

Abstract
The biological function of interleukin-10 (IL-10), a pleiotropic cytokine with an essential role in inflammatory processes, is known to be affected by glycosaminoglycans (GAGs). GAGs are highly negatively charged polysaccharides and integral components of the extracellular matrix with important functions in the biology of many growth factors and cytokines. The molecular mechanism of the IL-10/GAG interaction is unclear. In particular, experimental evidence about IL-10/GAG binding sites is lacking, despite its importance for understanding the biological role of the interaction. Here, we report the experimental determination of a GAG binding site of IL-10. Although no co-crystal structure of the IL-10·GAG complex could be obtained, its structural characterization was possible by NMR spectroscopy. Chemical shift perturbations of IL-10 induced by GAG binding were used to narrow down the location of the binding site and to assess the affinity for different GAG molecules. Subsequent observation of NMR pseudocontact shifts of IL-10 and its heparin ligand, as induced by a protein-attached lanthanide spin label, provided structural restraints for the protein·ligand complex. Using these restraints, pseudocontact shift-based rigid body docking together with molecular dynamics simulations yielded a GAG binding model. The heparin binding site is located at the C-terminal end of helix D and the adjacent DE loop and coincides with a patch of positively charged residues involving arginines 102, 104, 106, and 107 and lysines 117 and 119. This study represents the first experimental characterization of the IL-10·GAG complex structure and provides the starting point for revealing the biological significance of the interaction of IL-10 with GAGs.
Keywords: carbohydrate function, extracellular matrix, glycosaminoglycan, heparin, nuclear magnetic resonance (NMR), interleukin-10, paramagnetic NMR, pseudocontact shifts
Introduction
Interleukin-10 (IL-10) is a key regulator of the cellular immune system with both, pro- and anti-inflammatory functions. It prevents overwhelming immune responses by inhibiting the synthesis of inflammatory cytokines, such as IFN-γ, TNF-α, and IL-1β (1). Furthermore, it reduces the potential of macrophages and dendritic cells to act as antigen-presenting cells (2, 3). On the contrary, IL-10 also has stimulatory effects on certain immune cells, among them B lymphocytes (4, 5) and natural killer cells (6). Because of its broad range of functions in the immune system, deregulation of IL-10 can lead to situations of imbalanced immune reactions; IL-10-deficient mice develop spontaneously symptoms of acute and chronic inflammation, such as enteritis, asthma, and allergy (7–9). An overproduction of IL-10 has been shown to be involved in cases of autoimmunity (systemic lupus erythematodes) (10) and cancer (B cell lymphoma) (11).
IL-10 is a member of the class II family of cytokines, which show the same 6-helix bundle topology and share a common mode of receptor complex organization and signaling (12). In particular, IL-10 forms a special protein fold by intertwining the α-helix bundles from two identical peptide chains, which generates a symmetric 38-kDa homodimer (13). This domain-swapped architecture is shared with other cellular and viral homologues of IL-10 (14). Cell signaling of IL-10 requires concerted binding to two different cell surface receptor chains and formation of a ternary receptor complex to activate the intracellular JAK-STAT pathway (12).
When secreted from cells, IL-10 can interact with components of the extracellular matrix (ECM),2 and binding to glycosaminoglycans (GAGs) was shown experimentally (15, 16). GAGs are unbranched, negatively charged polysaccharides, which exist as covalent complexes with core proteins (i.e. proteoglycans) and are integral components of the ECM in the connecting tissue, in the basal membrane, and on cell surfaces (17). Based on different sugar building blocks as well as the extent and position of sulfate groups, GAGs are grouped into five classes: hyaluronan, chondroitin sulfate, dermatan sulfate, keratan sulfate, and heparan sulfate/heparin. The biological activity of GAGs arises from their ability to interact with numerous cytokines, chemokines, and growth factors (18–20), which in consequence regulates the physiological functions of those proteins. In the case of IL-10, GAG sulfation is a prerequisite for protein/ligand binding; in NMR studies, the highest sulfated GAG, heparin, was found to be the strongest binder, whereas the affinity gradually decreased when reducing the sulfation degree, with ultimately no binding being detected for unsulfated hyaluronan (16). Salek-Ardakani et al. (15) showed that GAGs also influence the biological activity of IL-10. On the one hand, soluble GAGs were observed to inhibit the IL-10-induced expression of CD16 and CD64 on monocytes and macrophages, whereas the strength of inhibition correlated with the extent of GAG sulfation (15). On the other hand, the same study revealed that sulfated cell surface-bound GAGs were necessary to trigger the full functional activity of IL-10 toward these cells. So far, the molecular mechanisms of these effects still remain unclear. Insights into the structural principles underlying the interaction between IL-10 and GAGs are needed to understand the biological role of this interaction.
Recently, we have characterized the IL-10·GAG binding properties and investigated GAG structures in complex with IL-10 using 1H saturation transfer difference (STD) and NOESY NMR spectroscopy along with molecular modeling (16). Predictions about the location of a GAG-binding site on IL-10 have been made previously using computational methods (21, 22), but experimental results are lacking. Here, we report the first experimental evidence of a GAG binding site of IL-10. The protein was crystallized, and its structure was solved at 2.1 Å resolution; however, no IL-10·GAG co-crystal structure could be obtained. To overcome the challenges of the studied system inherent to the low GAG binding affinity in the millimolar to micromolar KD range and the high molecular mobility of the GAG, a combination of protein-detected NMR experiments was used. First, the location of the binding site was narrowed down using chemical shift perturbation experiments, which indicated that all GAG ligands populate the same epitope in IL-10. Second, the lack of NOEs as structural constraints for the IL-10·GAG system motivated the application of paramagnetic NMR experiments because these take advantage of a strong electron/nucleus dipolar interaction that can be quantified over long distances (up to 30–40 Å). Recently, pseudocontact shifts (PCSs) were successfully applied to the structural investigation of proteins interacting with small ligands (23, 24), including carbohydrates (25, 26). PCSs, which are caused by paramagnetic metal ions with an anisotropic magnetic susceptibility (e.g. lanthanides), provide a rich source of long range distance and orientation information and are manifested in the NMR spectrum as large changes in chemical shifts (27). Lanthanide ions were attached to IL-10 using a codable peptide tag and provided PCS restraints for the protein and its GAG ligand. Subsequent PCS-based rigid body docking calculations and molecular dynamics (MD) simulations yielded a structural model of the IL-10·GAG complex showing ligand binding to a patch of cationic amino acid residues located at the central crevice formed by the IL-10 dimer. Knowledge about the IL-10/GAG binding site provides the starting point for revealing the biological role of the IL-10 interaction with GAGs.
Experimental Procedures
Materials
GAG oligosaccharides were purchased from Iduron (Manchester, UK). The proteins equine myoglobin, human carbonic anhydrase 1, and bovine carbonic anhydrase 2 were ordered from Sigma-Aldrich (Taufkirchen, Germany). All other chemicals were obtained either from Sigma-Aldrich or from Carl Roth (Karlsruhe, Germany). A persulfated HA tetrasaccharide (psHA) azide was obtained by a newly developed chemoenzymatic conversion from HA polysaccharide.3 Following the preparative digestion in acidic sodium acetate buffer using commercially available bovine testis hyaluronidase, the reducing anomeric position was fixed diastereoselectively as the stable glycosyl azide in the β configuration. The nine remaining hydroxyl groups were converted to sulfate esters using highly reactive sulfur trioxide pyridine complex followed by isolation of the nonasulfate by size exclusion chromatography. Purity of psHA tetrasaccharide azide was determined by 1H and 13C NMR spectroscopy.
Preparation of IL-10
Recombinant murine IL-10 was prepared and isotopically labeled with 2H and 15N as described previously (28). Briefly, IL-10 was expressed in Escherichia coli Rosetta (DE3) (Merck, Darmstadt, Germany) and was refolded from insoluble inclusion body material. For improved refolding yield, a C149Y mutant was used to decrease incorrect disulfide bond formation. Furthermore, a C-terminal histidine tag (LEHHHHHHHH) included in the pET41b(+) vector (Merck) was attached to IL-10 for purification.
Preparation of IL-10 with Lanthanide Binding Tag
For attaching lanthanide ions to IL-10, a lanthanide binding tag (LBT) was added to the protein's C terminus by polymerase chain reaction (PCR). The LBT amino acid sequence was YIDTNNDGWYEGDELLA (25, 29). No additional linker amino acids were incorporated to prevent increasing the tag's flexibility. Furthermore, to avoid elongation of the protein's C terminus, a stop codon was inserted behind the LBT sequence to prevent expression of the vector's own histidine tag. In contrast, an N-terminal StrepII tag (WSHPQFEK) was attached to IL-10 and used for purification. The sequences of the LBT and StrepII tag were introduced by three subsequent PCR steps using two forward and three reverse complementary oligonucleotide primers. These included 5′-GTGGAGCCACCCGCAGTTCGAAAAAAGCAGGGGCCAGTACAGCCG-3′ (firstforward primer), 5′-GATATACATATGTGGAGCCACCCGCAGTTCG-3′ (second forward primer), 5′-CAGCCATCGTTGTTGGTATCAATATAGCTTTTCATTTTGATCATCATGTATGC-3′ (first reverse primer), 5′-CGCCAGCAGTTCATCGCCTTCATACCAGCCATCGTTGTTGGTATCAATATAGC-3′ (second reverse primer), and 5′-ATATACTCGAGTTACGCCAGCAGTTCATCGCCTTC-3′ (third reverse primer). Forward primer 2 and reverse primer 3 contained an NdeI or XhoI restriction site, respectively, which was used for subsequent cloning of the PCR product into a pET41b(+) vector. Correctness of the entire coding sequence was confirmed by DNA sequencing.
The tagged protein was expressed in E. coli Rosetta (DE3), labeled with 2H and/or 15N, refolded, and purified from inclusion bodies as described previously (28). To simplify the PCS analysis and enable determination of the Δχ-tensor for one single lanthanide site, an IL-10 dimer variant carrying only one LBT was produced. That “mixed” variant was composed of one monomer modified with LBT and an N-terminal StrepII tag and a second monomer carrying only the C-terminal histidine tag. Furthermore, because each nucleus occurs twice in the IL-10 dimer, which would cause peak splitting and ambiguous PCS assignments, the 15N labeling was applied only to the histidine-tagged monomer, and the LBT-tagged monomer was left unlabeled. Production of the mixed dimer variant was accomplished by mixing equal amounts of both monomers prior to protein refolding. Separation of the mixed IL-10 dimer from those dimers with equal monomer composition that also form during refolding was achieved by applying a dual purification strategy with two affinity chromatography steps. First, the protein was loaded on a nickel-nitrilotriacetic acid column (Machery-Nagel, Düren, Germany) equilibrated in 20 mm sodium phosphate (pH 7.4), 50 mm NaCl and was eluted using the same buffer containing 600 mm imidazole. Second, the protein was loaded onto a StrepTactin column (GE Healthcare, Freiburg, Germany) in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 2 mm EDTA and eluted using the same buffer containing 2.5 mm d-desthiobiotin. Only the mixed dimer with both the histidine and the StrepII tag was retained. Finally, the dimeric protein was separated from higher molecular weight aggregates, which form during protein refolding, by size exclusion chromatography using an S-200 HR (GE Healthcare) preparative gel filtration column.
Furthermore, also a uniformly composed IL-10 dimer with two identical subunits, each carrying one LBT, was prepared. This “uniform” protein was expressed unlabeled (for ligand PCS measurements) or with 15N labeling (for protein PCS measurements) and purified solely by StrepTactin affinity chromatography and gel filtration.
Sample purity and attachment of the LBT were confirmed by SDS-PAGE. To ensure that the LBT does not change the protein structure significantly, 1H-15N HSQC NMR spectroscopy was performed to record the backbone amide chemical shifts. Lanthanide binding affinity of IL-10-LBT was measured for terbium as an example using fluorescence spectroscopy as described previously (30).
X-ray Structure Determination
Because crystallization of the full-length murine IL-10 protein was not successful, a shorter S1_E8del variant with deletion of the first eight N-terminal residues was produced. Secondary structure prediction on the basis of backbone chemical shifts (28) as well as relaxation rate measurements suggested that the N terminus is unstructured and exhibits a high flexibility, which is unfavorable for crystallization. The length of the truncation was confined to eight amino acids because one of the two disulfide bonds in IL-10 involves C12, and a short overhang of three residues was left. The S1_E8del deletion was introduced by PCR using the following forward and reverse primers: 5′-GATATACATATGGACAATAACTGCACCCACTTCC-3′ and 5′-GTGGTGCTCGAGGCTTTTCATTTTGATCATCATG-3′.
Crystals were grown under 30% (v/v) glycerolethoxylate, 0.2 m ammonium acetate, 0.1 m MES (pH 6.5) at 19 °C with the hanging drop method. The protein concentration inside the drop was 5–10 mg/ml. As cryoprotecting agent, 10% (v/v) glycerol was used. Diffraction data were collected with synchrotron radiation at a wavelength of 0.91841 Å at beamline BL14.1 of the Berlin Electron Storage Ring Society for Synchrotron Radiation (BESSY). A total of 180 diffraction images with an oscillation angle of 1° were recorded at cryogenic temperature (100 K) using a MarMosaic225 phosphor image plate/CCD detector (Rayonix, Evanston, IL). Data integration, intensity scaling, and conversion to structure factor amplitudes were performed with iMOSFLM (31, 32) and SCALA (33). The structure was solved by molecular replacement using MOLREP (34) and the structure of an engineered IL-10 monomer (PDB entry 1LK3) (35) as a search model. Refinement and model building were done with REFMAC5 (36) and COOT (37), respectively. Atomic coordinates of murine IL-10, S1_E8del, C149Y were deposited in the Protein Database under accession number 4X51.
NMR Experiments
NMR experiments were recorded on a Bruker Avance III 600-MHz spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) equipped with a 5-mm inverse triple resonance probe with z-gradient. The Bruker software TopspinTM version 2.1 was used for data acquisition and spectrum processing.
For chemical shift perturbation experiments, NMR samples contained 500–800 μm partially deuterated and fully 15N-labeled IL-10 in 20 mm potassium phosphate (pH 6.2), 50 mm NaCl, 90% H2O, 10% D2O to which increasing amounts of GAG oligosaccharides were titrated. For each titration point, a 1H-15N fast HSQC experiment (38) with improved sensitivity by preventing water saturation was performed. Each spectrum was collected with 32 scans/increment with spectral widths of 9.6 kHz for 1H and 1.6 kHz for 15N as well as 128 data points in the indirect dimension. Typical 1H and 15N 90° pulse lengths were 10 and 37 μs, respectively. The experiment utilized the 3-9-19 watergate sequence (39) for water suppression and GARP (40) for heteronuclear decoupling during acquisition. The weighted chemical shift change of each NH signal was calculated according to Equation 1,
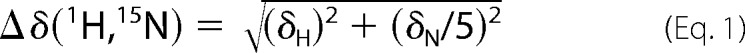 |
where δH and δN are the changes of the chemical shift in the 1H and 15N dimension, respectively.
STD control experiments were performed using a pseudo-two-dimensional NMR experiment with interleaved acquisition of on- and off-resonance spectra and selective pulses for protein saturation as described earlier (16). The binding affinity of heparin tetrasaccharide under physiological salt concentrations (10 mm sodium phosphate (pD 7.4), 150 mm NaCl in 99.8% D2O) was determined using the initial growth rate approach of the STD amplification factor as described previously (41).
For PCS measurements, lanthanide ions were added stepwise to the LBT-labeled protein while stirring the sample until an equimolar ratio was reached. Terbium, thulium, and dysprosium chloride were used to prepare paramagnetic samples, and lutetium chloride was used as a diamagnetic reference. PCSs of the protein were measured from 1H-15N spectra that were recorded in 10 mm HEPES (pH 7.2), 50 mm NaCl, 90% H2O, 10% D2O at 30 °C using a fast HSQC experiment (38). For the measurement of ligand PCSs, samples contained 380 μm IL-10-LBT and 1500 μm heparin tetrasaccharide (ΔUA-β(1–4)-GlcNS,6S-β(1–4)-IdoA,2S-β(1–4)-GlcNS,6S) in 10 mm [U-2H]HEPES (pD 7.2), 50 mm NaCl, 99.9% D2O. Because no labeled heparin ligand was available, it was used in excess (protein/ligand ratio of 1:4) to distinguish it from protein signals. That is why PCSs were the average over the free and bound state. Due to its moderate KD of 0.30 ± 0.04 mm, the heparin ligand undergoes exchange between both states, also causing PCS averaging. Thus, the PCS value was corrected for the amount of bound heparin on the basis of a known 1:1 binding stoichiometry (16) using the total protein ([P]) and ligand ([L]) concentrations as well as the KD value.
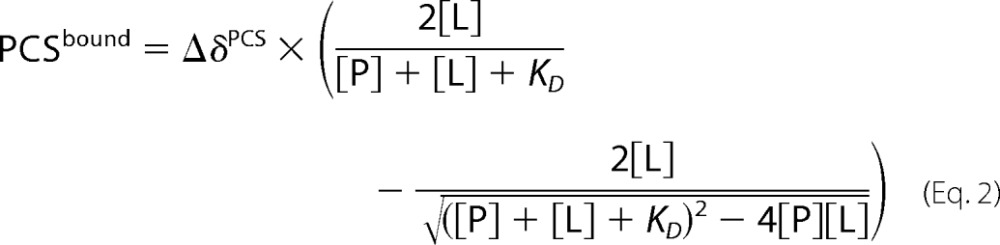 |
Heparin spectra were recorded using a 1H-13C HSQC experiment with sensitivity enhancement due to a double back INEPT transfer (42), with field gradients for coherence selection (43, 44) and GARP decoupling (40) during acquisition. Typical 1H and 13C 90° pulse lengths were 9 and 12.3 μs, respectively. Spectra were acquired with 1 k scans at spectral widths of 9.6 kHz for 1H and of 4.5 kHz for 13C as well as with 128 increments in the 13C dimension.
Magnetic Susceptibility (Δχ)-Tensor Determination
The Δχ-tensor values and lanthanide ion positions for one single LBT site were determined from experimental PCS values and from the structure of murine IL-10 (PDB entry 4X51) using the Numbat program (45). The magnitude of the PCS (ΔδPCS) is given by Equation 3,
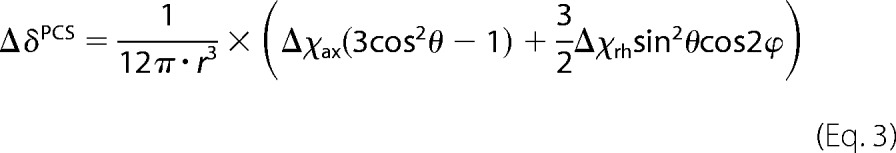 |
where r, θ, and ϕ are the polar coordinates of the nucleus with respect to the principal axes of the magnetic susceptibility tensor, and Δχax and Δχrh are its axial and rhombic component, respectively. In the case of mixed IL-10-LBT with only one 15N-labeled subunit, the structure of the IL-10 monomer was used for the Δχ-tensor fit. However, for uniform IL-10-LBT, Δχ-tensor values were determined by applying ensemble averaging in Numbat and by treating the IL-10 dimer as an ensemble of two structures.
PCS-based Heparin·IL-10 Docking
Rigid body docking calculations of the heparin tetrasaccharide and IL-10 based on PCS restraints were carried out using Xplor-NIH (version 2.34) (46). The positions of the individual lanthanide ions were obtained from the Δχ-tensor fit of the mixed IL-10-LBT protein. Their shared center of mass was used as the lanthanide ion binding site and fixed during the calculation. PCS restraints of the ligand were obtained with uniform IL-10-LBT; therefore, a second lanthanide ion was introduced into the calculation. Its position was symmetrical relative to the first ion and with respect to the 2-fold dimer rotation axis. The coordinates of the protein were also fixed, whereas the heparin ligand was allowed to rotate and translate. Each sugar ring was treated as a rigid body, and rotation around the glycosidic linkages was allowed. Starting structures of heparin were built from the NMR and MD structure of a heparin dodecasaccharide (PDB entry 1HPN) (47), and Xplor-NIH topology and parameter files were generated with the help of the HIC-Up-Server (48). Two heparin structures, the first having the iduronic acid ring in 1C4 chair conformation and the second in 2SO skew-boat conformation, were built. Because no appropriate parameterization of the unsaturated uronic acid ring, which arises from preparation of the heparin tetrasaccharide by lyase digestion, was available, the ring was left in a 1C4 or 2SO conformation, respectively. The error in the overall ligand position arising from the incorrect ring structure was expected to be negligible based on the 1/r3 distance dependence of the PCS and the long distance between the lanthanide ion and the ligand. That is why PCS data of the first iduronic acid ring were also included into the calculations. Those comprised a high temperature dynamics step at 3500 K for 1 ns, a simulated annealing to 25 K in 12.5-K steps, each with a 2-ps length, and a final Powell minimization with internal and Cartesian coordinates. Weighting factors for PCS restraints and other Xplor-NIH potentials (e.g. for molecule geometry) were linearly scaled up during the simulated annealing.
PCSs were calculated using the RDC module of Xplor-NIH, because both share the same tensor form, and this allowed treatment of both lanthanide ions at once. Calculations were tried with two types of protein PCS data sets, one obtained from mixed IL-10-LBT with one single lanthanide site and another one measured on uniform IL-10-LBT. Ligand PCSs were also measured in the presence of two lanthanide binding sites, and PCS sum averaging had to be considered. Highly similar solutions were obtained in both calculations, whereas the PCS data set obtained with the mixed IL-10-LBT showed fewer violations.
Because the symmetry of the PCS isosurfaces caused two degenerated solutions, including one model in which the ligand was placed away from the protein, a binding restraint was added to the calculation. This was set up as a loose distance restraint between both molecules with an upper border of 20 Å using the NOE potential of Xplor-NIH. Compliance of experimental PCS values with those calculated from the generated structure model was evaluated by calculating Cornilescu's quality factor (49) and by the number of violations outside a tolerance interval of 0.1 Å.
Molecular Dynamics Simulations
The five lowest energy docking structures were used as starting configuration for five 105-ns all-atom MD simulations at 30 °C. Each simulation consisted of one IL-10 molecule with the docked heparin structure in a hexagonal water box consisting of ∼9430 TIP3 water molecules and 12 sodium as well as 4 chloride ions to reach a salt concentration of 50 mm and neutralize the system. To avoid protein reorientation, backbone atoms in the subunit to which heparin was not bound were subjected to very weak harmonic constraints of 0.01 kcal mol−1 Å−2. The program NAMD (50) was employed for the simulation under conditions of normal pressure (1.013 bar), using the most recent CHARMM36 protein force field (51–53). The heparin force field values were taken from Refs. 54 and 55 with oxygen-linked sulfate groups parameterized according to Ref. 56 and nitrogen linked sulfate groups parameterized according to Ref. 57. The smooth particle mesh Ewald algorithm was used to compute the electrostatic forces (58), and the SHAKE (59) and SETTLE (60) algorithms were used to keep rigid all bonds involving hydrogen atoms, allowing a 2-fs time step.
Results
X-ray Structure Determination of Mouse IL-10
Initially, we attempted to solve the structure of the IL-10·GAG complex using x-ray crystallography. This led to the determination of the crystal structure of mouse IL-10 at a resolution of 2.1 Å, which was deposited in the PDB under accession number 4X51. Table 1 summarizes the statistics of the x-ray data collection and the refined structural model. Protein crystals were obtained only after truncation of the first nine N-terminal amino acids of IL-10, whereas the wild type protein did not form crystals. The lack of an ordered secondary structure of the IL-10 N terminus, which can be concluded from secondary chemical shifts (28), potentially prohibits crystallization in this case. Furthermore, also the subsequent protein residues up to Val-17 were not observed in the crystal structure, indicating indeed a high degree of structural disorder of this part of the protein.
TABLE 1.
Summary of the x-ray data collection and structure refinement statistics of IL-10
| Parameter | Value |
|---|---|
| Space group | P212121 |
| Resolution limits (Å) | 42.15–2.05 (2.16–2.05)a |
| Unit cell parameters | |
| a, b, c (Å) | 39.77, 80.82, 84.32 |
| α = β = γ (degrees) | 90 |
| No. of unique reflections | 17,734 |
| Multiplicity | 4.8 |
| Completeness (%) | 100 (99.9) |
| Mean I/σ(I) | 12.3 (2.9) |
| Rsym (Rmerge) (%) | 8.3 (55.6) |
| Rwork (%) | 18.9 |
| Rfree (%) | 24.6 |
| No. of protein residues | 283 |
| No. of water molecules | 57 |
| Mean protein B factor (Å2) | 30.8 |
| Mean solvent B factor (Å2) | 64.8 |
| Root mean square deviation bond lengths (Å) | 0.014 |
| Root mean square deviation bond angles (degrees) | 1.548 |
| Ramachandran plot statistics (%) | |
| Residues in most favored regions | 97.2 |
| Residues in additional allowed regions | 2.8 |
a Values in parentheses refer to the highest resolution shell.
Overall, mouse IL-10 shares a high structural similarity with human (PDB entry 2ILK) and viral (PDB entry 1VLK) IL-10 with Cα root mean square deviation values of 0.66 and 0.80 Å when comparing one 6-helix bundle domain. Interestingly, the mouse IL-10 dimer shows a smaller interdomain angle of ∼70° compared with ∼90° for human and viral IL-10. This effect is most likely caused by the different crystal packing, as described previously for different crystal forms of human IL-10 (13, 61), and indicates that some degree of domain movement is possible.
Despite extensive efforts to obtain an IL-10·GAG co-crystal, including excessive crystal soaking and the addition of GAGs to the crystallization buffer from the outset, the ligand could not be localized in the IL-10 crystal. As is known from the literature, obtaining protein·GAG co-crystals represents a very formidable challenge (62). In fact, only a few co-crystal structures of protein·GAG systems have been deposited in the PDB (62), although nearly 200 GAG-binding proteins are known as of today (63). The low GAG binding affinity and high conformational flexibility render them a difficult target for crystallization, and these are probably also the most limiting factors in the case of IL-10. Therefore, we used NMR spectroscopy, for which the extensive molecular dynamics is not problematic, to investigate the GAG-binding site of IL-10.
NMR Chemical Shift Perturbation Experiments
Detecting chemical shift changes of protein NMR signals and mapping them onto the protein structure represents a powerful and target-oriented approach extensively used to identify ligand binding sites by NMR. The detection of the amide group chemical shifts requires minimal isotopic labeling of the protein and can provide residue-specific resolution. Here, 1H and 15N chemical shift perturbations of the backbone amide groups of IL-10 were followed upon the addition of GAGs. Fig. 1 shows the chemical structure of GAGs used in this study.
FIGURE 1.

GAGs used for NMR chemical shift perturbation experiments. The structures of HA hexasaccharide (a), CS hexasaccharide (b), DS hexasaccharide (c), and psHA tetrasaccharide (d) are shown. For heparin, different chain lengths were tested: di- (e), tetra- (f), and hexasaccharide (g). CS, DS, and heparin molecules carried a 4,5-unsaturated uronic acid ring at the non-reducing end as a result of preparation by lyase digestion. CS had a non-uniform sulfation pattern with the residue R at the O4 or O6 position being either H or SO3−. psHA was synthesized with an azide group at the reducing end.
During the course of titration with GAGs, protein resonances were gradually shifted (Fig. 2), indicating a fast exchange of the ligand on the NMR time scale between the free and the bound state. This observation is in agreement with the low affinity of GAG ligands for IL-10 with KD values in the millimolar to micromolar range as measured by STD NMR spectroscopy (16). For the heparin tetrasaccharide (Figs. 2 and 3a), pronounced chemical shift changes (i.e. >0.07 ppm at a protein/ligand ratio of 1:2) were observed for residues Gln-54, Gly-59, Leu-60, and Gly-61, which are part of the BC-loop, and for Cys-114 and Glu-115, which are found in the loop that connects helix D and E and bridges both dimer subunits (Fig. 4). Additionally, high Δδ values were observed for residues of the N terminus (Tyr-5, Ser-6, Arg-7, Glu-8, Asp-9, Asn-10, and Gly-18) and C terminus (Lys-159, Lys-161, Glu-162, and His-163). Unfortunately, no chemical shift assignment was available for residues 12–16 and 107–113 (28), because their signals could not be detected in the HSQC spectrum under the chosen experimental conditions. Thus, no chemical shift mapping could be performed for those regions of the protein.
FIGURE 2.
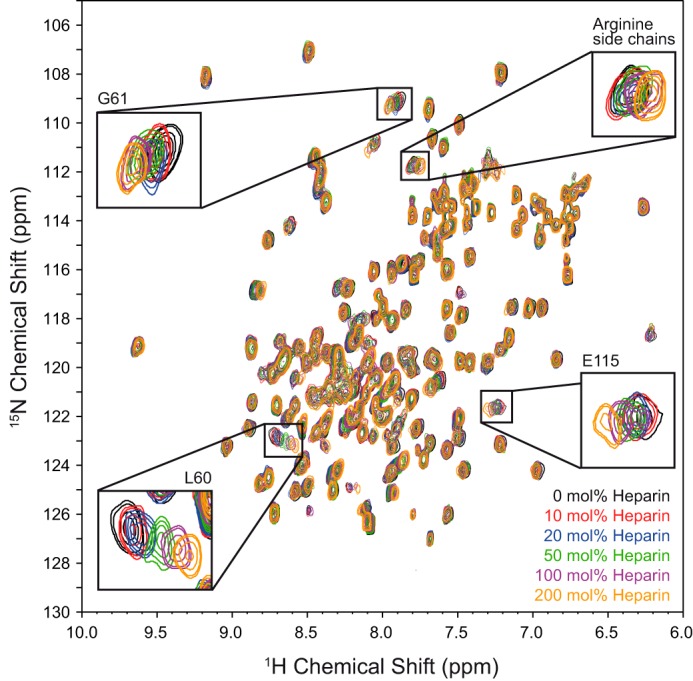
Heparin induced chemical shift perturbations of IL-10. A sequence of 1H-15N HSQC spectra of IL-10 at 600 MHz, a temperature of 30 °C, and pH 6.2 during a titration with heparin tetrasaccharide is shown. Selected protein signals with significant chemical shift changes are highlighted.
FIGURE 3.

Weighted NH chemical shift changes (Δδ) along the IL-10 primary sequence. Experiments were performed with heparins of increasing chain length (a) and different GAG oligosaccharides (b). The α-helix regions of IL-10 are indicated as gray cylinders. The protein/ligand ratio was 1:2 in each case. In b, hexasaccharides were used except for psHA tetrasaccharide. HP, heparin.
FIGURE 4.
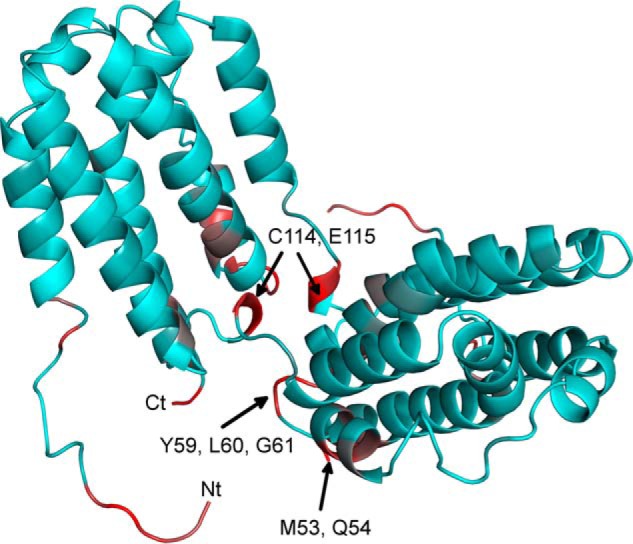
Observed NH chemical shift changes mapped onto the backbone structure of IL-10. The protein structure is shown as a schematic diagram. Residues are colored according to their NH Δδ value relative to that of Leu-60 (with the highest Δδ). Colors correspond to 0% (cyan) and 100% (red) Δδ relative to Leu-60. A possible conformation of the N-terminal region 5–17, which was not resolved in the crystal structure, was appended to the structural model here for visualization of their chemical shift changes.
A highly similar profile of Δδ values along the IL-10 primary sequence was observed when different GAG types (CS, DS, and heparin hexasaccharides and a persulfated HA tetrasaccharide) (Fig. 3b) were tested. This indicates that all GAGs bind to the same site in IL-10. Furthermore, the same observation was made for heparin fragments with varying chain length (Fig. 3a), suggesting that IL-10/GAG binding does not require a minimal ligand size. The magnitude of Δδ, obtained at the same protein/ligand ratio of 1:2, increases with the number of disaccharide repeating units (Δδdi < Δδtetra < Δδhexa) and with the degree of sulfation (ΔδCS ≤ ΔδDS < Δδheparin), which can reflect an increase in the bound protein content in the same order and agrees qualitatively with the previously determined binding affinities (16). In contrast, no significant chemical shift changes (>0.04 ppm) were observed for a HA hexasaccharide, indicating that HA up to that molecular size does not bind IL-10.
STD NMR Measurements of IL-10-Heparin Binding
For improved detection of amide signals in HSQC spectra of IL-10, chemical shift perturbation experiments were performed under slightly acidic conditions (pH 6.2) and low ionic strength (50 mm NaCl). To confirm that IL-10/GAG interaction can also occur under physiological salt concentration, we performed binding measurements of heparin tetrasaccharide using STD NMR spectroscopy. This ligand-detected NMR method is tolerable to a wide range of buffer conditions and particularly well suited for the detection of weak protein/ligand interactions. Fig. 5a shows the STD spectrum of heparin tetrasaccharide obtained at a protein/ligand molar ratio of 1:75 in buffer with 150 mm NaCl by accumulating only 32 scans. Considerable STD effects were observed for several ligand protons, indicating an interaction between heparin and IL-10. Furthermore, the binding affinity of heparin tetrasaccharide was measured using the initial growth rate approach of the STD amplification factor (Fig. 5b). A KD value of 0.41 ± 0.06 mm was determined, which is only slightly higher than the value obtained at 50 mm salt (0.30 ± 0.04 mm).
FIGURE 5.
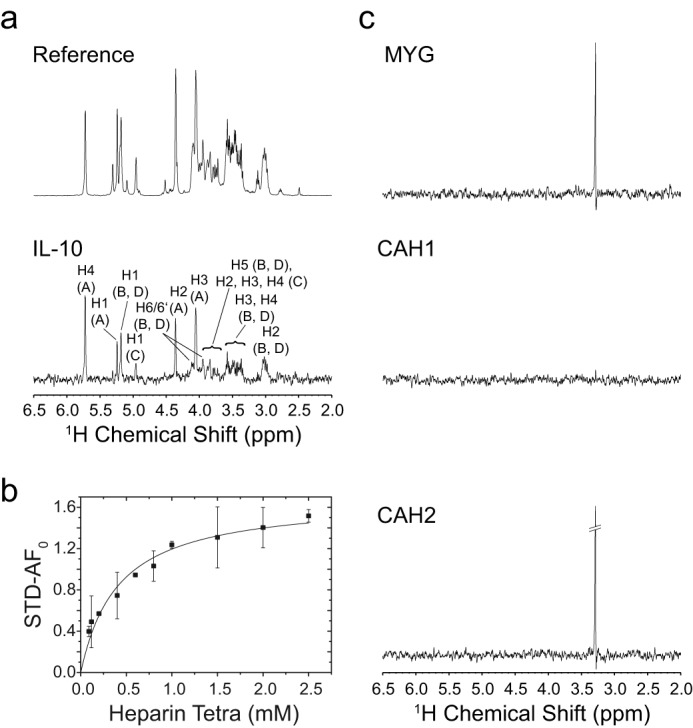
STD NMR measurement of IL-10-heparin binding. a, 600-MHz 1H reference and STD NMR spectrum (×15 magnification) of 3.0 mm heparin tetrasaccharide in the presence of IL-10 in 10 mm sodium phosphate (pD 7.4), 150 mm NaCl, 99.9% D2O at 20 °C. Spectra were obtained with 32 scans at a protein/ligand ratio of 1:75 and a saturation time of 5.0 s. STD signals are assigned to their respective heparin ring protons. The chemical structure of heparin with designation of the sugar rings is shown in Fig. 6. b, binding curve of heparin tetrasaccharide as obtained from the initial growth rates of the STD amplification factor (STD-AF). The STD signal of proton H4 of ring A at the non-reducing sugar end was analyzed. Error bars for each titration point were obtained from the fit of the STD buildup curve to an exponential function. The binding curve was obtained by fitting the initial growth rates (STD-AF0) to a one-site binding model. c, STD control experiments with non-heparin-binding proteins: equine myoglobin (MYG), human carbonic anhydrase 1 (CAH1), and bovine carbonic anhydrase 2 (CAH2). The same experimental conditions as for IL-10 were employed. All STD spectra are shown with ×15 magnification. For myoglobin and bovine carbonic anhydrase 2, an STD signal at around 3.25 ppm was detected, which very likely represents an additive of glycine in the commercially sold protein preparations.
To further confirm that this weak interaction is outside the realm of random and thus functionally unimportant binding events, we further performed STD control experiments with non-heparin-binding proteins. Because interaction with heparin is mainly of an electrostatic nature, proteins with different pI values and consequently net charge were tested: equine myoglobin (pI 7.2, 17 kDa), human carbonic anhydrase 1 (pI 6.6, 29 kDa), and bovine carbonic anhydrase 2 (pI 5.4, 29 kDa). Because their molecular weights were only slightly lower compared with IL-10 (38 kDa), a similar magnitude of spin diffusion and thus of the final STD effect was expected. Furthermore, STD measurements of the individual control proteins and heparin tetrasaccharide were performed under nearly the same experimental conditions as for IL-10. The selective radio frequency pulse for protein saturation was applied to a protein signal that was located near the frequency chosen for IL-10 saturation (around 0.2 ppm). No significant STD effects of heparin were observed for any control protein (Fig. 5c). For myoglobin and carbonic anhydrase 2, a STD signal at around 3.25 ppm was detected, which very likely represents an additive of glycine found in high abundance in the commercially sold protein preparations. This finding indicates that, although both proteins have a lower molecular mass than IL-10, they are large enough to induce a STD effect. However, the fact that significant STD signals of heparin were only observed in the presence of IL-10 indicates that its interaction with heparin stands out clearly from a random, nonspecific binding contact.
Introduction of a Lanthanide Ion Spin Label into IL-10
In a second NMR approach, structural information on the IL-10·GAG complex was obtained from measurement of PCSs using a protein-attached paramagnetic spin label. To this end, a 17-amino acid-long peptide tag (LBT), originally developed from the consensus sequence of EF-hand motifs (29) and capable of binding lanthanide ions, was attached to the C terminus of IL-10. No additional linker residues were used in order to minimize the internal motion between the tag and the protein, because this would lead to a reduction of the observed paramagnetic effects. The fact that high PCS and RDC values (±14 Hz at a magnetic field strength of 14.1 T) (data not shown) were observed indicates that the protein was oriented in the magnetic field by the paramagnetic ion and suggests that the tag was rather rigid.
To ensure that the tag did not significantly disturb the protein structure, 1H-15N HSQC spectra of IL-10-LBT were recorded and compared with the spectrum of the unmodified protein (data not shown). Only minor chemical shift changes were observed for mixed IL-10-LBT, which could be attributed to some residual tag mobility or differences in the buffer composition (e.g. the pH), because LBT-tagged and untagged protein samples were derived from different expressions and preparations. Furthermore, some additional peaks were observed in the spectrum of uniform IL-10-LBT (e.g. two tryptophan indole signals), which must belong to the LBT and StrepII tag, respectively.
The binding affinity of the LBT spin label for Tb3+ was studied using fluorescence spectroscopy (see Ref. 30 for methodological details). To this end, the fluorescence intensity of Tb3+ at 543 nm, which was caused by FRET from a nearby tryptophan residue in LBT, was measured. Alternatively, also the blue shift of the tryptophan fluorescence during titration with Tb3+ could be followed and used for quantification of the binding process. A high binding strength of Tb3+ was observed in both experiments, and low nanomolar KD values were obtained with both methods: 35 ± 2 nm (Tb3+ intensity) and 45 ± 2 nm (blue shift of tryptophan fluorescence). Remarkably, the lanthanide ion induced a very large wavelength shift for tryptophan of more than 5 nm. Furthermore, association between LBT and Tb3+ followed a clear 1:1 stoichiometry, as apparent from the position of the transition point in the binding curve (data not shown), which corresponds to identical concentrations of protein and Tb3+.
Measurement of Protein and Ligand PCSs and Δχ-Tensor Determination
NMR measurements with paramagnetic lanthanide ions (Tb3+, Tm3+, and Dy3+) immobilized to LBT allowed the observation of PCSs for IL-10 and its heparin tetrasaccharide ligand. Those values were subsequently used as distance and orientation restraints in the structure calculation of the IL-10·heparin complex. Protein PCS data were first measured on mixed IL-10-LBT with one 15N-labeled subunit and another unlabeled subunit carrying the LBT. Because dimerization of IL-10 occurs by domain swapping and intercalation of helices E and F into the opposite 4-helix bundle, dissociation of the dimer and demixing of both differently labeled subunits were not expected. Furthermore, the dimer was observed to be stable, as tested by gel filtration, and showed no dissociation for time courses of ≥3 months.
PCS assignments were made following the displacement of the individual HSQC cross-peaks along diagonal lines in the spectrum (Fig. 6a). A total of 172, 182, and 186 PCSs (1H and 15N) could be collected with Tb3+, Tm3+, and Dy3+, respectively. Δχ-Tensor parameters (Table 2) of each lanthanide ion were determined using the Numbat program as described under “Experimental Procedures” and were found to be comparable with those values reported in the literature (23, 64). The effect of residual chemical shift anisotropy, which leads to additional shifts between the diamagnetic and the paramagnetic sample, was also considered and corrected in Numbat. A good correlation between experimental and back-calculated PCSs was observed. The PCS Q-factor for all 1H and 15N PCSs was 0.14 (Tb3+), 0.16 (Tm3+), and 0.13 (Dy3+), respectively. The lanthanide ion was found to be located ∼13 Å away from the protein's C terminus, the LBT attachment point, whereas a slightly different position was found for Tm3+ than for Tb3+ and Dy3+ (Fig. 7a), which could reflect some residual tag mobility. Furthermore, all three lanthanide ions had similar Δχ-tensor orientations relative to the molecular frame except for the z-principal axis of Dy3+, which showed a deviation of around 30° compared with the tensor orientation of Tb3+ and Tm3+. Alternatively, protein PCS data were also obtained for uniform IL-10-LBT. However, due to stronger paramagnetic relaxation and signal broadening in the system with two lanthanide ions, fewer PCS effects could be assigned unambiguously. The Δχ-tensor parameters were determined considering that the PCSs were sum-averaged over both dimer subunits in that case. Similar Δχ-tensor values with nearly the same lanthanide ion position as compared with mixed IL-10-LBT were obtained.
FIGURE 6.
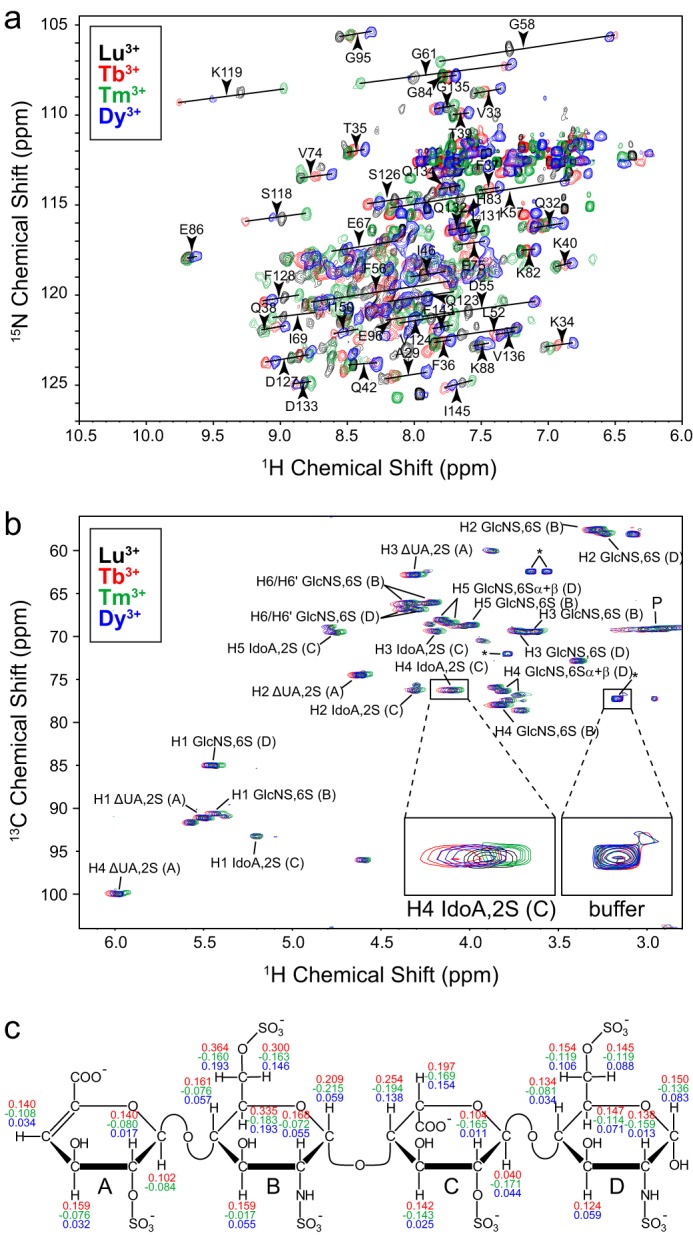
PCS measurement of IL-10 and its heparin ligand. a, 1H-15N HSQC spectra of mixed IL-10-LBT in the presence of 1 eq of Lu3+ (black), Tb3+ (red), Tm3+ (green), and Dy3+ (blue), respectively, at 600 MHz and 30 °C. PCS vectors of selected amino acid residues are indicated as black lines between corresponding diamagnetic and paramagnetic peaks. b, 1H-13C HSQC spectra of heparin tetrasaccharide in the presence of uniform IL-10-LBT complexed with Lu3+ (black), Tb3+ (red), Tm3+ (green), and Dy3+ (blue), respectively. The protein/ligand ratio was 1:4. The assignment of the heparin CH groups is shown. Sugar rings are labeled with capital letters beginning at the non-reducing end. Signals arising from residual not 2H-labeled HEPES buffer and from the protein are marked with asterisks and P, respectively. Buffer signals can serve as internal references, which show no shift compared with heparin signals. c, chemical structure of heparin tetrasaccharide with measured PCS values. The same color coding as in a and b is used.
TABLE 2.
Summary of Δχ-tensor parameters of lanthanide ions complexed with mixed IL-10-LBT
Error values were obtained from a Monte Carlo error analysis in which 30% of the PCS data were randomly deleted.
| Tb3+ | Tm3+ | Dy3+ | |
|---|---|---|---|
| Δχax (10−32 m3) | 24.6 ± 2.0 | −18.8 ± 2.3 | 24.6 ± 1.6 |
| Δχrh (10−32 m3) | 20.1 ± 1.3 | −17.6 ± 1.6 | 21.3 ± 1.5 |
| x (Å) | −26.4 ± 0.5 | −26.6 ± 0.7 | −27.5 ± 0.6 |
| y (Å) | −4.0 ± 0.7 | −8.7 ± 1.0 | −3.5 ± 0.7 |
| z (Å) | 57.3 ± 0.4 | 58.7 ± 0.7 | 58.5 ± 0.5 |
| α (degrees) | 50 ± 3 | 66 ± 2 | 53 ± 4 |
| β (degrees) | 310 ± 3 | 305 ± 3 | 334 ± 4 |
| γ (degrees) | 33 ± 1 | 42 ± 2 | 9 ± 4 |
FIGURE 7.

Lanthanide ion position and Δχ-tensor determination. a, IL-10 is represented as a schematic diagram, and the position of the lanthanide ions, as determined from the Δχ-tensor fit of mixed IL-10-LBT, is indicated by spheres. The lanthanide binding site is close to the protein's C terminus to which LBT was attached. b, representation of the Δχ-tensor of Tb3+ as PCS isosurfaces corresponding to PCS values of ±0.75 and ±0.2 ppm. Positive and negative PCSs are shown by blue and red isosurfaces, respectively.
Finally, PCS data for the heparin tetrasaccharide were collected in the presence of uniform IL-10-LBT at a protein/ligand ratio of 1:4. Again, the double LBT-labeled protein was used because the magnitude of the PCS was expected to be stronger. Because two heparin molecules can simultaneously bind to IL-10, as suggested by its highly symmetric structure and as confirmed experimentally (16), each ligand should have one nearby lanthanide ion in its vicinity in the case of two also symmetrical label positions. In contrast, if only one LBT site were to be used, one of the two heparin molecules could be too far away from the lanthanide ion and eventually experience no PCS.
For unambiguous assignment, PCSs of the ligand were measured from 1H-13C HSQC spectra (Fig. 6, b and c). Because the material had no 13C labeling, heparin was used in excess in order to distinguish it from the protein background. Under these circumstances, ligand PCSs are heavily weighted by the fraction in the free state, and measured values are much reduced from their bound values. Thus, a correction was made by the fraction of the bound ligand that was determined from the KD value of the tetrasaccharide, 0.30 ± 0.04 mm, along with total protein and ligand concentrations. Overall, 23, 22, and 22 PCSs could be collected with Tb3+, Tm3+, and Dy3+, respectively (Fig. 6c). A clear trend of the PCS values could be observed (i.e. they were all positive in the case of Tb3+ and Dy3+ and negative for Tm3+). From the topology of the PCS isosurfaces (Fig. 7b), one could identify two obvious areas satisfying the experimental PCS values: one at the dimer interface and the other one pointing away from the protein and lacking any intermolecular contact.
PCS-based Docking and MD Simulation of the Heparin·IL-10 Complex
PCS-restrained rigid body docking was performed to generate a structural model of the heparin·IL-10 complex, which was subsequently used as the starting point for an MD simulation of IL-10 and heparin. For docking calculations, PCSs were computed using the internal RDC module of Xplor-NIH because it offered the possibility to treat both lanthanide ions at the same time. RDCs and PCSs share the same form of the magnetic susceptibility tensor. In the case of PCSs, only introduction of a 1/r3 distance dependence and definition of the vector between the lanthanide and the nucleus had to be done. Because order tensors had a different definition in Xplor-NIH than in Numbat, the axial (Da) and rhombic (Dr) tensor components were recalculated and optimized during the simulation. Starting values of Da/Dr for Tb3+, Tm3+, and Dy3+ were 7979.28 Hz/0.650, 6556.45 Hz/0.337, and 6870.54 Hz/0.517, respectively. Docking calculations were performed with both protein PCS data sets (i.e. from mixed and uniform IL-10-LBT). Very similar solutions were obtained in both cases, whereas the first one produced fewer violations. Furthermore, independent docking calculations were done with heparin having the IdoA ring either in 1C4 or in 2SO conformation, yielding solutions that are indistinguishable with respect to the ligand position relative to IL-10. Fig. 8a displays the five lowest energy structures of 1C4 heparin in complex with IL-10. For clarity, the heparin binding site of only one dimer subunit is shown. A second symmetrical and indistinguishable docking solution was obtained for the second IL-10 subunit, which involves the same group of binding residues and is located on the opposite side of the central dimer crevice. In the docking model, the heparin molecule is located within a cluster of several positively charged amino acid residues, including Lys-99, Arg-102, Arg-104, Arg-106, Arg-107, Lys-117, and Lys-119. A good agreement between experimental and back-calculated PCSs was obtained. For the five lowest energy structures, the PCS Q-factor was on average 0.19 for both 1C4 heparin and 2SO heparin, respectively.
FIGURE 8.

Structural model of the heparin·IL-10 complex as obtained by PCS-based rigid body docking and subsequent MD simulation. a, ensemble of the five lowest energy structures of the 1C4 heparin ligand after PCS-based docking. The IL-10 molecule is represented with its van der Waals surface. Heparin is drawn in sticks and colored according to the atom type: carbon (cyan), oxygen (red), nitrogen (blue), and sulfur (gold). For clarity, only one heparin binding site is shown. For the second IL-10 dimer subunit on the opposite side of the central crevice, an identical docking pose was obtained, which involves the same group of basic residues. The best five docking solutions were used as the starting point for independent MD simulations from which snapshots after 105 ns are shown here (b–f). In the docking structure and during MD, the ligand position coincides with a cluster of positively charged amino acid residues of IL-10 as shown in sticks: arginines 102, 104, 106, and 107 are colored in blue, and lysines 99, 117, and 119 are shown in yellow.
To account for flexibility on the protein and ligand side, such as possible hinge movements around the central dimer crevice and flexibility of amino acid and sugar side chains, each of the five lowest energy docking structures was subjected to an all-atom MD simulation. Fig. 7, b–f, shows snapshots of the heparin·IL-10 complex after 105 ns of MD. During the simulation, the ligand was observed to remain flexible and did not adopt a single bound conformation. Comparing the individual MD simulations, differences in the ligand orientation can be recognized, which suggests that heparin may exist in an ensemble of conformations and that one fixed structure, as predicted by docking, is less likely. However, the heparin ligand was also observed to remain in the vicinity of the docked starting structure and to explore the space around the described basic amino acid cluster.
To test whether the MD simulations were in agreement with the experimental data, PCSs were back-calculated from the MD structures after some equilibration period at time points of 55, 80, and 105 ns using Numbat and the Xplor-NIH calcTensor function. PCSs were averaged along each MD trajectory and between all MD runs. A total PCS Q-factor of 0.26 was obtained showing that the MD structural models are in compliance with the experiment and can provide a description of the heparin·IL-10 complex. When only four MD simulations were compared, except for the one in Fig. 8b, in which the ligand is slightly outside the proposed binding region, the PCS Q-factor further improved to 0.24, suggesting that this binding pose of the MD solution is less likely.
Discussion
In this study, we have investigated the structure of the IL-10·GAG complex using NMR spectroscopy. Because the preparation of a IL-10·GAG co-crystal did not succeed, several NMR methods had to be employed to determine a structural model of the complex. First, in HSQC spectra of IL-10 (Fig. 2), several backbone sites showed pronounced shifts of their NMR signal indicative of an interaction with GAGs. For all tested GAGs (CS, DS, persulfated HA, and heparin) and for heparin molecules with variable chain lengths (di-, tetra-, and hexasaccharide), the same region experiencing the strongest chemical shift changes was found. This observation suggests the existence of one unique GAG binding site in IL-10 that is likewise occupied by different GAG molecules. Furthermore, the chemical shift perturbations were in agreement with the GAG binding affinities, which we have measured previously using STD NMR experiments (16), and reflected the order of the interaction strength between GAGs and IL-10. At comparable ligand concentrations, chemical shift changes were higher for GAG molecules bearing multiple sulfate groups and having a longer chain length. These findings highlight the importance of GAG sulfation and electrostatics in IL-10/GAG interactions.
The fast NMR chemical exchange, which we have observed here, and our STD NMR data show that GAG oligosaccharides have a weak binding affinity in the micromolar range. This result compares with a nanomolar affinity of polymeric heparin as reported by Salek-Ardakani et al. (15). These differences could possibly be attributed to the different kinds of experimental setups, because Salek-Ardakani et al. (15) performed binding affinity measurements with high molecular weight heparin in the absence of NaCl in the sample buffer and by using SPR spectroscopy (15). However, although IL-10/GAG interaction was found here to be weak, it may still become functionally important. The affinity of heparin tetrasaccharide is high enough to allow binding also under physiological salt concentrations, as shown by significant STD effects at 150 mm NaCl, which were not observed for non-GAG-binding proteins. The considerable STD effects correspond to a KD that is only slightly higher (0.41 ± 0.06 mm) than at the lower salt concentration investigated here (0.30 ± 0.04 mm). Thus, the studied interaction stands out clearly from a random, nonspecific binding, and the binding model described here for the tetrasaccharide is of functional relevance.
Although for many of the protein·GAG systems described in the literature, the determination of the GAG binding site was easily possible by measuring changes of the backbone NMR signals (65, 66), the chemical shift perturbation data presented here can only provide a rather unclear binding model. Most of the affected IL-10 residues identified via chemical shift perturbation analysis are not solvent-exposed and should therefore be taken with caution as direct indicators of the primary site of interaction. Furthermore, these amino acids are often uncharged. Positively charged protein segments, however, are expected to be a prerequisite for GAG binding due to the mostly electrostatic nature of the interaction (17). Nevertheless, residues with the highest chemical shift perturbations are found around the region identified by PCS-based docking and observed in the heparin·IL-10 MD simulation (Figs. 4 and 8). We therefore conclude that the IL-10/GAG interaction may trigger minor structural changes affecting the surrounding region within the cleft of the V shape of IL-10. Because shifts of the HSQC NMR signal report the response of the protein backbone to GAG binding, they are more likely for residues in flexible parts than for residues in, for example, a stable α-helix. This could explain why residues of the basic amino acid cluster found in the PCS-derived binding model showed only very small chemical shift changes. Furthermore, the long side chains of arginine and lysine residues might provide enough flexibility to accommodate the structural changes imposed by GAG binding and not transmit them to the protein backbone.
Thus, to gain a more accurate insight into the IL-10/GAG binding mode, PCS measurements using paramagnetic lanthanide ions were performed as a second NMR method. PCSs provided valuable long range distance information and at the same time gave more explicit structural information than chemical shift perturbation data, which were difficult to interpret due to their complex sensitivity to environmental and structural changes. To our knowledge, using PCSs also represents a novel approach in the field of protein·GAG systems in general. The low binding affinity and high flexibility of the model GAG ligands as well as the paucity of non-exchangeable hydrogens in close proximity to the protein surface limit the applicability of traditional approaches for molecular structure determination like NOE-based NMR methods or x-ray crystallography and therefore render paramagnetic NMR experiments very attractive.
The structure generation based on PCS-restrained docking and subsequent MD simulation yielded a binding model (Fig. 8), in which the heparin ligand is positioned in the area of a basic amino acid cluster located at the end of helices D and E. This finding compares closely with previous theoretical predictions of the IL-10/GAG binding interface as made by ab initio docking (21, 22). It is noteworthy that residues 101–108 (LRMRLRRC) within the identified cluster match a typical Cardin-Weintraub consensus sequence for a heparin-binding motif (67). This region of IL-10 is well conserved among species but is unique to IL-10 and not observed for other members of the IL-10 family of cytokines (14, 68). Already in the report of the crystal structure of IL-10, Zdanov et al. (13) described this region of densely packed basic residues as being a special feature of IL-10. Because of the protein's 2-fold rotational symmetry, the described region occurs twice in IL-10. It is located at both ends of the central dimer crevice within a distance of ∼30 Å. A sufficiently long GAG molecule could bridge the IL-10 dimer and simultaneously occupy both binding sites. This change in binding stoichiometry was observed for GAGs of eight sugar rings and longer (16) and could also be the mode in which native polymeric GAGs bind to IL-10.
Whereas the generated structural models can localize the GAG binding site of IL-10, the ligand orientation is less clear, and our MD simulations suggest that a GAG is flexible and can exist in multiple bound conformations. Both the static docked heparin structure and the multiple conformations sampled during MD are in agreement with our experimental PCS data. Using 3J proton scalar couplings, we have previously observed the simultaneous occurrence of 1C4 and 2SO IdoA ring structure in IL-10-bound heparin (16), which indicates ligand flexibility and does support the latter scenario. The non-directional forces of electrostatic interactions between the IL-10 basic residues and heparin could allow ligand binding in more than one orientation. In the end, more exhaustive MD sampling could help in exploring the ligand conformational space and identifying general binding poses.
The occurrence of a binding site for GAGs in IL-10 and its evolutionary conservation among species argue for a functional role of IL-10/GAG interaction. Salek-Ardakani et al. (15) observed an inhibitory effect of soluble GAGs on IL-10 function, whereas on the other hand, sulfated cell surface proteoglycans were also necessary for IL-10 to exert its full functional activity. Therefore, interaction with membrane-bound GAGs could be necessary for IL-10 binding to cells, which soluble GAGs could antagonize. ECM GAGs could lead to an enrichment and accumulation of IL-10 in the tissue, affecting its stability and function. Because of the noncovalent mode of dimerization, dissociation of the IL-10 dimer happens at low concentrations of a few μg/ml, generating the inactive monomer. Thus, in vivo functions of IL-10 are restricted to the microenvironment of the producing cell, and only a few endocrine effects over longer distances or through the blood circulation were reported (69). In this context, GAGs can slow down the diffusion of IL-10 and help to prolong its biological functions, an effect that was observed for other cytokines (e.g. IL-3 and GM-CSF) (70–72).
In a different scenario, GAGs could directly affect signal transduction of IL-10 by influencing recognition by its receptor. Our identification of a GAG binding site in IL-10 serves as an important starting point to evaluate whether GAGs of the ECM could possibly interfere with IL-10 receptor binding. Because the ternary IL-10 receptor complex could not be crystallized so far, only the structure of IL-10 in complex with its receptor chain 1 (IL-10R1) is known as of today. However, the structure of the related IL-20 receptor complex was solved recently (73). IL-10 and IL-20 belong to the same cytokine family and have a high structural similarity. Superimposing the structure of the IL-10·IL-10R1 complex (PDB entry 1Y6K) onto the IL-20·IL-20R1·IL-20R2 complex (PDB entry 4DOH) yields a very close match with Cα root mean square deviation values of 1.5 Å between IL-10 and IL-20 and of 2.2 Å between IL-10R1 and IL-20R1. That observation together with the fact that different IL-10 family cytokines share their receptor chains and have the same receptor stoichiometry suggests that the IL-10 and IL-20 receptor complex might have very similar structural arrangements of their receptor chains. According to that model, the IL-10R2 binding site would be located at helices A, C, and D next to the central crevice of the IL-10 dimer and possibly overlap with the GAG binding site reported here, producing a steric clash between GAG ligand and IL-10R2. Considering that GAGs have a binding affinity comparable with or even higher than that of IL-10R2, both molecules could compete for IL-10 binding. Such an inhibitory effect of GAGs was also observed for RANK (receptor activator of NF-κB) interacting with its ligand RANKL (74).
In conclusion, we show using a combination of NMR methods and MD simulations that binding of GAGs to IL-10 occurs via a common binding site involving a group of positively charged residues located at both ends of the IL-10 V-shaped crevice. Furthermore, we demonstrate here an innovative methodology for profound structural studies of weak protein/GAG interactions using paramagnetic lanthanide tags for PCS measurements. Future investigations of the IL-10·GAG system could concentrate on the energetic contributions of single amino acid residues within the identified binding region (e.g. using mutational studies along with computational analysis). The binding model presented here sheds light on a functional role of GAGs in the biology of IL-10 and provides the starting point for their functional characterization. Interaction with cell surface proteoglycans can increase local concentration of IL-10 and prolong its biological functions but also affect its receptor binding capacity.
Author Contributions
G. K. prepared the IL-10 protein, solved its x-ray structure, performed the NMR experiments, and wrote the paper. S. K. synthesized the persulfated HA tetrasaccharide. A. V. performed the MD simulation of heparin and IL-10. D. H. and J. R. conceived and coordinated the study and acquired funding.
Acknowledgment
We thank Professor Norbert Sträter (Center for Biotechnology and Biomedicine, Institute of Bioanalytical Chemistry, University of Leipzig), who enabled us to perform x-ray studies of murine IL-10 in his laboratory.
This work was supported by Deutsche Forschungsgemeinschaft Grants Transregio-SFB 67, A6, and A8 and Grant Do 187/11 from the Verband der Chemischen Industrie e.V. (to G. K.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4X51) have been deposited in the Protein Data Bank (http://wwpdb.org/).
S. Köhling, G. Künze, K. Lemnitzer, J. Blaszkiewicz, J. Schiller, D. Huster, and J. Rademann, manuscript in preparation.
- ECM
- extracellular matrix
- CS
- chondroitin sulfate
- DS
- dermatan sulfate
- HA
- hyaluronan
- psHA
- persulfated HA tetrasaccharide
- HSQC
- heteronuclear single quantum coherence
- IdoA
- l-iduronic acid
- LBT
- lanthanide binding tag
- MD
- molecular dynamics
- PCS
- pseudocontact shift
- RDC
- residual dipolar coupling
- SPR
- surface plasmon resonance
- GAG
- glycosaminoglycan
- STD
- saturation transfer difference
- PDB
- Protein Data Bank.
References
- 1.Fiorentino D. F., Bond M. W., and Mosmann T. R. (1989) Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 170, 2081–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiorentino D. F., Zlotnik A., Vieira P., Mosmann T. R., Howard M., Moore K. W., and O'Garra A. (1991) IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 146, 3444–3451 [PubMed] [Google Scholar]
- 3.Ding L., and Shevach E. M. (1992) IL-10 inhibits mitogen-induced T cell proliferation by selectively inhibiting macrophage costimulatory function. J. Immunol. 148, 3133–3139 [PubMed] [Google Scholar]
- 4.Ishida H., Hastings R., Kearney J., and Howard M. (1992) Continuous anti-interleukin 10 antibody administration depletes mice of Ly-1 B cells but not conventional B cells. J. Exp. Med. 175, 1213–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy Y., and Brouet J. C. (1994) Interleukin-10 prevents spontaneous death of germinal center B cells by induction of the bcl-2 protein. J. Clin. Invest. 93, 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai G., Kastelein R. A., and Hunter C. A. (1999) IL-10 enhances NK cell proliferation, cytotoxicity and production of IFN-γ when combined with IL-18. Eur. J. Immunol. 29, 2658–2665 [DOI] [PubMed] [Google Scholar]
- 7.Kühn R., Löhler J., Rennick D., Rajewsky K., and Müller W. (1993) Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 [DOI] [PubMed] [Google Scholar]
- 8.Tournoy K. G., Kips J. C., and Pauwels R. A. (2000) Endogenous interleukin-10 suppresses allergen-induced airway inflammation and nonspecific airway responsiveness. Clin. Exp. Allergy 30, 775–783 [DOI] [PubMed] [Google Scholar]
- 9.Grünig G., Corry D. B., Leach M. W., Seymour B. W., Kurup V. P., and Rennick D. M. (1997) Interleukin-10 is a natural suppressor of cytokine production and inflammation in a murine model of allergic bronchopulmonary aspergillosis. J. Exp. Med. 185, 1089–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Llorente L., Zou W., Levy Y., Richaud-Patin Y., Wijdenes J., Alcocer-Varela J., Morel-Fourrier B., Brouet J. C., Alarcon-Segovia D., Galanaud P., and Emilie D. (1995) Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J. Exp. Med. 181, 839–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta M., Han J. J., Stenson M., Maurer M., Wellik L., Hu G., Ziesmer S., Dogan A., and Witzig T. E. (2012) Elevated serum IL-10 levels in diffuse large B-cell lymphoma: a mechanism of aberrant JAK2 activation. Blood 119, 2844–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pestka S., Krause C. D., Sarkar D., Walter M. R., Shi Y., and Fisher P. B. (2004) Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 22, 929–979 [DOI] [PubMed] [Google Scholar]
- 13.Zdanov A., Schalk-Hihi C., Gustchina A., Tsang M., Weatherbee J., and Wlodawer A. (1995) Crystal structure of interleukin-10 reveals the functional dimer with an unexpected topological similarity to interferon γ. Structure 3, 591–601 [DOI] [PubMed] [Google Scholar]
- 14.Zdanov A. (2010) Structural analysis of cytokines comprising the IL-10 family. Cytokine Growth Factor Rev. 21, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salek-Ardakani S., Arrand J. R., Shaw D., and Mackett M. (2000) Heparin and heparan sulfate bind interleukin-10 and modulate its activity. Blood 96, 1879–1888 [PubMed] [Google Scholar]
- 16.Künze G., Gehrcke J. P., Pisabarro M. T., and Huster D. (2014) NMR characterization of the binding properties and conformation of glycosaminoglycans interacting with interleukin-10. Glycobiology 24, 1036–1049 [DOI] [PubMed] [Google Scholar]
- 17.Esko J. D., and Linhardt R. J. (2009) Proteins that bind sulfated glycosaminoglycans. in Essentials of Glycobiology (Varki A., Cummings R. D., and Esko J. D., eds), pp. 501–512, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 18.Pellegrini L., Burke D. F., von Delft F., Mulloy B., and Blundell T. L. (2000) Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 407, 1029–1034 [DOI] [PubMed] [Google Scholar]
- 19.Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., Linhardt R. J., and Mohammadi M. (2000) Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6, 743–750 [DOI] [PubMed] [Google Scholar]
- 20.Handel T. M., Johnson Z., Crown S. E., Lau E. K., and Proudfoot A. E. (2005) Regulation of protein function by glycosaminoglycans as exemplified by chemokines. Annu. Rev. Biochem. 74, 385–410 [DOI] [PubMed] [Google Scholar]
- 21.Mulloy B., and Forster M. J. (2008) Application of drug discovery software to the identification of heparin-binding sites on protein surfaces: a computational survey of the 4-helix cytokines. Mol. Simul. 34, 481–489 [Google Scholar]
- 22.Gehrcke J. P., and Pisabarro M. T. (2015) Identification and characterization of a glycosaminoglycan binding site on interleukin-10 via molecular simulation methods. J. Mol. Graph. Model. 62, 97–104 [DOI] [PubMed] [Google Scholar]
- 23.Saio T., Ogura K., Shimizu K., Yokochi M., Burke T. R. Jr., and Inagaki F. (2011) An NMR strategy for fragment-based ligand screening utilizing a paramagnetic lanthanide probe. J. Biomol. NMR 51, 395–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan J. Y., Keizers P. H., Liu W. M., Löhr F., Skinner S. P., Heeneman E. A., Schwalbe H., Ubbink M., and Siegal G. (2013) Small-molecule binding sites on proteins established by paramagnetic NMR spectroscopy. J. Am. Chem. Soc. 135, 5859–5868 [DOI] [PubMed] [Google Scholar]
- 25.Zhuang T., Lee H. S., Imperiali B., and Prestegard J. H. (2008) Structure determination of a galectin-3-carbohydrate complex using paramagnetism-based NMR constraints. Protein Sci. 17, 1220–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canales Á., Mallagaray Á., Berbis M. Á., Navarro-Vazquez A., Domínguez G., Cañada F. J., André S., Gabius H. J., Pérez-Castells J., and Jiménez-Barbero J. (2014) Lanthanide-chelating carbohydrate conjugates are useful tools to characterize carbohydrate conformation in solution and sensitive sensors to detect carbohydrate-protein interactions. J. Am. Chem. Soc. 136, 8011–8017 [DOI] [PubMed] [Google Scholar]
- 27.Otting G. (2010) Protein NMR using paramagnetic ions. Annu. Rev. Biophys. 39, 387–405 [DOI] [PubMed] [Google Scholar]
- 28.Künze G., Theisgen S., and Huster D. (2014) Backbone 1H, 15N, and 13C and side chain 13Cβ NMR chemical shift assignment of murine interleukin-10. Biomol. NMR Assign. 8, 375–378 [DOI] [PubMed] [Google Scholar]
- 29.Nitz M., Franz K. J., Maglathlin R. L., and Imperiali B. (2003) A powerful combinatorial screen to identify high-affinity terbium(III)-binding peptides. Chembiochem 4, 272–276 [DOI] [PubMed] [Google Scholar]
- 30.Barthelmes K., Reynolds A. M., Peisach E., Jonker H. R. A., DeNunzio N. J., Allen K. N., Imperiali B., and Schwalbe H. (2011) Engineering encodable lanthanide-binding tags into loop regions of proteins. J. Am. Chem. Soc. 133, 808–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leslie A. G. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 32.Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 34.Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 35.Josephson K., DiGiacomo R., Indelicato S. R., Iyo A. H., Nagabhushan T. L., Parker M. H., and Walter M. R. (2000) Design and analysis of an engineered human interleukin-10 monomer. J. Biol. Chem. 275, 13552–13557 [DOI] [PubMed] [Google Scholar]
- 36.Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 37.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38.Mori S., Abeygunawardana C., Johnson M. O., and van Zijl P. C. (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108, 94–98 [DOI] [PubMed] [Google Scholar]
- 39.Sklenar V., Piotto M., Leppik R., and Saudek V. (1993) Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. A 102, 241–245 [Google Scholar]
- 40.Shaka A. J., Barker P. B., and Freeman R. (1985) Computer-optimized decoupling scheme for wideband applications and low-level operation. J. Magn. Reson. 64, 547–552 [Google Scholar]
- 41.Angulo J., Enríquez-Navas P. M., and Nieto P. M. (2010) Ligand-receptor binding affinities from saturation transfer difference (STD) NMR spectroscopy: the binding isotherm of STD initial growth rates. Chemistry 16, 7803–7812 [DOI] [PubMed] [Google Scholar]
- 42.Cavanagh J., Palmer A. G. 3rd, Wright P. E., and Rance M. (1991) Sensitivity improvement in proton-detected two-dimensional heteronuclear relay spectroscopy. J. Magn. Reson. 91, 429–436 [Google Scholar]
- 43.Kay L., Keifer P., and Saarinen T. (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114, 10663–10665 [Google Scholar]
- 44.Schleucher J., Schwendinger M., Sattler M., Schmidt P., Schedletzky O., Glaser S. J., Sørensen O. W., and Griesinger C. (1994) A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 4, 301–306 [DOI] [PubMed] [Google Scholar]
- 45.Schmitz C., Stanton-Cook M. J., Su X. C., Otting G., and Huber T. (2008) Numbat: an interactive software tool for fitting Deltachi-tensors to molecular coordinates using pseudocontact shifts. J. Biomol. NMR 41, 179–189 [DOI] [PubMed] [Google Scholar]
- 46.Schwieters C. D., Kuszewski J. J., and Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 [DOI] [PubMed] [Google Scholar]
- 47.Mulloy B., Forster M. J., Jones C., and Davies D. B. (1993) NMR and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 293, 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleywegt G. J., and Jones T. A. (1998) Databases in protein Crystallography. Acta Crystallogr. D Biol. Crystallogr. 54, 1119–1131 [DOI] [PubMed] [Google Scholar]
- 49.Cornilescu G., Marquardt J. L., Ottiger M., and Bax A. (1998) Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 120, 6836–6837 [Google Scholar]
- 50.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., and Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 52.MacKerell A. D. Jr., Feig M., and Brooks C. L. 3rd (2004) Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699 [DOI] [PubMed] [Google Scholar]
- 53.Best R. B., Zhu X., Shim J., Lopes P. E. M., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guvench O., Greene S. N., Kamath G., Brady J. W., Venable R. M., Pastor R. W., and Mackerell A. D. Jr. (2008) Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 29, 2543–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guvench O., Hatcher E. R., Venable R. M., Pastor R. W., and Mackerell A. D. (2009) CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 5, 2353–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mallajosyula S. S., Guvench O., Hatcher E., and Mackerell A. D. Jr. (2012) CHARMM additive all-atom force field for phosphate and sulfate linked to carbohydrates. J. Chem. Theory. Comput. 8, 759–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huige C. J. M., and Altona C. (1995) Force-field parameters for sulfates and sulfamates based on ab-initio calculations: extensions of Amber and Charmm fields. J. Comput. Chem. 16, 56–79 [Google Scholar]
- 58.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 59.Van Gunsteren W. F., and Berendsen H. J. C. (1977) Algorithms for macromolecular dynamics and constraint dynamics. Mol. Phys. 34, 1311–1327 [Google Scholar]
- 60.Miyamoto S., and Kollman P. A. (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 61.Walter M. R., and Nagabhushan T. L. (1995) Crystal structure of interleukin 10 reveals an interferon γ-like fold. Biochemistry 34, 12118–12125 [DOI] [PubMed] [Google Scholar]
- 62.Imberty A., Lortat-Jacob H., and Pérez S. (2007) Structural view of glycosaminoglycan-protein interactions. Carbohydr. Res. 342, 430–439 [DOI] [PubMed] [Google Scholar]
- 63.Chautard E., Ballut L., Thierry-Mieg N., and Ricard-Blum S. (2009) MatrixDB, a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics 25, 690–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertini I., Janik M. B., Lee Y. M., Luchinat C., and Rosato A. (2001) Magnetic susceptibility tensor anisotropies for a lanthanide ion series in a fixed protein matrix. J. Am. Chem. Soc. 123, 4181–4188 [DOI] [PubMed] [Google Scholar]
- 65.Pichert A., Samsonov S. A., Theisgen S., Thomas L., Baumann L., Schiller J., Beck-Sickinger A. G., Huster D., and Pisabarro M. T. (2012) Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 22, 134–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.García-Mayoral M. F., Canales A., Diaz D., López-Prados J., Moussaoui M., de Paz J. L., Angulo J., Nieto P. M., Jiménez-Barbero J., Boix E., and Bruix M. (2013) Insights into the glycosaminoglycan-mediated cytotoxic mechanism of eosinophil cationic protein revealed by NMR. ACS Chem. Biol. 8, 144–151 [DOI] [PubMed] [Google Scholar]
- 67.Cardin A. D., and Weintraub H. J. (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9, 21–32 [DOI] [PubMed] [Google Scholar]
- 68.Sabat R. (2010) IL-10 family of cytokines. Cytokine Growth Factor Rev. 21, 315–324 [DOI] [PubMed] [Google Scholar]
- 69.Nagelkerken L., Blauw B., and Tielemans M. (1997) IL-4 abrogates the inhibitory effect of IL-10 on the development of experimental allergic encephalomyelitis in SJL mice. Int. Immunol. 9, 1243–1251 [DOI] [PubMed] [Google Scholar]
- 70.Roberts R., Gallagher J., Spooncer E., Allen T. D., Bloomfield F., and Dexter T. M. (1988) Heparan sulphate bound growth factors: a mechanism for stromal cell mediated haemopoiesis. Nature 332, 376–378 [DOI] [PubMed] [Google Scholar]
- 71.Bruno E., Luikart S. D., Long M. W., and Hoffman R. (1995) Marrow-derived heparan sulfate proteoglycan mediates the adhesion of hematopoietic progenitor cells to cytokines. Exp. Hematol. 23, 1212–1217 [PubMed] [Google Scholar]
- 72.Gupta P., Oegema T. R. Jr., Brazil J. J., Dudek A. Z., Slungaard A., and Verfaillie C. M. (1998) Structurally specific heparan sulfates support primitive human hematopoiesis by formation of a multimolecular stem cell niche. Blood 92, 4641–4651 [PubMed] [Google Scholar]
- 73.Logsdon N. J., Deshpande A., Harris B. D., Rajashankar K. R., and Walter M. R. (2012) Structural basis for receptor sharing and activation by interleukin-20 receptor-2 (IL-20R2) binding cytokines. Proc. Natl. Acad. Sci. U.S.A. 109, 12704–12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shinmyouzu K., Takahashi T., Ariyoshi W., Ichimiya H., Kanzaki S., and Nishihara T. (2007) Dermatan sulfate inhibits osteoclast formation by binding to receptor activator of NF-κB ligand. Biochem. Biophys. Res. Commun. 354, 447–452 [DOI] [PubMed] [Google Scholar]