

Abstract
Mutations in the Ras oncogene are one of the most frequent events in human cancer. Although Ras regulates numerous growth-promoting pathways to drive transformation, it can paradoxically promote an irreversible cell cycle arrest known as oncogene-induced senescence. Although senescence has clearly been implicated as a major defense mechanism against tumorigenesis, the mechanisms by which Ras can promote such a senescent phenotype remain poorly defined. We have shown recently that the Ras death effector NORE1A plays a critical role in promoting Ras-induced senescence and connects Ras to the regulation of the p53 tumor suppressor. We now show that NORE1A also connects Ras to the regulation of a second major prosenescent tumor suppressor, the retinoblastoma (Rb) protein. We show that Ras induces the formation of a complex between NORE1A and the phosphatase PP1A, promoting the activation of the Rb tumor suppressor by dephosphorylation. Furthermore, suppression of Rb reduces NORE1A senescence activity. These results, together with our previous findings, suggest that NORE1A acts as a critical tumor suppressor node, linking Ras to both the p53 and the Rb pathways to drive senescence.
Keywords: cancer; cellular senescence; protein phosphorylation; Ras protein; retinoblastoma protein (pRb, RB); NORE1A; RASSF
Introduction
Ras mutations are the most frequent oncogenic events in human cancer and can be found in ∼30% of all human cancers (1). In experimental systems, activated forms of Ras can be powerfully transforming, and transgenic animal models have validated the role of Ras activation in tumorigenesis (2, 3). However, despite the extensive evidence linking Ras to transformation and tumor development, activated Ras can also promote a state of irreversible cell cycle arrest called oncogene-induced senescence (4, 5). This tendency for deregulated Ras activity to provoke senescence can be observed in Ras-driven tumors (6). It appears that senescence provides a potent barrier to suppress the development of Ras-driven cancer because malignant tumors lose the senescence phenotype (6). The exact mechanisms by which Ras can promote senescence are not completely understood, but it appears that the main Ras senescence pathways involve the p53 and retinoblastoma (Rb)2 tumor suppressors (7). Initial evidence in mouse embryonic fibroblasts (MEFs) suggests that loss of functional p53 or Rb pathways alone is sufficient for Ras to bypass senescence (5). More recent studies have shown that, in vivo, suppression of p53 function (8) or Rb (9) enhances Ras-mediated transformation in murine systems. However, human systems may require inactivation of both p53 and Rb for full senescence evasion (10). Therefore, inactivation of the p53 and Rb senescence pathways may be essential for Ras-induced transformation.
In addition to the classic trio of growth-promoting Ras effector proteins, Raf, PI3K, and RalGDS, Ras also interacts with growth-suppressing effector proteins, including NORE1A (RASSF5) (11, 12). NORE1A is a member of the RASSF family of tumor suppressors that is frequently down-regulated during tumor development, and its inactivation has been linked to a rare familial cancer syndrome (13–15). NORE1A binds directly to Ras (16) and is thought to act as a scaffolding protein because it lacks any apparent enzymatic activity. NORE1A connects Ras to the proapoptotic Hippo pathway (16) and has apoptotic properties (17). NORE1A−/− MEFs are predisposed to Ras-induced transformation, unlike wild-type MEFs, which require inactivation of p53 or Rb to allow transformation by Ras (18). Furthermore, up-regulation of Ras activity in primary tumors is often correlated with inactivation of NORE1A (14, 19). Therefore, NORE1A may act as an important barrier against aberrant Ras signaling, and loss of NORE1A allows Ras to circumvent its own growth-inhibitory properties, shifting Ras toward transformation (20).
We have shown recently that NORE1A is a powerful Ras senescence effector that acts via p53 (20). However, although we found that NORE1A-induced senescence is heavily dependent upon p53, we noticed that NORE1A retained some senescence-inducing activity even when nearly all detectable p53 had been eliminated from the system (20). This suggested that additional mechanisms are required for the effects of NORE1A on senescence to manifest fully.
In addition to p53, Ras can promote senescence by activating the Rb pathway. The Rb gene was the first tumor suppressor gene identified and was initially linked to the formation of rare cases of pediatric tumors of the retina called retinoblastoma (21–23). Subsequent studies have identified alterations in the Rb gene or inactivation of the Rb protein in a variety of human cancers (24, 25), and it is now widely accepted that the inactivation of the Rb protein may be one of the most frequent events in cancer (26). In addition to its important function in regulating the cell cycle, Rb has critical functions in other biological processes, including chromosomal stability, regulation of apoptosis, and oncogene-induced senescence (5, 27, 28). Moreover, inactivation of Rb in vitro or in vivo suppresses Ras-induced senescence (5, 9). However, although it appears that Ras activates senescence in part via Rb, exactly how Ras modulates Rb activity remains unclear.
Rb regulation is complex and involves both inhibitoryphosphorylation and activating dephosphorylation events. Although the mechanisms of Rb phosphorylation by cyclin-dependent kinases are well characterized (29), the processes that activate Rb by dephosphorylation remain less so. Recent reports have shown that the phosphatases PP1 and PP2A play important roles in the mammalian cell cycle (30–32). Moreover, it has been shown that PP1 phosphatases can act on Rb to promote the formation of the active, hypophosphorylated form of the protein (33, 34). Intriguingly, PP1A enzymatic activity can be regulated by Ras (35). Therefore, PP1A might serve as the link between Ras and Rb, and Ras may, in part, promote senescence by activating PP1A, thereby promoting the dephosphorylation and activation of Rb (36). Exactly how Ras stimulates the activity of PP1A toward Rb remains obscure.
PP1A, a key regulator of Rb activity, has been detected in complex with NORE1A in a yeast two-hybrid system (37), and, because we have recently established that NORE1A-mediated, Ras-induced senescence is only partly driven by p53, we sought to determine whether NORE1A could also be modulating senescence by regulating Rb function. We now show that NORE1A regulates the dephosphorylation of Rb by forming an endogenous Ras-regulated complex with PP1A, scaffolding it to Rb and enhancing the Rb-PP1A complex. Moreover, suppression of Rb suppresses NORE1A-induced senescence. Therefore, we now identify a powerful new mechanism by which Ras can induce senescence via regulating the phosphorylation status of Rb. Therefore NORE1A acts as a critical node, linking Ras to both p53 and Rb. This may explain why Ras-driven tumors often exhibit reduced NORE1A expression (19).
Experimental Procedures
Plasmids
The human Rb expression construct (pGS5L-HA-Rb) was obtained from Addgene (plasmid 10720) (38) and digested with BamHI/NheI and NheI/EcoRI in two separate digests to generate 1.9- and 0.8-kb fragments, respectively. The two fragments were cloned into pEGFP-C1 (Clontech, Paolo Alto, CA) digested with BglII/EcoR1 to generate a GFP-tagged, full-length Rb expression construct. pcDNA-HA, KATE, GFP, and FLAG-NORE1A have been described previously (20, 39). KATE-tagged H- and K-Ras12V were generated by subcloning a BamHI fragment from pCGN-H- and K-Ras12V into pmKate2C (Evrogen, Moscow, Russia). shRNAs for human NORE1A have been described previously (20). pEGFP-(C1)-PP1A, pLKO-Rb1-shRNA-19, and pLKO-Rb1-shRNA-63 were purchased from Addgene (plasmids 44224, 25640, and 25641, respectively) (40, 41).
Tissue Culture and Transfections
HEK-293, HEK-293T, A549, HepG2, and COS-7 cells were obtained from the ATCC and grown in DMEM supplemented with 10% fetal bovine serum (Valley Biologicals) and 1% penicillin/streptomycin (Corning). NCI-H1299 cells stably expressing NORE1A have been described previously (42) and were cultured in RPMI medium. Wild-type and Rb−/− MEFs were provided by B. Clem (University of Louisville). HEBEC-3KT−/+ NORE1A cells were grown as described previously (20). Cells were grown in a 5% CO2 humidified incubator at 37 °C. Transient transfections were performed using jetPRIME® (Polyplus, Illkirch, France) or Lipofectamine® 3000 as described by the manufacturers. siRNA transfections were performed using DharmaFECTTM transfection reagent according to the protocol of the manufacturer (Dharmacon, Lafayette, CO), with final concentrations of 25 nm control (catalog no. sc-37007) or PP1A (catalog no. 36299) siRNA (Santa Cruz Biotechnology). For siRNA transfection experiments, cells were transfected with siRNA 24 h prior to transfection with expression constructs. Cycloheximide (Sigma) treatments were performed 24 h after transfection at a concentration of 20 μg/ml.
Immunoprecipitation and Western Blotting Analysis
Total cell lysates were prepared by harvesting the cells in modified radioimmune precipitation assay buffer (150 mm Tris (pH 7.5), 150 mm NaCl, and 1% Nonidet P-40) supplemented with a protease inhibitor mixture (Sigma) and 1 mm sodium orthovanadate. Immunoprecipitations were performed using GFP-conjugated Sepharose beads (Allele Biotechnology, San Diego, CA). FLAG and β-Actin antibodies were obtained from Sigma, anti-HA antibody from Covance, anti-GFP and anti-PP1A (catalog no. sc-6104) antibodies from Santa Cruz Biotechnology, and anti-RFP antibody from Evrogen. Phospho-Rb-Ser-795 (catalog no. 9301) and total Rb (catalog no. 9309) antibodies were purchased from Cell Signaling Technology (Boston, MA). Rabbit polyclonal NORE1A antibodies have been described previously (20). HRP-conjugated Trueblot secondary antibodies were purchased from eBioscience (San Diego, CA), and Western blotting analyses were developed using the West Pico or West Femto enhanced ECL detection system (Thermo Scientific, Rockford, IL). All Western blotting and immunoprecipitation experiments were repeated twice.
Luciferase Assays
Luciferase assays were performed in triplicate using the LightSwitchTM luciferase assay kit with a corresponding IL-6 promoter construct (stock keeping unit S721728) generated by Active Motif (Carlsbad, CA). Cells were transfected 48 h before lysis, and data were acquired using a Lumat LB 9507 from Berthold Technologies (Oak Ridge, TN).
Image Acquisition and Processing
A Pharos FX plus molecular imager (Bio-Rad, Hercules, CA) was used to digitize images prior to quantification using Quantity One software (Bio-Rad). Figures were compiled using Photoshop software (Adobe).
Immunofluorescence
Fluorescence microscopy was performed on cells grown in glass-bottom dishes (MatTek Corp., Ashland, MA), and images were captured with an Olympus IX50-FLA inverted fluorescent microscope (Optical Elements Corp., Dulles, VA) with an attached SPOT Junior digital camera (Diagnostic Instruments Inc., Sterling Heights, MI).
Senescence Assays
MEFs or A549 cells were transfected using Lipofectamine® 3000 and incubated for 72 h. Senescence was measured by staining the cells for β-galactosidase using a BioVision kit (BioVision, Milpitas, CA) as described by the manufacturer. Representative images of β-gal-positive cells and NORE1A expression measured by GFP were taken using an Olympus IX50 inverted microscope and a SPOT camera. All senescence assays were repeated twice in duplicate.
Statistical Analysis
Data are reported as mean ± S.D. Differences between treatment groups were tested using a two-sided Student's t test as appropriate. Data were considered statistically significant when p ≤ 0.05.
Results
NORE1A Forms an Endogenous, Ras-regulated Complex with PP1A
NORE1A is localized primarily to the nucleus (20) and has been observed to shuttle between the cytoplasmic and nuclear cell fractions (43). However, the localization pattern of PP1A is more complex. PP1A exhibits diffuse expression in the cytoplasm and the nucleoplasm but also accumulates in unidentified nuclear bodies (41). Because NORE1A also occurs in nuclear speckles, we determined whether NORE1A and PP1A co-localized in mammalian cells using transient transfections of the fluorescently tagged proteins. We found that, in cells expressing both PP1A and NORE1A, ∼55% of the cells contained a pool of PP1A specifically co-localized with NORE1A in the nucleus. A representative image is shown in Fig. 1A. To determine whether NORE1A and PP1A can be found in a complex, we performed co-immunoprecipitations in HEK-293T cells co-transfected with NORE1A and PP1A in the presence or absence of activated Ras. We found that NORE1A does complex with PP1A. Furthermore, the results show that the interaction of NORE1A and PP1A is enhanced significantly in the presence of activated Ras (Fig. 1B). Further analysis confirmed that endogenous NORE1A could be co-immunoprecipitated with endogenous PP1A from the HepG2 human liver hepatocellular carcinoma cell line, which contains an activated Ras (44) (Fig. 1C), suggesting that the NORE1A/PP1A interaction is physiologically relevant.
FIGURE 1.
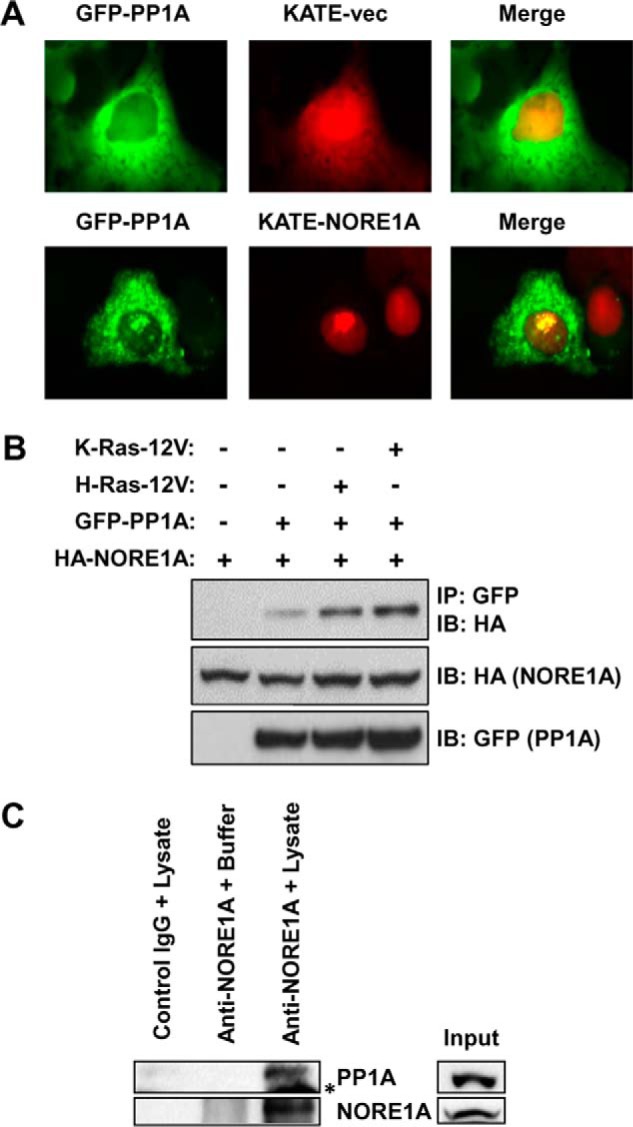
NORE1A forms an endogenous, Ras-regulated complex with PP1A. A, exogenously expressed NORE1A and PP1A co-localize in the nucleus. COS-7 cells were transfected with GFP-PP1A in the presence or absence of KATE-NORE1A. A pool of GFP-PP1A co-localized with RFP-NORE1A within the nuclei. B, activated Ras enhances the interaction between NORE1A and PP1A. HEK-293T cells were co-transfected with expression constructs for PP1A, NORE1A, and activated H- or K-Ras for 24 h and lysed, and equal amounts of protein were immunoprecipitated (IP) with anti-GFP. The immunoprecipitates were analyzed by Western blotting with anti-HA and anti-GFP antibodies. IB, immunoblot. C, NORE1A and PP1A are found in an endogenous complex. HepG2 cells were immunoprecipitated for NORE1A and immunoblotted for PP1A. IgG incubated with HepG2 lysate and Ig/NORE1A antibody incubated with lysis buffer served as negative controls. Asterisk, nonspecific IgG band.
Ras/NORE1A Stabilize PP1A
Although studying the effects of Ras on the NORE1A/PP1A interaction, we observed an increase in the levels of PP1A in whole cell lysates in the presence of Ras and NORE1A. We hypothesized that Ras/NORE1A could be promoting PP1A stability. To address this, we analyzed the effects of NORE1A and Ras on PP1A protein stability by using cycloheximide treatment after transient transfections in HEK-293 cells. The experiment was repeated three times, and a representative blot is shown in Fig. 2A. Although the results show that Ras or NORE1A individually did not seem to promote the stability of PP1A, the presence of both Ras and NORE1A together lead to a statistically significant (p < 0.05) increase in PP1A expression, even 24 h after cycloheximide treatment (Fig. 2B). To confirm these results on endogenous PP1A, we analyzed the expression of endogenous PP1A in HBEC-3KT cells stably knocked down for NORE1A in the presence of activated Ras (20) and found that PP1A levels were decreased in the NORE1A knockdown cells compared with control cells (Fig. 2C). Therefore, it seems that Ras and NORE1A cooperate to promote PP1A stability.
FIGURE 2.

Ras/NORE1A stabilizes PP1A. A, HEK-293 cells were transfected with PP1A, NORE1A, and activated H-Ras expression constructs for 24 h. The cells were treated with cycloheximide (20 μg/ml) and lysed at the indicated times after addition of cycloheximide. Levels of PP1A protein were measured by Western blotting analysis. Shown is a representative blot of three independent experiments. IB, immunoblot. B, the density of the bands was quantitated using ImageJ software, and relative PP1A expression was calculated after normalizing to β-Actin expression. No significant difference in PP1A expression levels was found in cells transfected with NORE1A or Ras individually. However, in cells transfected with both NORE1A and Ras, there was a significant increase in the levels of PP1A. C, HBEC-3KT cells stably knocked down for NORE1A were induced to express activated Ras. 3 days after induction, cell lysates were prepared and analyzed for PP1A expression by Western blotting analysis.
NORE1A Complexes with Rb and Cooperates with Ras to Scaffold PP1A to Rb
PP1A is a phosphatase that binds to Rb and modulates its activity by dephosphorylation (45). Because we established an endogenous interaction between NORE1A and PP1A, it was plausible that NORE1A could be found in a complex with Rb. We co-transfected HEK-293T cells with NORE1A and Rb in the presence and absence of activated Ras and found that NORE1A co-immunoprecipitated with overexpressed Rb and that this interaction was enhanced in the presence of Ras. A representative blot is shown in Fig. 3A, and quantitation of three independent experiments is shown in Fig. 3B. To confirm that the NORE1A/Rb interaction is physiologically relevant, we analyzed the same immunoprecipitates described in Fig. 1C for the presence of Rb and found that NORE1A and Rb do indeed interact endogenously (Fig. 3C), although weakly, suggesting that their interaction may be transient or cell cycle-dependent.
FIGURE 3.

NORE1A complexes with Rb and cooperates with Ras to scaffold PP1A to Rb. A and B, NORE1A forms a Ras-regulated complex with Rb. HEK-293T cells were co-transfected with expression constructs for Rb, NORE1A, and activated H-Ras for 24 h, lysed, and immunoprecipitated (IP) with anti-GFP. The immunoprecipitates were analyzed by Western blotting with anti-HA and anti-GFP antibodies. The blots were quantitated densitometrically using ImageJ software, and the amount of NORE1A bound to the immunoprecipitated Rb was determined after normalization to the NORE1A input levels. *, p < 0.05 compared with NORE1A alone. IB, immunoblot. C, NORE1A forms an endogenous complex with Rb. The HepG2 immunoprecipitates described in Fig. 1C were immunoblotted for the presence of Rb. D and E, Ras regulates the interaction between PP1A and Rb via NORE1A. HEK-293T cells were co-transfected with expression constructs for PP1A, Rb, NORE1A, and activated H-Ras. Cells were lysed and immunoprecipitated for GFP-PP1A, and the immunoprecipitates were analyzed by Western blotting with anti-HA, GFP, and FLAG antibodies. A representative blot is shown in D. E, the blots were quantified densitometrically to calculate the relative amount of Rb found in complex with PP1A after normalizing to the amount of Rb in the input. *, p ≤ 0.05 compared with cells transfected with empty vector. **, p ≤ 0.05 compared with cells transfected with either NORE1A or activated Ras alone.
NORE1A is a tumor suppressor that is thought to act as a scaffolding molecule because it has no apparent enzymatic activity (14, 39, 42). We recently identified a Ras-mediated mechanism by which NORE1A scaffolds HIPK2 to p53 to promote the prosenescent functions of p53 (20). Therefore, a similar mechanism may be occurring here where NORE1A acts to scaffold PP1A onto Rb in a Ras-dependent manner. To test this, we transfected HEK-293T cells with Rb and PP1A in the presence and absence of NORE1A and activated Ras and examined the effects of Ras and NORE1A on complex formation between PP1A and Rb. The results show that the Rb-PP1A complex is enhanced by NORE1A and that this effect is increased further in the presence of activated Ras. A representative blot is shown in Fig. 3D, and quantification of three independent experiments is shown in Fig. 3E. Exactly how Ras facilitates the interaction between NORE1A and PP1A and Rb is not entirely clear. However, binding of Ras to NORE1A induces a conformational change (46), and this may promote the interaction between NORE1A and its binding partners.
NORE1A Promotes the Dephosphorylation of Rb at Serine 795
The activity of Rb is primarily regulated by its phosphorylation status at several Ser/Thr residues, and Rb can be activated when Ser/Thr phosphatases promote its dephosphorylation (29). Because NORE1A forms an endogenous complex with PP1A, a key mediator of Rb dephosphorylation, and scaffolds it to Rb, we sought to determine whether NORE1A could promote the dephosphorylation of Rb. By using a phospho-specific antibody to Ser-795, a residue known to be regulated in part by PP1A (47), as a surrogate marker for the effects of PP1A on Rb, we examined the effects of NORE1A on Rb phosphorylation. In transient transfections of A549 cells (a mutant K-Ras, NORE1A-negative, p53-positive lung tumor cell line (48)), we observed that NORE1A decreased the phosphorylation of endogenous Rb at serine 795 (Fig. 4A). In addition, we examined the phosphorylation status of endogenous Rb in NCI-H1299 lung cancer cells (mutant Ras-positive, NORE1A-negative, p53-negative (49)) stably expressing exogenous NORE1A at more physiological levels (42) and found similar results (Fig. 4B). To confirm the link between NORE1A and Rb phosphorylation, we transiently knocked down NORE1A in HEK-293 cells using two previously validated shRNA constructs to NORE1A (20) and found that loss of NORE1A enhanced the phosphorylation of Rb at serine 795 (Fig. 4C). Therefore, it seems that NORE1A may be a crucial mediator of Rb function by regulating its phosphorylation status.
FIGURE 4.
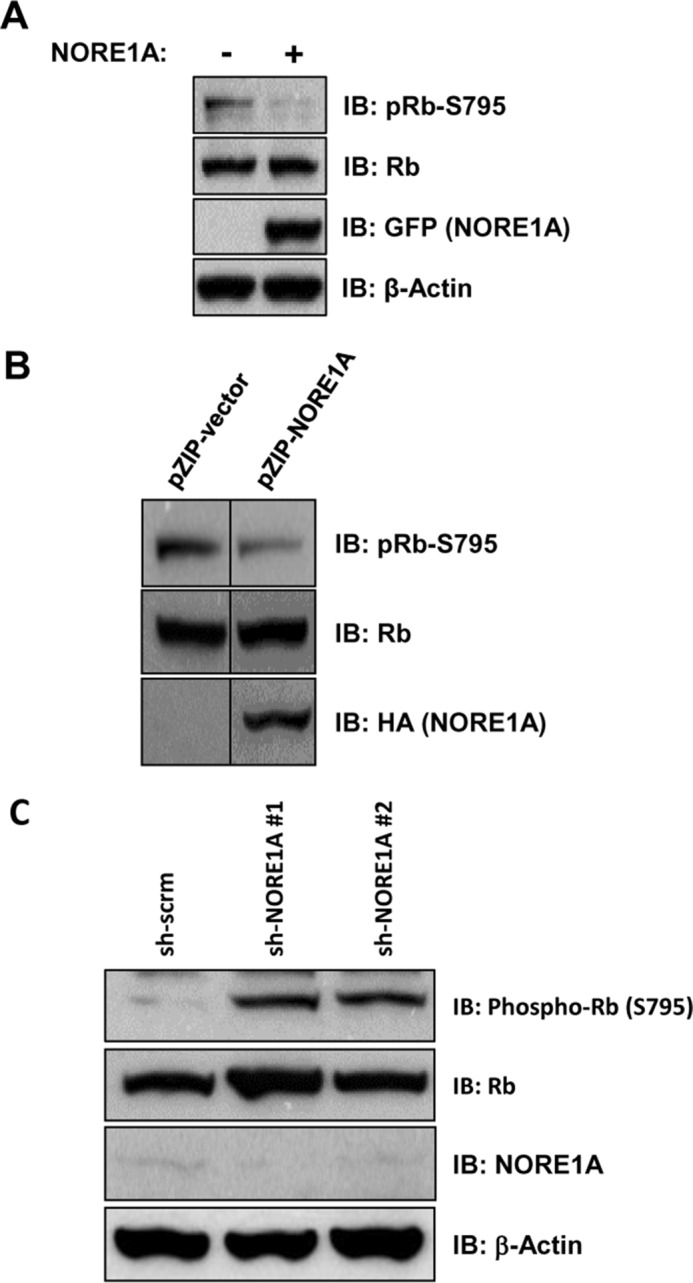
NORE1A promotes Rb-dephosphorylation. A and B, NORE1A suppresses Rb phosphorylation. A549 cells, which do not express NORE1A, were transiently transfected with GFP-NORE1A for 24 h. Cells were lysed and immunoblotted (IB) for Rb phosphorylated at serine 795 using a Ser-795-specific antibody (A). NCI-H1299 cells stably expressing NORE1A or an empty vector were lysed and immunoblotted for phospho-Rb at serine 795 (B). Shown are representative blots of two independent experiments. C, loss of NORE1A enhances Rb phosphorylation. HEK-293 cells were transiently knocked down for NORE1A expression using two different shRNA constructs to NORE1A. Cells were lysed and immunoblotted for phospho-Rb at serine 795.
Rb Is a Downstream Effector of NORE1A-induced Senescence
We have shown recently that NORE1A is a critical Ras senescence effector that acts by forming a Ras-regulated complex with p53 (20). However, although suppression of p53 strongly impaired the NORE1A senescence phenotype, we found that it did not completely abolish it. This suggests that NORE1A may also be able to promote senescence via additional mechanisms. Because the Rb pathway is one of the most powerful effector pathways of Ras-induced senescence and because we have now shown that NORE1A can regulate the dephosphorylation of Rb, we sought to determine whether NORE1A can promote senescence through Rb in addition to p53. To address this, we transfected NORE1A into wild-type and Rb−/− MEFs and measured senescence by β-galactosidase activity, the most widely used and well accepted marker of senescence (6). As we have shown previously, NORE1A induces senescence in wild-type MEFs, but, in the absence of Rb, the ability of NORE1A to induce senescence was almost abolished (Fig. 5, A and B). This suggests that NORE1A requires Rb to induce senescence in primary murine fibroblasts.
FIGURE 5.

Rb is required for NORE1A-induced senescence. A, loss of Rb significantly impairs NORE1A-mediated senescence in MEFs. Wild-type and Rb−/− MEFs were transfected with 1 μg of pcDNA-HA-vector or NORE1A. Cells were incubated for 72 h before assaying for β-galactosidase activity. *, p ≤ 0.05 compared with wild-type-transfected MEFs. Representative images are shown in B. C, Rb is a downstream effector of NORE1A-induced senescence in human cells. Stable scrambled or sh-Rb transduced A549 human lung cancer cells were transfected with 1 μg of GFP-NORE1A. After 72 h, cells were assayed for β-galactosidase activity. *, p ≤ 0.05 compared with scrambled control-transfected cells. Inset, Rb protein levels in A549 cells stably transfected with the two Rb shRNA constructs and scrambled control. D, representative images of NORE1A expression (top panels) and β-galactosidase-stained cells (bottom panels). E, Rb is an effector of NORE1A-induced IL-6 expression. Control and A549 cells stably knocked down for Rb as described above were co-transfected with GFP-NORE1A and an IL-6 promoter luciferase reporter construct. 48 h after transfection, luciferase activity was measured using a LightSwitch luciferase assay system. *, p ≤ 0.05. Representative images of NORE1A expression are shown in the right panels.
A459 is a human lung adenocarcinoma cell line that expresses wild-type p53 and Rb but not NORE1A (48). To confirm the role of Rb in NORE1A-mediated senescence in human cells, we generated A549 cells that were knocked down for Rb using two different shRNAs to Rb. We then transiently transfected NORE1A into these cells and assayed senescence by β-galactosidase activity. As expected, NORE1A was able to promote senescence in cells stably transfected with the scrambled control, but its ability to drive senescence was severely, although not completely, suppressed in cells knocked down for Rb expression (Fig. 5, C and D). To substantiate that the elevated levels of β-galactosidase staining observed in these cells was senescence-related, we measured the effects on IL-6 expression, an additional well established marker of senescence (6). NORE1A induced a significant (p < 0.05) increase in IL-6 promoter activity in A549 cells that was abrogated in the absence of Rb (Fig. 5E). Therefore, in addition to p53, NORE1A also appears to act via Rb to fully promote senescence.
PP1A-mediated Dephosphorylation of Rb Is Required for NORE1A-induced Senescence
We have established that NORE1A interacts with PP1A, enhances the complex formation of PP1A with Rb, promotes the dephosphorylation of Rb, and requires Rb for senescence. To fully substantiate that the NORE1A-mediated dephosphorylation of Rb by PP1A is required for NORE1A-induced senescence, we examined the ability of NORE1A to promote the dephosphorylation of Rb and promote senescence in the absence of PP1A. In the absence of PP1A, both NORE1A-mediated Rb dephosphorylation (Fig. 6A) and senescence (Fig. 6B) were impaired severely, confirming that dephosphorylation of Rb via PP1A is required for NORE1A-mediated senescence.
FIGURE 6.
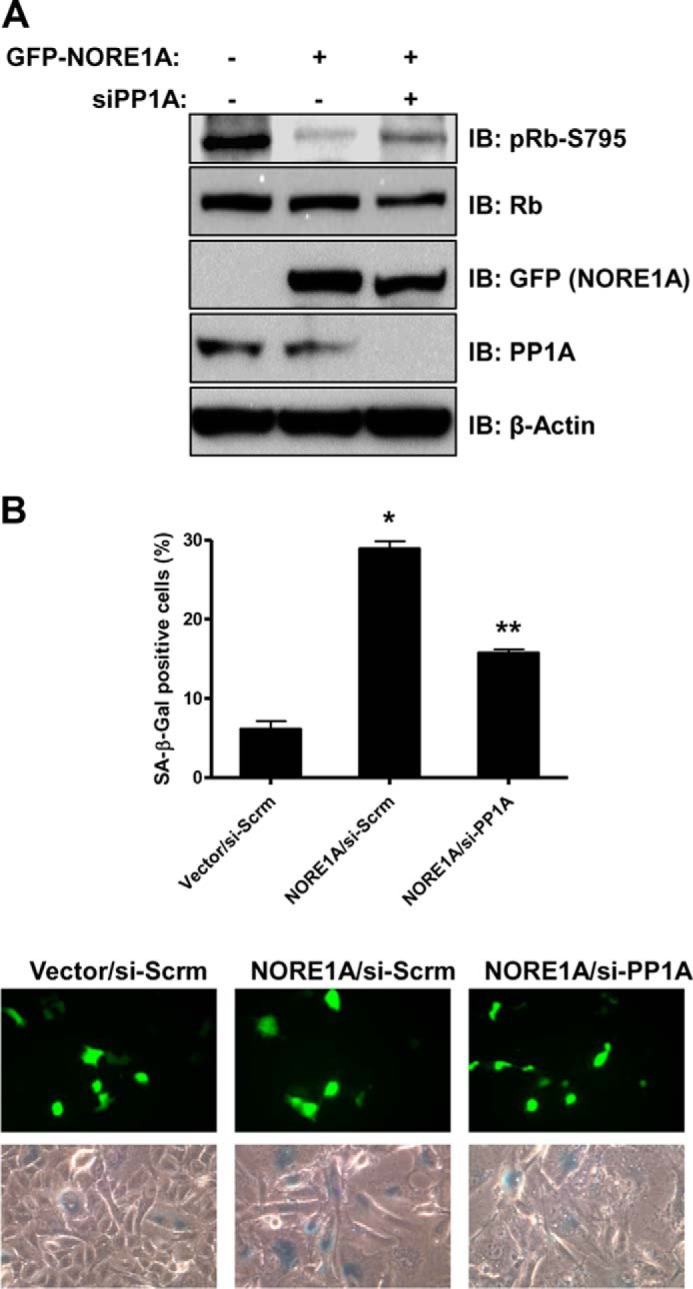
NORE1A-induced Rb dephosphorylation and senescence require PP1A. A, loss of PP1A impairs NORE1A-induced dephosphorylation of Rb. A549 cells were transiently knocked down for PP1A expression using a validated pool of PP1A siRNA. The cells were transfected with GFP-NORE1A and, 24 h later, lysed and immunoblotted (IB) for phospho-Rb at serine 795, Rb, PP1A, and NORE1A. B, PP1A is required for NORE1A-induced senescence. A549 cells were transiently knocked down for PP1A as described above and transfected with GFP-NORE1A. 72 h later, cells were assayed for β-galactosidase activity. *, p ≤ 0.05 compared with control cells transfected with vector. **, p ≤ 0.05 compared with si-Scrm cells transfected with NORE1A. Representative images of NORE1A expression and β-galactosidase staining are shown in the bottom panels.
Discussion
Oncogenic Ras mutations are critical drivers of transformation via promotion of mitogenic signaling pathways (11). Paradoxically, Ras also regulates growth-inhibitory pathways such as apoptosis and senescence (5, 50, 51). Ras-induced senescence is a major defense mechanism suppressing Ras-driven transformation, and loss of functional senescence pathways is necessary for Ras to manifest its full transforming potential (6, 52). Although recent studies have confirmed the significance of Ras-induced senescence in vivo, the mechanisms by which Ras can promote a senescent phenotype both in vitro and in vivo remain poorly defined (6, 53, 54).
Two pathways that have been identified as key players in oncogene-induced senescence involve the p53 and Rb tumor suppressors. Early studies in primary rodent cells have suggested that loss of either the p53 or the Rb pathway was sufficient for Ras to bypass senescence and promote transformation (5). In contrast, the loss of one of these pathways typically only delays the onset of senescence in human cells. Indeed, recent studies have shown that the activation of both the p53 and the Rb pathways is essential for induction of senescence in a variety of human cell lines (5, 10, 55–57). Furthermore, cross-talk between the p53 and Rb pathways could allow for additional protection against oncogenic Ras bypassing senescence and promoting tumorigenesis (7). Until now, the link between Ras and the p53/Rb pathways and how this drives senescence have been unclear.
NORE1A (RASSF5) is a member of the RASSF family of tumor suppressors (14). Like other members, it binds directly to Ras and serves as an effector to promote its growth-inhibitory properties (16, 17). The best characterized member of the RASSF family is RASSF1A, which shares considerable homology with NORE1A. RASSF1A binds and activates MST kinases, which then feed into the Hippo pathway to regulate the transcriptional coactivators YAP1, YAP2, and TAZ. However, although NORE1A also binds MST kinases (16), it does not seem to activate them (58). Moreover, deletion mutagenesis has shown that the interaction of NORE1A with MST kinases is not required for its ability to inhibit cellular growth (48). This suggests that NORE1A may function differently than RASSF1A and act via non-canonical Hippo components.
By inducing physiological expression levels of NORE1A in cells, we found that NORE1A plays a key role in p53-mediated cell cycle arrest (39). This led us to determine that NORE1A is a potent senescence effector of Ras that precisely regulates the posttranslational modification code of p53 (20). NORE1A forms a Ras-regulated complex with p53 and the kinase HIPK2. This scaffolding event is an essential component of Ras-induced senescence and results in the enhanced acetylation of p53 at the Lys-320 and Lys-382 residues. Acetylation of p53 at these residues activates prosenescent transcriptional programs (20, 59). However, we noted that, although suppression of p53 severely reduced NORE1A-induced senescence, it did not completely abolish it (20). Therefore, the role of NORE1A in Ras-induced senescence is likely more complex and may work through additional senescence pathways.
We hypothesized that NORE1A may also be able to promote senescence by regulating the Rb pathway, the other powerful senescence pathway mediating oncogene-induced senescence. Indeed, we found that loss of Rb in both MEFs and human cells suppressed the powerful senescence phenotype promoted by NORE1A. Interestingly, the suppression of either Rb or p53 alone in A549 cells did not completely inhibit NORE1A-induced senescence, consistent with the notion that human cells require the loss of both the p53 and Rb pathways to fully bypass senescence.
One of the main regulators of Rb activity is the PP1A phosphatase (60, 61). PP1A has been implicated in Ras-induced senescence (36), and Ras has been shown to control the catalytic activity of PP1A (35). Because PP1A specificity is often controlled by targeting proteins (62), we wondered whether NORE1A might serve as a direct connection between Ras, PP1A, and Rb. We found that NORE1A forms an endogenous, Ras-regulated complex with the phosphatase PP1A. Because PP1A has been detected in complex with NORE1A in a yeast two-hybrid system, the interaction is likely to be direct (37). Furthermore, we found that NORE1A appears to scaffold PP1A to Rb in a Ras-dependent manner because NORE1A could be co-precipitated with Rb and Ras/NORE1A enhanced the interaction between PP1A and Rb. The scaffolding of Rb to its phosphatase results in Rb dephosphorylation, a prosenescent event (63). The exact mechanism by which Ras activates this NORE1A-PP1A-Rb axis is not entirely clear because Ras is found predominantly on the cell membrane, and the NORE1A-PP1A-Rb complex is located primarily in the nucleus. One possibility involves a Ras-induced conformational change of NORE1A (46) that enables it to interact with PP1A and/or Rb and then shuttle them as a complex into the nucleus via various nuclear transport proteins (64). Another possible mechanism may involve additional Ras signaling pathways that act upon nuclear NORE1A to activate it, thereby inducing it to complex with PP1A and Rb. Interestingly, a pool of Ras has also been found to be located in the nucleus (65, 66), raising the possibility that NORE1A/Ras stimulation of PP1A/Rb is entirely nuclear.
During these studies, we also noticed that NORE1A seems to modulate the stability of PP1A. Recent studies have shown that the ubiquitin ligase mdm2, a negative regulator of Rb that contributes to tumorigenesis in part by destabilizing Rb (67), can be found in an endogenous complex with PP1A (68), although the effects of mdm2 on PP1A stability have not been elucidated. Interestingly, NORE1A has been shown to regulate the degradation of specific mdm2 targets (69). Therefore, NORE1A could be regulating PP1A stability through its interaction with mdm2, potentially by antagonizing the ubiquitination/degradation properties of mdm2.
PP1A is not an Rb-specific phosphatase and can regulate the functions of a variety of proteins in the cell by modulating their phosphorylation status. The specificity of PP1A for its various substrates is dictated by other targeting proteins (62). Our data suggest that NORE1A may serve as a PP1A-targeting protein, directing PP1A to a specific set of substrates, such as Rb. This may provide a novel mechanism whereby NORE1A mediates its tumor suppressor function through modulating specific protein phosphorylation. Further studies will be necessary to identify any additional NORE1A-targeted PP1A substrates.
Our data provide evidence that NORE1A provides a major link between Ras and Rb. Indeed, we can detect a faint but distinct endogenous complex between NORE1A and Rb in mutant Ras-containing cells. Activated Ras signaling promotes the association of PP1A to Rb via NORE1A, resulting in activation of Rb and senescence. In the absence of NORE1A, PP1A cannot effectively scaffold to Rb and activate it, resulting in senescence bypass and allowing the growth-promoting effects of aberrant Ras signaling to predominate. Therefore, NORE1A acts as a double-barreled Ras senescence effector that connects Ras to the two major senescence effectors in human cells, p53 (20) and Rb. This may explain why NORE1A is such a powerful senescence effector and why it is so frequently down-regulated during tumorigenesis (14), particularly in tumors with up-regulated Ras activity (19).
Author Contributions
T. B. conducted the experiments, analyzed the data, and wrote the paper. H. D. analyzed the data and wrote the paper. G. J. C. conceived the idea for the project and wrote the paper with T. B. and H. D.
This work was supported by National Institutes of Health Grant R01 CA133171–01A2 (to G. J. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Rb
- retinoblastoma protein
- MEF
- mouse embryonic fibroblast
- RASSF
- Ras association domain family.
References
- 1.DeNicola G. M., and Tuveson D. A. (2009) RAS in cellular transformation and senescence. Eur. J. Cancer 45, 211–216 [DOI] [PubMed] [Google Scholar]
- 2.Campbell P. M., and Der C. J. (2004) Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 14, 105–114 [DOI] [PubMed] [Google Scholar]
- 3.Malumbres M., and Barbacid M. (2003) RAS oncogenes: the first 30 years. Nat. Rev. Cancer 3, 459–465 [DOI] [PubMed] [Google Scholar]
- 4.Ferbeyre G., de Stanchina E., Lin A. W., Querido E., McCurrach M. E., Hannon G. J., and Lowe S. W. (2002) Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell Biol. 22, 3497–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serrano M., Lin A. W., McCurrach M. E., Beach D., and Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 6.Collado M., and Serrano M. (2010) Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Porath I., and Weinberg R. A. (2005) The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 37, 961–976 [DOI] [PubMed] [Google Scholar]
- 8.Gao J., Huang H. Y., Pak J., Cheng J., Zhang Z. T., Shapiro E., Pellicer A., Sun T. T., and Wu X. R. (2004) p53 deficiency provokes urothelial proliferation and synergizes with activated Ha-ras in promoting urothelial tumorigenesis. Oncogene 23, 687–696 [DOI] [PubMed] [Google Scholar]
- 9.Carrière C., Gore A. J., Norris A. M., Gunn J. R., Young A. L., Longnecker D. S., and Korc M. (2011) Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterology 141, 1091–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smogorzewska A., and de Lange T. (2002) Different telomere damage signaling pathways in human and mouse cells. EMBO J. 21, 4338–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pylayeva-Gupta Y., Grabocka E., and Bar-Sagi D. (2011) RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto T., Taya S., and Kaibuchi K. (1999) Ras-induced transformation and signaling pathway. J. Biochem. 126, 799–803 [DOI] [PubMed] [Google Scholar]
- 13.Chen J., Lui W. O., Vos M. D., Clark G. J., Takahashi M., Schoumans J., Khoo S. K., Petillo D., Lavery T., Sugimura J., Astuti D., Zhang C., Kagawa S., Maher E. R., Larsson C., Alberts A. S., Kanayama H. O., and Teh B. T. (2003) The t(1;3) breakpoint-spanning genes LSAMP and NORE1 are involved in clear cell renal cell carcinomas. Cancer Cell 4, 405–413 [DOI] [PubMed] [Google Scholar]
- 14.Donninger H., Vos M. D., and Clark G. J. (2007) The RASSF1A tumor suppressor. J. Cell Sci. 120, 3163–3172 [DOI] [PubMed] [Google Scholar]
- 15.van der Weyden L., and Adams D. J. (2007) The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 1776, 58–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khokhlatchev A., Rabizadeh S., Xavier R., Nedwidek M., Chen T., Zhang X. F., Seed B., and Avruch J. (2002) Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 12, 253–265 [DOI] [PubMed] [Google Scholar]
- 17.Vos M. D., Martinez A., Ellis C. A., Vallecorsa T., and Clark G. J. (2003) The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J. Biol. Chem. 278, 21938–21943 [DOI] [PubMed] [Google Scholar]
- 18.Park J., Kang S. I., Lee S. Y., Zhang X. F., Kim M. S., Beers L. F., Lim D. S., Avruch J., Kim H. S., and Lee S. B. (2010) Tumor suppressor ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J. Biol. Chem. 285, 35029–35038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calvisi D. F., Pinna F., Pellegrino R., Sanna V., Sini M., Daino L., Simile M. M., De Miglio M. R., Frau M., Tomasi M. L., Seddaiu M. A., Muroni M. R., Feo F., and Pascale R. M. (2008) Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int. J. Cancer 123, 2057–2064 [DOI] [PubMed] [Google Scholar]
- 20.Donninger H., Calvisi D. F., Barnoud T., Clark J., Schmidt M. L., Vos M. D., and Clark G. J. (2015) NORE1A is a Ras senescence effector that controls the apoptotic/senescent balance of p53 via HIPK2. J. Cell Biol. 208, 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benavente C. A., and Dyer M. A. (2015) Genetics and epigenetics of human retinoblastoma. Annu. Rev. Pathol. 10, 547–562 [DOI] [PubMed] [Google Scholar]
- 22.Friend S. H., Bernards R., Rogelj S., Weinberg R. A., Rapaport J. M., Albert D. M., and Dryja T. P. (1986) A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646 [DOI] [PubMed] [Google Scholar]
- 23.Giacinti C., and Giordano A. (2006) RB and cell cycle progression. Oncogene 25, 5220–5227 [DOI] [PubMed] [Google Scholar]
- 24.Doorbar J. (2006) Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 110, 525–541 [DOI] [PubMed] [Google Scholar]
- 25.Meuwissen R., Linn S. C., Linnoila R. I., Zevenhoven J., Mooi W. J., and Berns A. (2003) Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189 [DOI] [PubMed] [Google Scholar]
- 26.Dick F. A., and Rubin S. M. (2013) Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dannenberg J. H., and te Riele H. P. (2006) The retinoblastoma gene family in cell cycle regulation and suppression of tumorigenesis. Results Probl. Cell Differ. 42, 183–225 [DOI] [PubMed] [Google Scholar]
- 28.Zheng L., and Lee W. H. (2001) The retinoblastoma gene: a prototypic and multifunctional tumor suppressor. Exp. Cell Res. 264, 2–18 [DOI] [PubMed] [Google Scholar]
- 29.Kolupaeva V., and Janssens V. (2013) PP1 and PP2A phosphatases: cooperating partners in modulating retinoblastoma protein activation. FEBS J. 280, 627–643 [DOI] [PubMed] [Google Scholar]
- 30.Barr F. A., Elliott P. R., and Gruneberg U. (2011) Protein phosphatases and the regulation of mitosis. J. Cell Sci. 124, 2323–2334 [DOI] [PubMed] [Google Scholar]
- 31.Bollen M., Gerlich D. W., and Lesage B. (2009) Mitotic phosphatases: from entry guards to exit guides. Trends Cell Biol. 19, 531–541 [DOI] [PubMed] [Google Scholar]
- 32.Wurzenberger C., and Gerlich D. W. (2011) Phosphatases: providing safe passage through mitotic exit. Nat. Rev. Mol. Cell Biol. 12, 469–482 [DOI] [PubMed] [Google Scholar]
- 33.Alberts A. S., Thorburn A. M., Shenolikar S., Mumby M. C., and Feramisco J. R. (1993) Regulation of cell cycle progression and nuclear affinity of the retinoblastoma protein by protein phosphatases. Proc. Natl. Acad. Sci. U.S.A. 90, 388–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang R. H., Liu C. W., Avramis V. I., and Berndt N. (2001) Protein phosphatase 1α-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene 20, 6111–6122 [DOI] [PubMed] [Google Scholar]
- 35.Ayllón V., Martínez A. C., García A., Cayla X., and Rebollo A. (2000) Protein phosphatase 1α is a Ras-activated Bad phosphatase that regulates interleukin-2 deprivation-induced apoptosis. EMBO J. 19, 2237–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castro M. E., Ferrer I., Cascón A., Guijarro M. V., Lleonart M., Ramón y Cajal S., Leal J. F., Robledo M., and Carnero A. (2008) PPP1CA contributes to the senescence program induced by oncogenic Ras. Carcinogenesis 29, 491–499 [DOI] [PubMed] [Google Scholar]
- 37.Schwarz D. (2002) Novel Ras effector 1 (Nore1). Ph.D. thesis, Ruhr-Universität Bochum [Google Scholar]
- 38.Sellers W. R., Novitch B. G., Miyake S., Heith A., Otterson G. A., Kaye F. J., Lassar A. B., and Kaelin W. G. Jr. (1998) Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 12, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvisi D. F., Donninger H., Vos M. D., Birrer M. J., Gordon L., Leaner V., and Clark G. J. (2009) NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 69, 4629–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michaud K., Solomon D. A., Oermann E., Kim J. S., Zhong W. Z., Prados M. D., Ozawa T., James C. D., and Waldman T. (2010) Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 70, 3228–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trinkle-Mulcahy L., Sleeman J. E., and Lamond A. I. (2001) Dynamic targeting of protein phosphatase 1 within the nuclei of living mammalian cells. J. Cell Sci. 114, 4219–4228 [DOI] [PubMed] [Google Scholar]
- 42.Schmidt M. L., Donninger H., and Clark G. J. (2014) Ras regulates SCF(β-TrCP) protein activity and specificity via its effector protein NORE1A. J. Biol. Chem. 289, 31102–31110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suryaraja R., Anitha M., Anbarasu K., Kumari G., and Mahalingam S. (2013) The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner. Cell Death Dis. 4, e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards C. A., Short S. A., Thorgeirsson S. S., and Huber B. E. (1990) Characterization of a transforming N-ras gene in the human hepatoma cell line Hep G2: additional evidence for the importance of c-myc and ras cooperation in hepatocarcinogenesis. Cancer Res. 50, 1521–1527 [PubMed] [Google Scholar]
- 45.Durfee T., Becherer K., Chen P. L., Yeh S. H., Yang Y., Kilburn A. E., Lee W. H., and Elledge S. J. (1993) The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev. 7, 555–569 [DOI] [PubMed] [Google Scholar]
- 46.Klumpers F., Götz U., Kurtz T., Herrmann C., and Gronewold T. M. A. (2014) Conformational changes at protein-protein interaction followed online with an SAW biosensor. Sensors and Actuators B: Chemical 203, 904–908 [Google Scholar]
- 47.De Leon G., Sherry T. C., and Krucher N. A. (2008) Reduced expression of PNUTS leads to activation of Rb-phosphatase and caspase-mediated apoptosis. Cancer Biol. Ther. 7, 833–841 [DOI] [PubMed] [Google Scholar]
- 48.Aoyama Y., Avruch J., and Zhang X. F. (2004) Nore1 inhibits tumor cell growth independent of Ras or the MST1/2 kinases. Oncogene 23, 3426–3433 [DOI] [PubMed] [Google Scholar]
- 49.Shinmura K., Tao H., Nagura K., Goto M., Matsuura S., Mochizuki T., Suzuki K., Tanahashi M., Niwa H., Ogawa H., and Sugimura H. (2011) Suppression of hydroxyurea-induced centrosome amplification by NORE1A and down-regulation of NORE1A mRNA expression in non-small cell lung carcinoma. Lung Cancer 71, 19–27 [DOI] [PubMed] [Google Scholar]
- 50.Cox A. D., and Der C. J. (2003) The dark side of Ras: regulation of apoptosis. Oncogene 22, 8999–9006 [DOI] [PubMed] [Google Scholar]
- 51.Lowe S. W., Cepero E., and Evan G. (2004) Intrinsic tumour suppression. Nature 432, 307–315 [DOI] [PubMed] [Google Scholar]
- 52.Overmeyer J. H., and Maltese W. A. (2011) Death pathways triggered by activated Ras in cancer cells. Front. Biosci. 16, 1693–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X., Mitsutake N., LaPerle K., Akeno N., Zanzonico P., Longo V. A., Mitsutake S., Kimura E. T., Geiger H., Santos E., Wendel H. G., Franco A., Knauf J. A., and Fagin J. A. (2009) Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc. Natl. Acad. Sci. U.S.A. 106, 7979–7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morton J. P., Timpson P., Karim S. A., Ridgway R. A., Athineos D., Doyle B., Jamieson N. B., Oien K. A., Lowy A. M., Brunton V. G., Frame M. C., Evans T. R., and Sansom O. J. (2010) Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 107, 246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Q. M., Bartholomew J. C., Campisi J., Acosta M., Reagan J. D., and Ames B. N. (1998) Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 332, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuilman T., Michaloglou C., Mooi W. J., and Peeper D. S. (2010) The essence of senescence. Genes Dev. 24, 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shay J. W., Pereira-Smith O. M., and Wright W. E. (1991) A role for both RB and p53 in the regulation of human cellular senescence. Exp. Cell Res. 196, 33–39 [DOI] [PubMed] [Google Scholar]
- 58.Praskova M., Khoklatchev A., Ortiz-Vega S., and Avruch J. (2004) Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 381, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearson M., Carbone R., Sebastiani C., Cioce M., Fagioli M., Saito S., Higashimoto Y., Appella E., Minucci S., Pandolfi P. P., and Pelicci P. G. (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210 [DOI] [PubMed] [Google Scholar]
- 60.Tamrakar S., and Ludlow J. W. (2000) The carboxyl-terminal region of the retinoblastoma protein binds non-competitively to protein phosphatase type 1α and inhibits catalytic activity. J. Biol. Chem. 275, 27784–27789 [DOI] [PubMed] [Google Scholar]
- 61.Tamrakar S., Mittnacht S., and Ludlow J. W. (1999) Binding of select forms of pRB to protein phosphatase type 1 independent of catalytic activity. Oncogene 18, 7803–7809 [DOI] [PubMed] [Google Scholar]
- 62.Bollen M., Peti W., Ragusa M. J., and Beullens M. (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Futreal P. A., and Barrett J. C. (1991) Failure of senescent cells to phosphorylate the RB protein. Oncogene 6, 1109–1113 [PubMed] [Google Scholar]
- 64.Kumari G., Singhal P. K., Rao M. R., and Mahalingam S. (2007) Nuclear transport of Ras-associated tumor suppressor proteins: different transport receptor binding specificities for arginine-rich nuclear targeting signals. J. Mol. Biol. 367, 1294–1311 [DOI] [PubMed] [Google Scholar]
- 65.Contente S., Yeh T. J., and Friedman R. M. (2011) H-ras localizes to cell nuclei and varies with the cell cycle. Genes Cancer 2, 166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wurzer G., Mosgoeller W., Chabicovsky M., Cerni C., and Wesierska-Gadek J. (2001) Nuclear Ras: unexpected subcellular distribution of oncogenic forms. J. Cell Biochem. 36, 1–11 [DOI] [PubMed] [Google Scholar]
- 67.Sdek P., Ying H., Chang D. L., Qiu W., Zheng H., Touitou R., Allday M. J., and Xiao Z. X. (2005) MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 20, 699–708 [DOI] [PubMed] [Google Scholar]
- 68.Lee S. J., Lim C. J., Min J. K., Lee J. K., Kim Y. M., Lee J. Y., Won M. H., and Kwon Y. G. (2007) Protein phosphatase 1 nuclear targeting subunit is a hypoxia inducible gene: its role in post-translational modification of p53 and MDM2. Cell Death Differ. 14, 1106–1116 [DOI] [PubMed] [Google Scholar]
- 69.Lee D., Park S. J., Sung K. S., Park J., Lee S. B., Park S. Y., Lee H. J., Ahn J. W., Choi S. J., Lee S. G., Kim S. H., Kim D. H., Kim J., Kim Y., and Choi C. Y. (2012) Mdm2 associates with Ras effector NORE1 to induce the degradation of oncoprotein HIPK1. EMBO Rep. 13, 163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]