. C. and Q. L. designed the research and drafted the manuscript. H. A., K. A., and W. N. performed the research. H. A., K. D., J. C., and Q. L. analyzed the data. All authors reviewed the manuscript.
Acknowledgments
We thank our colleagues for a supportive research environment and the Wiper-Bergeron Lab for help with mouse primary myoblast cultures.
This work was supported in part by an operating grant from Natural Sciences and Engineering Research Council of Canada (to Q. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- RXR
- retinoid X receptor
- GM
- growth medium
- qPCR
- quantitative PCR.
References
- 1.Fanzani A., Conraads V. M., Penna F., and Martinet W. (2012) Molecular and cellular mechanisms of skeletal muscle atrophy: an update. J. Cachexia Sarcopenia Muscle 3, 163–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berkes C. A., and Tapscott S. J. (2005) MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 16, 585–595 [DOI] [PubMed] [Google Scholar]
- 3.Tajbakhsh S., Rocancourt D., Cossu G., and Buckingham M. (1997) Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell 89, 127–138 [DOI] [PubMed] [Google Scholar]
- 4.Manning B. D., and Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J., Halappanavar S. S., St-Germain J. R., Tsang B. K., and Li Q. (2004) Role of Akt/protein kinase B in the activity of transcriptional coactivator p300. Cell. Mol. Life Sci. 61, 1675–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang W. C., and Chen C. C. (2005) Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell. Biol. 25, 6592–6602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaneko S., Feldman R. I., Yu L., Wu Z., Gritsko T., Shelley S. A., Nicosia S. V., Nobori T., and Cheng J. Q. (2002) Positive feedback regulation between Akt2 and MyoD during muscle differentiation. Cloning of Akt2 promoter. J. Biol. Chem. 277, 23230–23235 [DOI] [PubMed] [Google Scholar]
- 8.Kandel E. S., and Hay N. (1999) The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 253, 210–229 [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez E., and McGraw T. E. (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8, 2502–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho H., Mu J., Kim J. K., Thorvaldsen J. L., Chu Q., Crenshaw E. B. 3rd, Kaestner K. H., Bartolomei M. S., Shulman G. I., and Birnbaum M. J. (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292, 1728–1731 [DOI] [PubMed] [Google Scholar]
- 11.Sykes S. M., Lane S. W., Bullinger L., Kalaitzidis D., Yusuf R., Saez B., Ferraro F., Mercier F., Singh H., Brumme K. M., Acharya S. S., Schöll C., Tothova Z., Attar E. C., Fröhling S., et al. (2011) AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell 146, 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garofalo R. S., Orena S. J., Rafidi K., Torchia A. J., Stock J. L., Hildebrandt A. L., Coskran T., Black S. C., Brees D. J., Wicks J. R., McNeish J. D., and Coleman K. G. (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBβ. J. Clin. Invest. 112, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schultze S. M., Jensen J., Hemmings B. A., Tschopp O., and Niessen M. (2011) Promiscuous affairs of PKB/AKT isoforms in metabolism. Arch. Physiol. Biochem. 117, 70–77 [DOI] [PubMed] [Google Scholar]
- 14.Tschopp O., Yang Z. Z., Brodbeck D., Dummler B. A., Hemmings-Mieszczak M., Watanabe T., Michaelis T., Frahm J., and Hemmings B. A. (2005) Essential role of protein kinase Bγ (PKBγ/Akt3) in postnatal brain development but not in glucose homeostasis. Development 132, 2943–2954 [DOI] [PubMed] [Google Scholar]
- 15.Chambon P. (2005) The nuclear receptor superfamily: a personal retrospect on the first two decades. Mol. Endocrinol. 19, 1418–1428 [DOI] [PubMed] [Google Scholar]
- 16.Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., and Evans R. M. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gampe R. T. Jr., Montana V. G., Lambert M. H., Wisely G. B., Milburn M. V., and Xu H. E. (2000) Structural basis for autorepression of retinoid X receptor by tetramer formation and the AF-2 helix. Genes Dev. 14, 2229–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Umesono K., and Evans R. M. (1989) Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 57, 1139–1146 [DOI] [PubMed] [Google Scholar]
- 19.Leid M., Kastner P., and Chambon P. (1992) Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem. Sci. 17, 427–433 [DOI] [PubMed] [Google Scholar]
- 20.Wu K., Kim H. T., Rodriquez J. L., Hilsenbeck S. G., Mohsin S. K., Xu X. C., Lamph W. W., Kuhn J. G., Green J. E., and Brown P. H. (2002) Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol. Biomarkers Prev. 11, 467–474 [PubMed] [Google Scholar]
- 21.Gniadecki R., Assaf C., Bagot M., Dummer R., Duvic M., Knobler R., Ranki A., Schwandt P., and Whittaker S. (2007) The optimal use of bexarotene in cutaneous T-cell lymphoma. Br. J. Dermatol. 157, 433–440 [DOI] [PubMed] [Google Scholar]
- 22.Le May M., Mach H., Lacroix N., Hou C., Chen J., and Li Q. (2011) Contribution of retinoid X receptor signaling to the specification of skeletal muscle lineage. J. Biol. Chem. 286, 26806–26812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boergesen M., Pedersen T. Å., Gross B., van Heeringen S. J., Hagenbeek D., Bindesbøll C., Caron S., Lalloyer F., Steffensen K. R., Nebb H. I., Gustafsson J. Å., Stunnenberg H. G., Staels B., and Mandrup S. (2012) Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor α in mouse liver reveals extensive sharing of binding sites. Mol. Cell. Biol. 32, 852–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Megeney L. A., Kablar B., Garrett K., Anderson J. E., and Rudnicki M. A. (1996) MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 10, 1173–1183 [DOI] [PubMed] [Google Scholar]
- 25.Hamed M., Khilji S., Chen J., and Li Q. (2013) Stepwise acetyltransferase association and histone acetylation at the Myod1 locus during myogenic differentiation. Sci. Rep. 3, 2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J., St-Germain J. R., and Li Q. (2005) B56 regulatory subunit of protein phosphatase 2A mediates valproic acid-induced p300 degradation. Mol. Cell. Biol. 25, 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan R., Gan X., Menke J. G., Wright S. D., and Cai T. Q. (2002) Bacterial lipopolysaccharide induces expression of ABCA1 but not ABCG1 via an LXR-independent pathway. J. Lipid Res. 43, 952–959 [PubMed] [Google Scholar]
- 28.Langmead B., Trapnell C., Pop M., and Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., and Liu X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sapin V., Dollé P., Hindelang C., Kastner P., and Chambon P. (1997) Defects of the chorioallantoic placenta in mouse RXRα null fetuses. Dev. Biol. 191, 29–41 [DOI] [PubMed] [Google Scholar]
- 31.Sucov H. M., Dyson E., Gumeringer C. L., Price J., Chien K. R., and Evans R. M. (1994) RXRα mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 8, 1007–1018 [DOI] [PubMed] [Google Scholar]
- 32.Kastner P., Grondona J. M., Mark M., Gansmuller A., LeMeur M., Decimo D., Vonesch J. L., Dollé P., and Chambon P. (1994) Genetic analysis of RXRα developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 78, 987–1003 [DOI] [PubMed] [Google Scholar]
- 33.Silberstein L., Webster S. G., Travis M., and Blau H. M. (1986) Developmental progression of myosin gene expression in cultured muscle cells. Cell 46, 1075–1081 [DOI] [PubMed] [Google Scholar]
- 34.Nahoum V., Pérez E., Germain P., Rodríguez-Barrios F., Manzo F., Kammerer S., Lemaire G., Hirsch O., Royer C. A., Gronemeyer H., de Lera A. R., and Bourguet W. (2007) Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc. Natl. Acad. Sci. U.S.A. 104, 17323–17328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barbosa-Morais N. L., Irimia M., Pan Q., Xiong H. Y., Gueroussov S., Lee L. J., Slobodeniuc V., Kutter C., Watt S., Colak R., Kim T., Misquitta-Ali C. M., Wilson M. D., Kim P. M., Odom D. T., et al. (2012) The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593 [DOI] [PubMed] [Google Scholar]
- 36.Fujio Y., Guo K., Mano T., Mitsuuchi Y., Testa J. R., and Walsh K. (1999) Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol. Cell. Biol. 19, 5073–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin Q., Yu L. R., Wang L., Zhang Z., Kasper L. H., Lee J. E., Wang C., Brindle P. K., Dent S. Y., and Ge K. (2011) Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asp P., Blum R., Vethantham V., Parisi F., Micsinai M., Cheng J., Bowman C., Kluger Y., and Dynlacht B. D. (2011) Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc. Natl. Acad. Sci. U.S.A. 108, E149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blum R., Vethantham V., Bowman C., Rudnicki M., and Dynlacht B. D. (2012) Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 26, 2763–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daniel B., Nagy G., Hah N., Horvath A., Czimmerer Z., Poliska S., Gyuris T., Keirsse J., Gysemans C., Van Ginderachter J. A., Balint B. L., Evans R. M., Barta E., and Nagy L. (2014) The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 28, 1562–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Z., and Clemens P. R. (2006) Cellular caspase-8-like inhibitory protein (cFLIP) prevents inhibition of muscle cell differentiation induced by cancer cells. FASEB J. 20, 2570–2572 [DOI] [PubMed] [Google Scholar]
- 42.Krezel W., Dupé V., Mark M., Dierich A., Kastner P., and Chambon P. (1996) RXR γ null mice are apparently normal and compound RXRα +/−/RXRβ−/−/RXRγ−/− mutant mice are viable. Proc. Natl. Acad. Sci. U.S.A. 93, 9010–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen R., Pedersen T. A., Hagenbeek D., Moulos P., Siersbaek R., Megens E., Denissov S., Børgesen M., Francoijs K. J., Mandrup S., and Stunnenberg H. G. (2008) Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 22, 2953–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajagopal N., Ernst J., Ray P., Wu J., Zhang M., Kellis M., and Ren B. (2014) Distinct and predictive histone lysine acetylation patterns at promoters, enhancers, and gene bodies. G3 4, 2051–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folkers G. E., van der Burg B., and van der Saag P. T. (1998) Promoter architecture, cofactors, and orphan receptors contribute to cell-specific activation of the retinoic acid receptor β2 promoter. J. Biol. Chem. 273, 32200–32212 [DOI] [PubMed] [Google Scholar]
- 46.Higazi A., Abed M., Chen J., and Li Q. (2011) Promoter context determines the role of proteasome in ligand-dependent occupancy of retinoic acid responsive elements. Epigenetics 6, 202–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datta S. R., Brunet A., and Greenberg M. E. (1999) Cellular survival: a play in three Akts. Genes Dev. 13, 2905–2927 [DOI] [PubMed] [Google Scholar]
- 48.Gardner S., Anguiano M., and Rotwein P. (2012) Defining Akt actions in muscle differentiation. Am. J. Physiol. Cell Physiol 303, C1292–C1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Acharyya S., and Guttridge D. C. (2007) Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin. Cancer Res. 13, 1356–1361 [DOI] [PubMed] [Google Scholar]
- 50.Dodson S., Baracos V. E., Jatoi A., Evans W. J., Cella D., Dalton J. T., and Steiner M. S. (2011) Muscle wasting in cancer cachexia: clinical implications, diagnosis, and emerging treatment strategies. Annu. Rev. Med. 62, 265–279 [DOI] [PubMed] [Google Scholar]
- 51.Lokireddy S., Wijesoma I. W., Bonala S., Wei M., Sze S. K., McFarlane C., Kambadur R., and Sharma M. (2012) Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem. J. 446, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Acharyya S., Ladner K. J., Nelsen L. L., Damrauer J., Reiser P. J., Swoap S., and Guttridge D. C. (2004) Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Invest. 114, 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guttridge D. C., Mayo M. W., Madrid L. V., Wang C. Y., and Baldwin A. S. Jr. (2000) NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 [DOI] [PubMed] [Google Scholar]
- 54.Langen R. C., Van Der Velden J. L., Schols A. M., Kelders M. C., Wouters E. F., and Janssen-Heininger Y. M. (2004) Tumor necrosis factor-α inhibits myogenic differentiation through MyoD protein destabilization. FASEB J. 18, 227–237 [DOI] [PubMed] [Google Scholar]
- 55.Szalay K., Rázga Z., and Duda E. (1997) TNF inhibits myogenesis and downregulates the expression of myogenic regulatory factors myoD and myogenin. Eur. J. Cell Biol. 74, 391–398 [PubMed] [Google Scholar]
- 56.Nagy L., Szanto A., Szatmari I., and Széles L. (2012) Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 92, 739–789 [DOI] [PubMed] [Google Scholar]