

Abstract
Aims: Impairment in adenosine monophosphate-activated protein kinase (AMPK) activity and NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome activation are associated with several metabolic and inflammatory diseases. In this study, we investigated the role of AMPK/NLRP3 inflammasome axis in the molecular mechanism underlying pain perception. Results: Impairment in AMPK activation induced by compound C or sunitinib, two AMPK inhibitors, provoked hyperalgesia in mice (p<0.001) associated with marked NLRP3 inflammasome protein activation and increased serum levels of interleukin-1β (IL-1β) (24.56±0.82 pg/ml) and IL-18 (23.83±1.882 pg/ml) compared with vehicle groups (IL-1β: 8.15±0.44; IL-18: 4.92±0.4). This effect was rescued by increasing AMPK phosphorylation via metformin treatment (p<0.001), caloric restriction diet (p<0.001), or NLRP3 inflammasome genetic inactivation using NLRP3 knockout (nlrp3−/−) mice (p<0.001). Deficient AMPK activation and overactivation of NLRP3 inflammasome axis were also observed in blood cells from patients with fibromyalgia (FM), a prevalent human chronic pain disease. In addition, metformin treatment (200 mg/daily), which increased AMPK activation, restored all biochemical alterations examined by us in blood cells and significantly improved clinical symptoms, such as, pain, fatigue, depression, disturbed sleep, and tender points, in patients with FM. Innovation and Conclusions: These data suggest that AMPK/NLRP3 inflammasome axis participates in chronic pain and that NLRP3 inflammasome inhibition by AMPK modulation may be a novel therapeutic target to fight against chronic pain and inflammatory diseases as FM. Antioxid. Redox Signal. 24, 157–170.
Introduction
Chronic pain is a multifaceted and highly variable condition with a high prevalence in the general population worldwide. Several mitochondrial functions and inflammation have been implicated in pain (11). Adenosine monophosphate-activated protein kinase (AMPK) has been shown to be involved in the control of peripheral sensitization of nociceptors and inflammatory nociception. Based on this evidence, manipulations that provoke AMPK activation may be a novel effective treatment for acute and chronic pain states (12, 25, 29). The AMPK cascade is one of the systems that have evolved to ensure that energy homeostasis is maintained even under pathological conditions or stress (10). Recent findings have implicated AMPK as an important contributor in NOD-like receptor family, pyrin domain containing 3 (NLRP3)-dependent inflammation and immunity (22, 28). The NLRP3 inflammasome complex is a molecular platform, which activates innate immune defenses through the maturation of proinflammatory cytokines (interleukins IL-1β and IL-18) that are activated by a wide variety of danger signals (2, 26). NLRP3 inflammasome has been related with some pain conditions, such as neuropathic pain, fibromyalgia (FM), and complex regional pain syndrome (6, 16, 15). Since AMPK and the NLRP3 inflammasome pathway have been related with nociception, we propose that AMPK downregulation could be associated with pain symptoms through NLRP3 inflammasome activation. Our hypothesis also suggests that both AMPK and NLRP3 inflammasome might serve as novel therapeutic targets in the fight against hyperalgesia and allodynia.
Innovation.
In recent years, inflammasome and AMPK have attracted increasing interest among basic and clinical researchers. Its implication in the pathophysiology of several diseases generates new therapeutic strategies. Our study shows an interesting role of AMPK/NLRP3-inflammasome axis in chronic pain and that AMPK modulation can induce a NLRP3-inflammasome inhibition which could be a novel therapeutic target to fight against chronic pain and inflammatory diseases such as FM.
Results
To establish the causal role of AMPK in nociception, we first performed a subchronic administration of compound C, an AMPK inhibitor (20 mg/kg/day), in mice. After 5 days of administration, we evaluated the phosphorylation status of AMPK as well as inflammasome activation in blood mononuclear cells (BMCs) from vehicle and compound C-treated mice. Compound C administration in mice inhibited AMPK phosphorylation and induced inflammasome activation, as indicated by upregulation expression of NRLP3 protein in BMCs, and increased serum levels of IL-1β and IL-18 (Fig. 1A–C). To verify the effect of AMPK inactivation on inflammasome modulation, we then examined the effect of compound C in THP1 cells, which represent the most commonly used model cell line to study inflammasome activation, and the effect of compound C in skin fibroblasts from AMPK knockout mice. Compound C induced activation of inflammasome in THP1 cells, as indicated by increasing expression levels of IL-1β (p17) (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars), which was not observed in AMPK knockout fibroblasts (Supplementary Fig. S2). Moreover, compound C administration produced a marked thermal nociception in mice in the hot plate test when thermal stimuli were above 50°C (latencies for compound C-treated mice were 7.36±1.06 [t (40)=5.77; p<0.001], 6.04±0.54 [t (8)=5.7; p<0.001], and 3.96±0.31 [t (33)=7.87; p<0.001] seconds faster than vehicle-injected mice at 50°C, 52.5°C, and 55°C, respectively; Fig. 1D). Interestingly, cotreatment with metformin (100 ng/kg/day), a potent AMPK stimulator (27), during a period of 6 days induced partial AMPK activation (AMPK phosphorylation) and reverted the effects of compound C to values found in vehicle-injected mice, both at the biochemical level (Fig. 1A–C) and thermal nociceptive behavior (Fig. 1E, F). Metformin treatment that induced partial AMPK activation (Fig. 2A) significantly attenuated the decreased adenosine triphosphate (ATP) levels, reduced mitochondrial mass (citrate synthase activity), and increased levels of lipid peroxidation in BMCs from mice treated with compound C (Fig. 2B–D).
FIG. 1.

Inflammasome activation induced by AMPK inhibition in mice and effect of metformin treatment. N=10 per group. (A) Protein levels of phosphorylated and nonphosphorylated AMPK (anti-AMPK and anti-pAMPK; Cell Signaling) and NLRP3 (anti-NLRP3; Adipogen) from vehicle- and compound C-treated mice compared with GADPH protein in BMCs. *p<0.001, **p<0.05 between vehicle and compound C-treated mice; ap<0.001 and aap<0.001 between compound C-treated mice and metformin. (B, C) IL-1β and IL-18 in serum levels from mice treated with the vehicle or compound C and the reduction after metformin treatment. *p<0.001 between vehicle and compound C-treated mice; ap<0.001 between compound C-treated mice and metformin. (D) Pain sensitivity in vehicle- and compound C-treated mice was evaluated in the hot plate test at 50–52.5 and 55°C±0.5°C. *p<0.05. (E, F) Evolution of pain sensitivity in vehicle- and compound C-treated mice treated with metformin evaluated in the hot plate test at 50°C and 55°C. Data are shown as change of withdrawal latency with respect to the control group. Negative and positive values represent hyperalgesia and analgesia, respectively.*p<0.001 between vehicle and compound C-treated wild-type mice. AMPK, adenosine monophosphate-activated protein kinase; BMCs, blood mononuclear cells; IL-1β, interleukin-1β; NLRP3: NOD-like receptor family, pyrin domain containing 3. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 2.
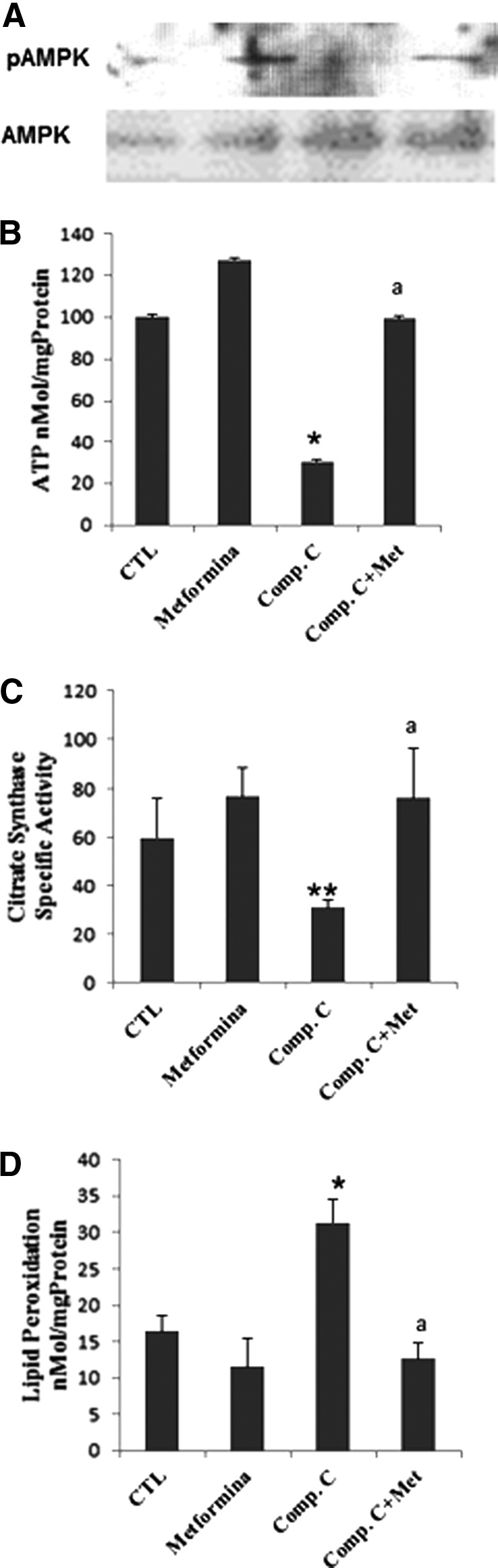
pAMPK phosphorylation levels, ATP, mitochondrial mass (citrate synthase), and lipid peroxidation in BMCs from mice treated with vehicle or compound C for 10 days (n=8 per group). (A) pAMPK phosphorylation levels, (B) ATP, (C) mitochondrial mass (citrate synthase), and (D) lipid peroxidation in BMCs from mice treated with vehicle or compound C for 10 days (n58 per group). All parameters were restored after metformin treatment. Data represent the mean±SD of three separate experiments. *p<0.001; **p<0.01 between control and compound C; ap<0.001 between compound C and metformin. ATP, adenosine triphosphate.
Then, to examine the role of NLRP3 inflammasome in compound C-induced hyperalgesia, we compared the response to AMPK inhibition between wild-type mice and mice lacking NLRP3 (nlrp3−/− mice). Knockout mice of NLRP3 were not affected by compound C treatment, despite the inhibition of phosphorylated AMPK (Fig. 3A). In fact, NLRP3 mutant mice did not show thermal algesia changes after compound C treatment compared with mutant mice treated with vehicle (Fig. 3B, C). In addition, wild-type mice treated with 16673-34-0 (5-chloro-2-methoxy-N-[2-(4-sulfamoylphenyl)ethyl] benzamide) (Fig. 3D), a new specific inhibitor of NLRP3 (18), reproduced nociceptive behavior found in the nlrp3 ko mice. In addition, compound C-dependent increase in IL-1β in wild-type mice was completely inhibited in the NLRP3 ko mice (Fig. 3E).
FIG. 3.

AMPK inhibition in NLRP3 inflammasome-depleted mice. N=10 per group. (A) Protein levels of phosphorylated AMPK in BMCs from vehicle- and compound C-treated wt and Nlrp3(−/−) mice. ap<0.001 between wt and Nlrp3(−/−) mice.*p<0.001 between vehicle and compound C-treated wild-type mice. Western blot image was quantified using ImageJ software (http://rsb.info.nih.gov/ij/download.html). (B, C) Depletion of NLRP3 in mice induced reduced levels of pain in Nlrp3(−/−) mice compared with wild-type mice after compound C treatment. ***p<0.001 between control and control treated with compound C in the hot plate test at 50°C and 52°C, respectively. (D) Pharmacological inhibition of NLRP3 by 166673-34-0 in mice induced reduced levels of pain after compound C-treated mice compared with saline and compound C. Data are shown as change of withdrawal latency with respect to the control group. Negative and positive values represent hyperalgesia and analgesia, respectively. (E) IL-1β in serum levels from wild-type and Nlrp3(−/−) mice treated with the vehicle or compound C. *p<0.001 between vehicle and compound C-treated wild-type mice. #p<0.001 between Nlrp3(−/−) mice and wild-type mice.
Caloric restriction (CR) has previously shown to have analgesic and anti-inflammatory effects in mice (7, 8). It has been speculated that AMPK might mediate the beneficial effects of CR (30). To assess the role of AMPK and inflammasome activation in pain modulation by CR, mice were subjected to CR and ad libitum (AL) diets and pain assays were performed. Mice subjected to CR diet during a period of 1 month developed a marked analgesia when compared with AL-fed mice (Fig. 4A). Then, AL- and CR-fed mice were injected with sunitinib (20 mg/kg/day, i.p.), an efficient and selective inhibitor of AMPK (13). The analgesic effect induced by CR was reverted to control algesia levels in AL mice after 7 days of treatment with sunitinib (Fig. 4A). In addition, no changes in body weight were observed after sunitinib treatment (Fig. 4B). Protein analysis in BMCs confirmed that CR increased phosphorylated AMPK levels compared with the AL group, but did not induce either NLRP3 inflammasome activation or IL-1β protein maturation (Fig. 4C), and increased serum IL-1β levels (Fig. 4D). All beneficial effects observed by us in CR mice were reverted after a week of sunitinib treatment (Fig. 4A–C). Sunitinib is more potent than compound C, the commonly used chemical inhibitor of AMPK (IC50 ranging between 45 and 62 nM for sunitinib vs. 1.3 and 2.4 μM for compound C) (13). So, the repetition of the experiment of CR showed a more significant effect of sunitinib than compound C (Fig. 4E, F) and supported the analgesic effect of CR by AMPK modulation.
FIG. 4.

AMPK and NLRP3 inflammasome modulation by CR. N=10 per group. (A) Evolution of pain sensitivity in AL and CR mice treated with sunitinib evaluated in the hot plate test at 55°C. Sunitinib treatment started after 1 month of AL and CR. Data are shown as change of withdrawal latency with respect to the control group. Negative and positive values represent hyperalgesia and analgesia, respectively. (B) Evolution of weight in AL and CR mice treated with sunitinib. (C) Protein levels of phosphorylated AMPK, NLRP3, and matured IL-1β (p17) from sunitinib-treated mice after 15 days of treatment. (D) IL-1β in serum levels from mice treated with sunitinib after 15 days of treatment in AL or CR conditions. *p<0.001 between vehicle and sunitinib-treated mice. (E) Evolution of pain sensitivity in AL and CR mice treated with compound C evaluated in the hot plate test at 55°C. (F) IL-1β in serum levels from mice treated with compound C after 15 days of treatment in AL or CR conditions. *p<0.001 between vehicle and compound C-treated mice. AL, ad libitum; CR, caloric restriction.
Next, we studied the relevance of the AMPK/NLRP3 inflammasome axis in FM, a prevalent human pain disease (Table 1). We found reduced levels of phosphorylated AMPK (Fig. 5A, B), decreased ATP levels (Fig. 5C), and increased mitochondrial reactive oxygen species (ROS) in BMCs (Fig. 5D), as well as high levels of IL-1β and IL-18 (Fig. 5E, F) in serum from patients with FM. Interestingly, all these biochemical alterations were restored to control values when BMCs from patients with FM were treated with metformin. Since mitochondrial dysfunction has been previously reported in BMCs, platelets, and muscle from patients with FM (3, 5, 9), we then decided to confirm these biochemical alterations in fibroblasts derived from patients with FM. As in BMCs, FM fibroblasts displayed a significant reduction of ATP levels (Fig. 6A) associated with reduced expression levels of active phosphorylated AMPK and tumor suppressor kinase (LKB1), an activator of AMPK (Fig. 6B, C). As mitochondrial ROS have been described to activate AMPK, which subsequently induces a PGC-1α-dependent increase of antioxidant defense, mainly MnSOD and catalase (4), and NLRP3 inflammasome activation (27, 31), we next explored the expression levels of proteins involved in these pathways. We found that reduced AMPK activation was associated with low expression levels of PGC-1α, TFAM, NRF1 and, interestingly, of both MnSOD and catalase in FM fibroblasts (Fig. 6B, C). In addition, mitochondrial superoxide production was significantly increased in FM fibroblasts compared with controls (p<0.001) (Fig. 6D).
Table 1.
Anthropometric and Symptomatic Parameters in Healthy Volunteers and Patients with FM
| Parameter | Controls | FM patients |
|---|---|---|
| Age (years) | 45.5±6.1 | 46.1±8 |
| Tender points | — | 14.5±1.8 |
| Disease duration (years) | — | 8.1±3.3 |
| Sex (male/female) | 0/20 | 0/20 |
| BMI (kg/m2) | 23.2±2.5 | 22.9±1.2 |
| FIQ total score, range 0–80 | 2.7±1.5 | 56.6±8.3a |
| Pain | 0.6±0.2 | 6.9±2.1b |
| Fatigue | 1.2±0.5 | 7.1±1.2a |
| Morning tiredness | 1±0.3 | 5.5±1b |
| Stiffness | 0.4±0.1 | 6.2±2.2b |
| Anxiety | 1±0.5 | 5.8±1.2b |
| Depression | 0.2±0.6 | 5.6±1.2a |
| VAS pain total score 0–10 | 0.7±0.2 | 7.5±2.1a |
n=20 and 30 for control and fibromyalgia groups, respectively.
p<0.001.
p<0.01.
BMI, body–mass index; FIQ, fibromyalgia impact questionnaire; FM, fibromyalgia; VAS, visual analogue scale.
FIG. 5.

Mitochondrial bioenergetic inflammasome and AMPK activation pathway in BMCs from patients with FM and the effect of metformin. (A) Protein expression levels of phosphorylated AMPK determined in control and FM BMCs from seven patients. (B) Protein levels were determined by densitometric analysis (IOD, integrated optical density) of three different Western blots and normalized to total AMPK signal, using BMCs from patients with FM, compared with a pool of five healthy age- and sex-matched control subjects. (C) ATP levels in control and FM BMCs with metformin. (D) Effect of metformin in mitochondrial ROS production analyzed in BMCs from control and patients with FM. (E, F) IL-1β and IL-18 levels in the culture media of BMCs from FM patients incubated with metformin for 24 h and analyzed by ELISA. Data represent the mean±SD of three separate experiments. *p<0.001 between control and FM patients cells. ap<0.001 between patients with FM cells with and without metformin. FM, fibromyalgia; ROS, reactive oxygen species.
FIG. 6.

Mitochondrial bioenergetic and AMPK activation pathway in fibroblasts from patients with FM. (A) ATP levels in control and FM fibroblasts. Data represent the mean±SD of three separate experiments. *p<0.001; **p<0.01 between control and patients with FM. (B) Protein expression levels of antioxidants (MnSOD and catalase), mitochondrial biogenesis, and phosphorylated and nonphosphorylated AMPK determined in control and FM fibroblast cultures. (C) Protein levels were determined by densitometric analysis (IOD, integrated optical density) of three different Western blots and normalized to GADPH signal, using fibroblasts from patients with FM, compared with healthy age- and sex-matched control subjects. (D) Mitochondrial ROS production was analyzed in fibroblasts from control and three patients with FM by flow cytometry as described in the Materials and Methods section. Data represent the mean±SD of three separate experiments.*p<0.001, ap<0.01 between control and patients with FM.
The upregulation of antioxidant enzymes by AMPK activation has been described as a defensive mechanism to protect cells against oxidative damage. However, high levels of oxidative stress and low levels of antioxidant enzymes have been reported in FM (1, 5). Our results suggest that AMPK inactivation or insensibility that impairs antioxidant defense response could be involved in the pathological mechanisms leading to high oxidative stress in FM.
As our results suggested that AMPK and NLRP3 inflammasome may be involved in the molecular mechanism underlying pain perception and activation of AMPK by metformin or CR-modulated nociception in mice, we next wondered if activation of AMPK had an analgesic effect on FM patients. As a proof of concept, a small pilot trial was designed: six women with FM (Table 2) received metformin (200 mg/daily), a reduced diabetic therapeutic dose. A significant improvement in clinical symptoms was observed after 1 month of metformin treatment. The clinical improvement was assessed by a reduction in the Fibromyalgia Impact Questionnaire (FIQ) (p<0.001), with reduction in pain, fatigue, morning tiredness, stiffness, anxiety, and depression, all subscales of FIQ (Fig. 7A, B). Furthermore, we observed a marked reduction in tender points (p<0.001), pain visual scale (p<0.001), fatigue visual scale (p<0.01), depression by Beck Depression Inventory (BDI) total score, and sleep quality determined by the sleep visual scale and Pittsburgh Sleep Quality Index (PSQI) (p<0.001) (Fig. 7C–H). No adverse effects were observed during metformin treatment. The clinical improvement induced by metformin was associated with increased AMPK activation and reduced NLRP3 expression levels in BMCs and decreased IL-1β and IL-18 serum levels (Fig. 8A–C). Furthermore, a sustained improvement in pain and other common FM symptoms was observed in a long-term treatment with metformin after 7 months (Fig. 9A–G).
Table 2.
Anthropometric, Body Composition, and Biochemical Parameters in Patients with FM Pre- and Post-Treatment with Metformin
| Parameter | Pretreatment (N=6) | Post-treatment (N=6) | Control (N=10) |
|---|---|---|---|
| Age (years) | 49.1±9 | — | 44.3±9.7 |
| Duration of diseases (years) | 6.9±3.3 | 6.6±3 | 7.1±3.8 |
| Systol BP (mmHg) | 134.2±19.4 | 136.4±23.5 | 126.5±21.7 |
| Diastol BP (mmHg) | 76.9±9.6 | 78±13.7 | 80.5±12.5 |
| Pulse rate (bpm) | 74.2±10.3 | 69±6.8 | 78.8±14.5 |
| Weight (kg) | 70.5±11.2 | 73.2±13.8 | 67.8±13.7 |
| BMI (kg/m2) | 27.6±4 | 27.3±4.8 | 26±4.8 |
| Body fat (%) | 36.6±6.1 | 41.4±6 | 38±6 |
| Muscle mass | 42±4.6 | 40.1±4.8 | 39.5±4.7 |
| Bone mass | 2.2±0.2 | 2.1±0.2 | 2.1±0.2 |
| Visceral fat rating | 7.9±2.6 | 10±2.4 | 7.1±2.5 |
| Metabolic age | 53.9±13.6 | 60.4±0.5 | 47.1±1.2 |
Values are mean±SD.
FIG. 7.

Metformin improves clinical symptoms in patients with FM after 1 month of treatment. n=6 patients were treated with metformin. Clinical response of the patients after treatment by FIQ total score punctuation (A), several subitems from the FIQ as pain, fatigue, morning tiredness, stiffness, and depression (B), tender points (C), VAS about pain (D), fatigue (E), BDI (F), PSQI (G), and VAS sleep quality (H). Data represent the mean±SD.*p<0.001 between before and after metformin treatment in patients with FM. **p<0.01 between before and after metformin treatment in patients with FM. ***p<0.05 between before and after metformin treatment in patients with FM. BDI, Beck Depression Inventory; FIQ, Fibromyalgia Impact Questionnaire; PSQI, Pittsburgh Sleep Quality Index; VAS, visual analogue scale.
FIG. 8.

Biochemical improvement in FM patients after 1 month of treatment with metformin. (A, B) IL-1β and IL-18 levels in serum from patients after metformin treatment. (C) Protein expression levels of pAMPK and NLRP3 in BMCs from patients after oral treatment. Data represent the mean±SD. #p<0.001 between control and before metformin treatment in patients with FM. *p<0.001 between before and after metformin treatment in patients with FM.
FIG. 9.

Metformin long-term treatment after 7 months. (A) FIQ total score punctuation, (B) tender points, (C) VAS about pain, (D) fatigue, (E) PSQI, (F) VAS sleep quality, (G) BDI.
Discussion
Chronic pain is the most common symptom in FM. The special characteristic of FM is pain perception in the absence of any damage. In this study, we show that AMPK activation impairment may induce pain through the NLRP3 inflammasome pathway. In mice models of pain, an inverse correlation between pain sensation and AMPK activation was also found. In contrast, increased AMPK phosphorylation by pharmacological treatment with metformin or CR decreased the hyperalgesia induced by AMPK dysregulation. Impairment of AMPK activation was associated with activation of the NLRP3 inflammasome pathway. However, the causal relationship between nociception and inflammasome is unknown. In this study, we demonstrated that the levels of NLRP3 inflammasome were increased in pain models where AMPK activation was reduced. These data suggest that AMPK activation and NLRP3 inflammasome are central components in pain modulation. Nevertheless, the molecular mechanisms involved in the AMPK-NLRP3 axis nociception regulation need further investigation. AMPK seems to work on nociceptors also at the sensory neuron level in the spinal cord (23) through mTorc and MAPK pathways, respectively, two kinases which play an important role in pain plasticity. Additionally, it has been reported that inflammasome is involved in periphery response to acute inflammatory pain (17), although its role in the central nervous system remains unknown. On the other hand, mitochondrial dysfunction is a high defined alteration in FM, which is not restored by the impairment of AMPK in this study. In this sense, dysfunctional mitochondria are selectively degraded by an autophagic process named mitophagy and, interestingly, mitophagy has been shown to be involved in FM in a previous study by our group (6). So, the clearance of damaged ROS-producing mitochondria through mitophagy is important for limiting excess NLRP3 signaling (14). Accordingly, antioxidant treatment has shown to inhibit NLRP3 stimulation (5, 14), and in the same sense, autophagy induction should decrease NLRP3. Interestingly, rapamycin, a known promoter of autophagy, has been shown to inhibit IL-1β (23), so autophagy induction could decrease pain perception.
Metformin is a known antidiabetic drug that upregulates AMPK phosphorylation. Previous works (12, 21, 25, 27) and our results here support the hypothesis that metformin can work as an analgesic drug in rodent models of pain. In fact, these studies suggest that AMPK activators can be an effective treatment for human chronic pain diseases. Long-term treatment with metformin mimics some of the benefits of CR through AMPK activation and antioxidant protection by inducing a decrease of both oxidative damage and inflammation (19). In this regard, our results showed that patients with FM presenting with low levels of phosphorylated AMPK and inflammasome activation in BMCs, subjected to long-term treatment with metformin, improved both pain and FM-associated symptoms. As expected, AMPK and inflammasome activation levels were restored to control values after metformin treatment. Our findings reinforce the hypothesis that AMPK activators or NLRP3 inflammasome inhibitor may be a potentially effective therapy in the fight against chronic pain.
However, we are aware of the limitations of our work. Compound C is a cell-permeable pyrazolopyrimidine compound that can act as a reversible and ATP-competitive inhibitor of AMPK, widely used in in vitro and in vivo assays to study the role of AMPK. However, it has been shown that compound C could have other effects. Recently, sunitinib, a tyrosine kinase inhibitor (TKI) that binds to at least eight receptor tyrosine kinases, has been shown to be a more effective inhibitor of AMPK (13). Therefore, we used both compounds to study the role of AMPK from different perspectives. Of course, a better approach will be to carry out the study in AMPK knockout mice. Furthermore, to avoid the variability due to the estral cycle in female mice, only male mice were used in this study. Working with rodents in general is a factor that could restrict extrapolating these results to humans, which is an evident limitation.
Furthermore, despite the fact that we explored the feasibility of our hypothesis through a pilot trial with six patients with FM, further analysis involving more patients in double-blind placebo-controlled clinical trials is required to confirm these observations. Indeed, our research group is currently working in this direction on the basis of the conclusions discussed in this study.
Materials and Methods
Ethics statement
Approval by the ethics committee of the University of Seville was obtained according to the principles of the Declaration of Helsinki and all the International Conferences on Harmonization and Good Clinical Practice Guidelines. All the participants of the study gave their written informed consent before initiating it.
Pain studies in mice were performed in accordance with the European Union guidelines (86/609/EU) and Spanish regulations for the use of laboratory animals in chronic experiments (BOE 67/8509-12, 1988). All experiments were approved by the local institutional animal care committee.
Patients
Briefly, 20 patients from the register of the Andalusian Federation of Fibromyalgia (ALBA ANDALUCÍA) and 20 healthy matched controls were enrolled in the study, having previously obtained informed consent and the approval of the local ethics committee. The inclusion criterion was FM, based on current American College of Rheumatology (ACR) diagnostic criteria, diagnosed 2–3 years ago. The clinical characteristics of each group are shown in Table 1. Exclusion criteria were acute infectious disease within the previous 3 weeks; past or present neurological, psychiatric, metabolic, autoimmune, allergy-related, dermal, or chronic inflammatory disease; undesired habits (e.g., smoking, alcohol consumption); medical conditions that required glucocorticoid treatment, analgesics, or antidepressant drugs; past or current substance abuse or dependence; and pregnancy or current breastfeeding. Twenty healthy women volunteers were included in the study and matched with the recruited female patients with FM for age range, gender, ethnicity, and demographic features (completion of at least 9 years of education and member of the middle socioeconomic class). Healthy controls had no signs or symptoms of FM and had not taken any medication for at least 3 weeks before commencing the study. None of the patients or controls had taken any drug or vitamin/nutritional supplements during the 3 weeks before blood sample collection. All patients and controls followed a standard balanced diet (carbohydrate 50%–60%, protein 10%–20%, and fat 20%–30%) for 3 weeks before blood collection, as established by a diet program. Clinical data were obtained by physical examination and the subjects were evaluated using the FIQ (total score ranged from 0 to 100, with higher scores indicating more severe symptoms), visual analogue scale (VAS) for pain, fatigue, and sleep quality (range, 0–10, with higher scores indicating greater symptom), BDI (range, 0–63, with higher scores indicating greater symptom), and PSQI (range, 0–21, with higher scores indicating worse sleep quality). Tender points were identified by digital pressure at the 18 locations recommended by ACR, which included a minimum of 11 of 18. Heparinized and coagulated blood samples were collected from patients and controls after 12 h of fasting, centrifuged at 3800×g for 5 min, and the plasma and serum were stored at −80°C until testing. Serum biochemical parameters were assayed by routine analytical methods. A routine laboratory test yielded normal results for glucose, uric acid, creatine kinase, aspartate aminotransferase, alanine aminotransferase, cholesterol, and triglycerides (data not shown).
Reagents
Trypsin and metformin (in vitro assay) were purchased from Sigma Chemical Co. MitoSOX™ was purchased from Invitrogen/Molecular Probes. Anti-GAPDH monoclonal antibody, clone 6C5, was purchased from Research Diagnostic, Inc. Anti-NLRP3 antibody was purchased from Adipogen. Anti-PGC-1α and OGG-1 antibodies were from Abcam; anti-AMPK and anti-p-AMPK antibodies were from Cell Signaling; and anti-coq antibodies and TFAM, NRF1, MnSOD, and catalase antibodies were purchased from Santa Cruz Biotechnology. A cocktail of protease inhibitors (complete cocktail) was purchased from Boehringer Mannheim. The Immun-Star HRP substrate kit was from Bio-Rad Laboratories, Inc.
Cell cultures
Peripheral BMCs were purified from heparinized blood by isopycnic centrifugation using Histopaque-1119 and Histopaque-1077 (Sigma Chemical Co.). BMCs were cultured at 37°C in a 5% CO2 atmosphere in RPMI-1640 medium supplemented with l-glutamine, an antibiotic/antimycotic solution (Sigma Chemical Co.), and 10% fetal bovine serum (FBS).
Murine fibroblasts were extracted from 12-week-old mice ears. The ears were sterilized with ethanol, washed with phosphate-buffered saline (PBS), and triturated with razor blades. The resulting samples were incubated with 600 μl of 4 mg/ml collagenase D (Roche) and 4 mg/ml of dispase II (Roche) in Dulbecco's modified Eagle's medium (DMEM) for 45 min in 5% CO2 at 37°C. After filtering and washing, 6 ml of DMEM with 10% FBS and 1% antibiotic–antimycotic were added, and the mixture was incubated in 5% CO2 at 37°C. Approximately 106 cells were passed in a 10-cm plate every 3 days and cultured under standard conditions.
Treatment
Two millimolars of metformin (Sigma Aldrich) and/or 100 μM of H2O2 at 48 h were used for in vitro experiments.
Western blotting
Whole cellular lysate from fibroblasts was prepared by gentle shaking with a buffer containing 0.9% NaCl, 20 mM Tris–HCl, pH 7.6, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and 0.01% leupeptine. Electrophoresis was carried out in a 10%–15% acrylamide sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS/PAGE). Proteins were transferred to Immobilon membranes (Amersham Pharmacia). AMPK, AMPK-P, PGC-1α, NRF1, TFAM, MnSOD, and catalase antibodies were used to detect proteins by Western blotting. Proteins were electrophoresed, transferred to nitrocellulose membranes, and, after blocking overnight at 4°C, incubated with the respective antibody solution diluted at 1:1000. Membranes were then probed with their respective secondary antibody (1:2500). Immunolabeled proteins were detected by using a chemiluminescence method (Immun-Star HRP substrate kit; Bio-Rad Laboratories, Inc.). Protein was determined by the Bradford method. Western blot image was quantified using ImageJ software (http://rsb.info.nih.gov/ij/download.html).
IL-1β and IL-18 levels
IL-1β (GenWay) and IL-18 (Biosensis) levels in serum or culture media were assayed in duplicates by commercial ELISA kits.
ATP levels
ATP levels were determined by a bioluminescence assay using an ATP determination kit from Invitrogen/Molecular Probes according to the instructions of the manufacturer.
Mitochondrial ROS production
Mitochondrial ROS generation in BMCs and fibroblasts were assessed by MitoSOX Red, a red mitochondrial superoxide indicator. MitoSOX Red is a novel fluorogenic dye recently developed and validated for highly selective detection of superoxide in the mitochondria of live cells. MitoSOX Red reagent is live-cell permeant and is rapidly and selectively targeted to the mitochondria. Once in the mitochondria, MitoSOX Red reagent is oxidized by superoxide and exhibits red fluorescence. Approximately 1×106 cells were incubated with 1 μM MitoSOX Red for 30 min at 37°C, washed twice with PBS, resuspended in 500 μl of PBS, and analyzed by flow cytometry in an Epics XL cytometer, Beckman Coultier, Brea, CA (excitation at 510 nm and fluorescence detection at 580 nm).
Animals and drug administration
For all experiments, to avoid the variability due to the estral cycle in female mice, only male mice were used in this study. Nlrp3−/− transgenic mice (C57BL/6 background), provided by FG mice, have been previously described (20). Ten-week-old male C57/BL6/J mice weighing 25–30 g were maintained on a 12-h light/12-h dark cycle. Behavioral studies were performed in accordance with the European Union guidelines (86/609/EU) and Spanish regulations for the use of laboratory animals in chronic experiments (BOE 67/8509-12, 1988). All experiments were approved by the local institutional animal care committee. Compound C, a specific inhibitor of AMPK, was dissolved in saline (vehicle) and intraperitoneally administered at a dose of 20 mg/kg/day for 15 days. Sunitinib (BioVision), a more specific inhibitor of AMPK, was dissolved in DMSO (vehicle) and intraperitoneally administered at a dose of 20 mg/kg/day for 15 days in 60 μl as total volume per day. 5-Chloro-2-methoxy-N-[2-(4-sulfamoylphenyl)ethyl]benzamide (16673-34-0) (Sigma Aldrich) was dissolved in DMSO (0.05–0.1 ml). Mice were treated with 100 mg/kg/day for 15 days. Behavioral tests were performed 5 days after the first drug administration. After testing, mice were anesthetized with CO2 and sacrificed by decapitation. Blood samples were collected for immediate biochemical analysis and BMCs were isolated. Serum samples were frozen at −80°C for further analyses.
Calorie restriction was a progressive process that was initiated with 10% restriction during the first week, followed by 20% and 30% during the second and third weeks, respectively, and maintained at 30% until the end of the treatment.
Behavioral assays
Behavioral analyses were performed in a testing room with homogeneous noise and light levels. The testing apparatus was cleaned with 70% ethanol (Panreac Química S.A.U.) between trials to eliminate any influence of animal odor on the exploratory behavior.
Pain assay
For the hot plate test, a glass cylinder (16 cm high, 16 cm in diameter) was used to constrain the mice to the heated surface of the plate. The plate surface was maintained at 50°C–55°C±0.5°C and the latency to commence paw licking was measured, with a cutoff of 30 s. Data are shown as change of withdrawal latency compared with the control group. Negative and positive values represent hyperalgesia and analgesia, respectively.
Metformin treatment in patients
Six volunteer patients who had not taken any drug or vitamin/nutritional supplements were supplemented with metformin (Merck & Co., Inc.) for 1 month (200 mg/day in the morning). After 1 month of treatment, heparinized blood samples were collected after 12-h fasting and 24 h after the last dose, and clinical symptoms were evaluated. All patients had a sedentary lifestyle. All patients and controls followed a standard balanced diet (carbohydrate 50%–60%, protein 15%–20%, and fat 25%–30%) for 3 weeks before blood collection and during treatment with five meals per day to avoid reduction in glucose, as established by a diet program supervised by a clinical dietitian. Patient routine laboratory tests yielded normal results (data not shown).
Outcome measurements
The primary outcome measurement was the change in the FIQ score from baseline to the end of the intervention. The FIQ is a well-validated multidimensional measurement of the overall severity of FM as rated by patients. Categories included the intensity of pain, physical functioning, fatigue, morning tiredness, stiffness, depression, anxiety, job difficulty, and overall well being. The total score ranged from 0 to 100, with higher scores indicating more severe symptoms. Secondary efficacy measurements included reduction in the number of positive tender points, a VAS (range, 0–10, with higher scores indicating greater pain), and PSQI (range, 0–21, with higher scores indicating worse sleep quality).
Body composition
Several parameters related to body composition (weight, BMI, body fat, muscle mass, bone mass, visceral fat) were determined using the TANITA BC-601 machine (TANITA, Middlesex, United Kingdom).
Statistical analyses
Statistical analyses were performed using the SPSS package for Windows (SPSS). Unless otherwise indicated, data represent the mean±SEM. The unpaired Student's t-test was used to evaluate the significance of differences between groups, accepting p<0.05 as the level of significance. Two-way analysis of variance (ANOVA) was used to compare the behavioral results from animals treated with vehicle alone or with compound C. Chi-squared tests were used for statistical analysis in cases in which qualitative variables were compared.
Supplementary Material
Abbreviations Used
- AL
ad libitum
- AMPK
adenosine monophosphate-activated protein kinase
- ATP
adenosine triphosphate
- BDI
Beck Depression Inventory
- BMCs
blood mononuclear cells
- CR
caloric restriction
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- FIQ
Fibromyalgia Impact Questionnaire
- FM
fibromyalgia
- IL
interleukin
- NLRP3
NOD-like receptor family, pyrin domain containing 3
- PBS
phosphate-buffered saline
- PSQI
Pittsburgh Sleep Quality Index
- ROS
reactive oxygen species
- VAS
visual analogue scale
Acknowledgments
This work has been supported by Federación Andaluza de Fibromialgia y Fatiga Crónica (ALBA Andalucía) and Grupo de Investigacion Junta de Andalucia CTS113. Authors are indebted to Ms. Monica Glebocki for extensive editing of the manuscript and Drs. López Otin, J.A. Enriquez, and Benoit Viollet for the scientific advice.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Bagis S, Tamer L, Sahin G, et al. Free radicals and antioxidants in primary fibromyalgia: an oxidative stress disorder? Rheumatol Int 25: 188–190, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, Barberà-Cremades M, Yagüe J, Ruiz-Ortiz E, Antón J, Buján S, Couillin I, Brough D, Arostegui JI, and Pelegrín P. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15: 738–748, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Bazzichi L, Giannaccini G, Betti L, Fabbrini L, Schmid L, Palego L, Giacomelli C, Rossi A, Giusti L, De Feo F, Giuliano T, Mascia G, Bombardieri S, and Lucacchini A. ATP, calcium and magnesium levels in platelets of patients with primary fibromyalgia. Clin Biochem 41: 1084–1090, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Colombo SL. and Moncada S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem J 421: 163–169, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Cordero MD, Alcocer-Gómez E, Culic O, Carrión AM, de Miguel M, Díaz-Parrado E, Pérez-Villegas EM, Bullón P, Battino M, and Sánchez-Alcazar JA. NLRP3 inflammasome is activated in fibromyalgia: the effect of coenzyme Q10. Antioxid Redox Signal 20: 1169–1180, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cordero MD, De Miguel M, Moreno Fernández AM, Carmona López IM, Garrido Maraver J, Cotán D, Gómez Izquierdo L, Bonal P, Campa F, Bullon P, Navas P, and Sánchez Alcázar JA. Mitochondrial dysfunction and mitophagy activation in blood mononuclear cells of fibromyalgia patients: implication in the pathogenesis of the disease. Arthritis Res Ther 12: R17, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de los Santos-Arteaga M, Sierra-Domínguez SA, Fontanella GH, Delgado-García JM, and Carrión AM. Analgesia induced by dietary restriction is mediated by the kappa-opioid system. J Neurosci 23: 11120–11126, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fontana L. Neuroendocrine factors in the regulation of inflammation: excessive adiposity and calorie restriction. Exp Gerontol 44: 41–45, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerdle B, Forsgren MF, Bengtsson A, Leinhard OD, Sören B, Karlsson A, Brandejsky V, Lund E, and Lundberg P. Decreased muscle concentrations of ATP and PCR in the quadriceps muscle of fibromyalgia patients—a 31P-MRS study. Eur J Pain 17: 1205–1215, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Hardie DG, Ross FA, and Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuner R. Central mechanisms of pathological pain. Nat Med 16: 1258–1266, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Labuzek K, Liber S, Suchy D, and Okopieå BA. A successful case of pain management using metformin in a patient with adiposis dolorosa. Int J Clin Pharmacol Ther 51: 517–524, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Laderoute KR, Calaoagan JM, Madrid PB, Klon AE, and Ehrlich PJ. SU11248 (sunitinib) directly inhibits the activity of mammalian 5'-AMP-activated protein kinase (AMPK). Cancer Biol Ther 10: 68–76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawlor KE. and Vince JE. Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta 1840: 1433–1440, 2014 [DOI] [PubMed] [Google Scholar]
- 15.Li Q, Tian Y, Wang ZF, Liu SB, Mi WL, Ma HJ, Wu GC, Wang J, Yu J, and Wang YQ. Involvement of the spinal NALP1 inflammasome in neuropathic pain and aspirin-triggered-15-epi-lipoxin A4 induced analgesia. Neuroscience 254: 230–240, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Li WW, Guo TZ, Liang D, Shi X, Wei T, Kingery WS, and Clark JD. The NALP1 inflammasome controls cytokine production and nociception in a rat fracture model of complex regional pain syndrome. Pain 147: 277–286, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopes AH, Talbot J, Silva RL, Lima JB, Franca RO.VErri WA, Jr, Mascarenhas DP, Ryffel B, Cunha FQ, Zamboni DS, and Cunha TM. Peripheral NLCR4 inflammasome participates in the genesis of acute inflammatory pain. Pain 156: 451–459, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, and Abbate A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol 63: 316–322, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, and de Cabo R. Metformin improves healthspan and lifespan in mice. Nat Commun 4: 2192, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, Sonenberg N, Dussor G, and Price TJ. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain 7: 70, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Neill LA. and Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493: 346–355, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Price TJ. and Dussor G. AMPK: an emerging target for modification of injury-induced pain plasticity. Neurosci Lett 557 Pt A: 9–18, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodgers MA, Bowman JW, Liang Q, and Jung JU. Regulation where autophagy intersects the inflammasome. Antioxid Redox Signal 20: 495–506, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russel OQ, Möser CV, Kynast KL, King TS, Stephan H, Geisslinger G, and Niederberger E. Activation of the AMP-activated protein kinase reduces inflammatory nociception. J Pain 14: 1330–1340, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Schroder K, Zhou R, and Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 327: 296–300, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, and Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinberg GR. and Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol 92: 340–345, 2014 [DOI] [PubMed] [Google Scholar]
- 29.Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, and Price TJ. Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol Pain 8: 5, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viollet B. and Andreelli F. AMP-activated protein kinase and metabolic control. Handb Exp Pharmacol 203: 303–330, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou R, Yazdi AS, Menu P, and Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.