

The multidomain zinc finger protein PRDM9 is thought to influence the locations of recombination hot spots during meiosis by sequence-specific DNA binding and trimethylation of histone H3 Lys4. Patel et al. cocrystallized zinc fingers 8–12 of the most common variant of human PRDM9, allele A (hPRDM9A), with an oligonucleotide representing a known hot spot sequence and report the structure here.
Keywords: PRDM9, crystallography, recombination hot spots, zinc finger arrays
Abstract
The multidomain zinc finger (ZnF) protein PRDM9 (PRD1–BF1–RIZ1 homologous domain-containing 9) is thought to influence the locations of recombination hot spots during meiosis by sequence-specific DNA binding and trimethylation of histone H3 Lys4. The most common variant of human PRDM9, allele A (hPRDM9A), recognizes the consensus sequence 5′-NCCNCCNTNNCCNCN-3′. We cocrystallized ZnF8–12 of hPRDM9A with an oligonucleotide representing a known hot spot sequence and report the structure here. ZnF12 was not visible, but ZnF8–11, like other ZnF arrays, follows the right-handed twist of the DNA, with the α helices occupying the major groove. Each α helix makes hydrogen-bond (H-bond) contacts with up to four adjacent bases, most of which are purines of the complementary DNA strand. The consensus C:G base pairs H-bond with conserved His or Arg residues in ZnF8, ZnF9, and ZnF11, and the consensus T:A base pair H-bonds with an Asn that replaces His in ZnF10. Most of the variable base pairs (N) also engage in H bonds with the protein. These interactions appear to compensate to some extent for changes from the consensus sequence, implying an adaptability of PRDM9 to sequence variations. We investigated the binding of various alleles of hPRDM9 to different hot spot sequences. Allele C was found to bind a C-specific hot spot with higher affinity than allele A bound A-specific hot spots, perhaps explaining why the former is dominant in A/C heterozygotes. Allele L13 displayed higher affinity for several A-specific sequences, allele L9/L24 displayed lower affinity, and allele L20 displayed an altered sequence preference. These differences can be rationalized structurally and might contribute to the variation observed in the locations and activities of meiotic recombination hot spots.
Homologous recombination is a key event during meiosis, the chromosome-partitioning process that produces gametes, and it drives genetic diversification by shuffling of parental chromosomes (Nachman 2001). Recombination does not occur randomly throughout the genome but rather clusters at specific loci called hot spots (International HapMap 2005; Jeffreys et al. 2005; Myers et al. 2005). Here we investigate the role of hPRDM9 (human PRD1–BF1–RIZ1 homologous domain-containing 9), which expresses specifically in gametocytes, in determining the locations of recombination hot spots (Baudat et al. 2010).
PRDM9 comprises an N-terminal Krüppel-associated box (KRAB) domain; a central PR-SET domain, known for catalyzing histone H3 Lys4 (H3K4) trimethylation (Hayashi et al. 2005; Wu et al. 2013; Eram et al. 2014; Koh-Stenta et al. 2014); and a C-terminal tandem array of multiple Cys2–His2 (C2H2) zinc fingers (ZnFs) (Fig. 1A; Supplemental Fig. S1). PRDM9 is thought to influence the locations of recombination hot spots by sequence-specific DNA binding and H3K4 trimethylation of neighboring nucleosomes (Hayashi et al. 2005; Pratto et al. 2014). This signals recruitment of a recombination–initiation complex that stimulates homologous recombination by introducing DNA double-strand breaks (Pratto et al. 2014).
Figure 1.
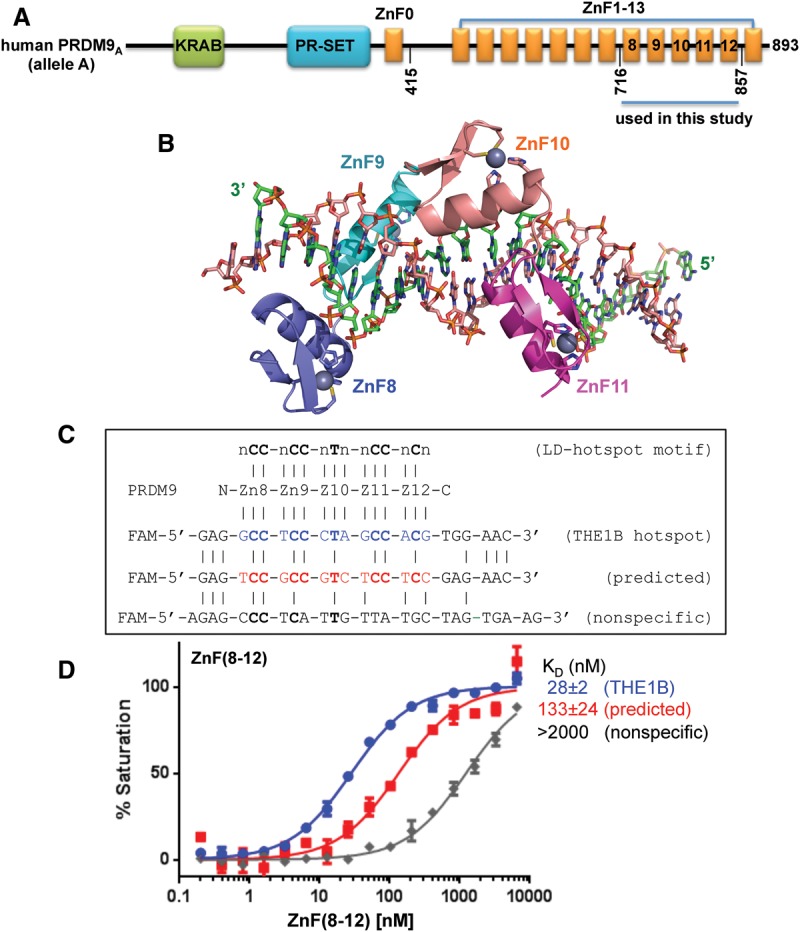
PRDM9A DNA binding. (A) Domain organization of the hPRDM9 protein (accession no. Q9NQV7). (B) Crystal structure of hPRDM9A (hPRDM9, allele A) ZnF8–12 in complex with the THE1B recombination hot spot sequence. ZnF8–11 (color-coded blue [ZnF8], cyan [ZnF9], orange [ZnF10], and magenta [ZnF11]) are shown in cartoon representation. ZnF12 was not visible in the structure. The helix of each finger forms specific H bonds in the major groove mainly with bases in the DNA strand (green) that is complementary to the consensus sequence (orange). The ZnF array, oriented N to C from left to right, interacts with this DNA strand oriented 3′ to 5′. (C) DNA sequences of THE1B hot spot (Myers et al. 2008), a predicted sequence (Baudat et al. 2010), and an arbitrary negative control that partially overlapped the consensus. The top line indicates the LD-hot spot motif (Myers et al. 2008), which was derived from recombination hot spots that are intrinsically sex- and population-averaged (Pratto et al. 2014). (D) PRDM9A ZnF(8–12) binds the THE1B hot spot sequence.
PRDM9 is conserved in overall domain architecture, but the ZnF array is highly polymorphic both within and between species (Oliver et al. 2009; Thomas et al. 2009; Segurel et al. 2011; Groeneveld et al. 2012). This polymorphism implies variation in DNA-binding specificity, in agreement with the finding that hot spot positions and activities vary (Groeneveld et al. 2012). More than 40 allelic variants of hPRDM9 have been documented, which display marked differences in recombination profile and crossover frequency (Baudat et al. 2010; Berg et al. 2010, 2011; Kong et al. 2010; Hinch et al. 2011).
In conventional C2H2 ZnF proteins, each finger comprises two β strands and a helix and interacts with three or four adjacent DNA base pairs (Wolfe et al. 2000; Klug 2010), which we term the “triplet element.” Characteristically, two histidines in the helix together with one cysteine in each β strand coordinate a zinc ion, forming a tetrahedral Cys2–Zn–His2 structural unit that confers rigidity to fingers. When bound to DNA, the helix of each ZnF lies in the DNA major groove, and the β strands and the C2–Zn–H2 unit lie on the outside (Fig. 1B). Side chains from specific amino acids within the N-terminal portion of each helix and the preceding loop make major groove contacts with the bases of primarily one DNA strand. The identities of these amino acids are the principle determinants of the DNA sequence recognized (Supplemental Fig. S2a,b; Gupta et al. 2014; Persikov and Singh 2014).
Allele A of hPRDM9 (hPRDM9A) is the most common form of PRDM9, found in ∼86% of European and ∼50% of African populations (Berg et al. 2010). The predicted DNA-binding specificity of ZnF8–12 of hPRDM9A matches a five-triplet consensus sequence, NCCNCCNTNNCCNCN, that is enriched in ∼40% of recombination hot spots (Myers et al. 2008; Baudat et al. 2010). Using in vitro DNA-binding assays, we show that the purified hPRDM9A ZnF8–12 peptide (residues 716–857) bind to this sequence and that significant differences in binding affinity occur among natural allelic variants. We also show, by means of X-ray crystallography, the way in which hPRDM9A ZnF8–11 recognizes this sequence and how amino acid substitutions within the fingers enhance or impair binding and switch hot spot preference.
Results
Binding affinities
We compared the binding of hPRDM9A ZnF8–12 with three double-stranded oligonucleotides (oligos): a positive control based on an actual hot spot in the THE1B retrotransposon (Myers et al. 2008), a test sequence predicted using the Zinc Finger Consortium Database (Baudat et al. 2010), and an arbitrary negative control that partially overlapped the consensus (Fig. 1C). Fluorescence polarization was used to measure the dissociation constants (KD) toward these oligos (see the Materials and Methods). ZnF8–12 displayed approximately fivefold higher affinity for the actual hot spot than for the predicted sequence and >70-fold higher affinity than for the negative control (Fig. 1D). These findings confirm the presumed specificity of ZnF8–12.
Structural investigations
To investigate the molecular mechanism of sequence recognition, we crystallized the ZnF8–12 peptide with oligos containing the THE1B hot spot sequence. The oligos were synthesized with a 5′-terminal thymine extension on one strand and a 5′-terminal adenine extension on the other strand (Fig. 2; Supplemental Fig. S3). Three structures were solved to a resolution of ∼2 Å in two space groups, P21 and P1 (Supplemental Table S1). These structures were closely similar, with a root mean squared deviation of <1 Å over 98 pairs of Cα atoms. The main differences concerned crystal packing interactions (Supplemental Fig. S4). Here we describe the structure of P21. The DNA molecules were coaxially stacked, with the terminal A and T bases of neighboring DNA molecules pairing to form a pseudo-continuous duplex. ZnF8–12 were used for crystallization, but the last finger, ZnF12, could not be seen in the structure.
Figure 2.

PRDM9A binds the THE1B hot spot. (A) Schematic representation of the ZnF8–12 DNA-binding domain. The top line indicates the LD-hot spot motif. The second line indicates the base pair positions (1–20). The third and the fourth lines are the sequence of the oligos used for this study, shown with the top strand (orange) oriented left to right from 5′ to 3′, matching the 13-mer LD-hot spot motif sequence. The complementary G-rich strand (green) has the base-specific interactions with each ZnF. Two cysteine and two histidine residues (C2H2) in each finger are responsible for Zn2+ ligand binding (bottom connecting lines). Amino acids at positions −1, −4, and −7 (or −8) relative to the first histidine interact specifically with the DNA bases shown below. (B,E,H,K) DNA base interactions involve a residue at position −7 or −8 of each ZnF. (C,F,I,L) DNA base interactions involve a residue at position −4 of each ZnF. (D,G,J,M) DNA base interactions involve a residue at position −1 of each ZnF. (N) Superimposed ZnF9 (cyan) and ZnF10 (orange). Arg757 of ZnF9 at the −8 position makes a DNA–phosphate interaction, while Arg785 of ZnF10 at the −8 position makes a DNA base interaction. (O) Comparison of ZnF8–11, ZnF8–12, and ZnF8–13 with oligos containing THE1B hot spot sequence. (P) Comparison of ZnF8–12 with oligos containing unmodified C or 5-methyl-cytosine (M) at the G:C base pair of position 17.
ZnF8–11 interacts with DNA exclusively in the DNA major groove (Fig. 1B). Most of the hydrogen-bond (H-bond) contacts are to purine bases (G or A) in the strand that is complementary to the consensus sequence (colored green in Fig. 2A–M). ZnF8 interacts only with bases of the first triplet (NCC). The interactions of the remaining fingers overlap triplets: ZnF9 interacts with triplet 2 and the last base of triplet 1 (C-NCC), ZnF10 interacts with triplet 3 and the last base of triplet 2 (C-NTN), and ZnF11 interacts with triplet 4 and the last base of triplet 3 (N-NCC) (Fig. 2A). The invariant base pairs in the consensus sequence are discriminated by a combination of H-bond patterns and steric complementarity that distinguishes them unambiguously. These involve juxtapositions between guanine and arginine or histidine and between adenine and asparagine (Fig. 2) and serve to define the sequence recognized. Most of the bases at the variable (“N”) positions in the consensus also engage in H bonds with amino acids in our structure. These latter interactions appear to be “versatile” contacts that enhance binding when they can form—as they can with the THE1B sequence—but are not critical to sequence recognition, and do not impair it if they cannot form.
Specific interactions
The convention that we used for numbering nucleotides and amino acids is shown in Figure 2A. Base pairs of the crystallization oligo are numbered 1–20, with the consensus sequence as the “top” strand. The first zinc-coordinating histidine in the α helix of each finger is assigned reference position 0. Residues prior to this, at positions −1, −4 and −5, −7, and −8, lie on the inside face of the helix or the preceding loop and form H bonds with the exposed edges of the DNA bases in the major groove. Most of the differences between the ZnFs of PRDM9 involve changes in the residues at just four of these positions (−1, −4, −7, and −8 in Supplemental Fig. S2b) (Baudat et al. 2010).
The six invariant C:G base pairs in consensus triplets 1–4 (Fig. 1C) are recognized primarily by H bonds between the guanines and conserved histidine residues at position −4 (Fig. 2C,F,L) or conserved arginine residues at position −1 (Fig. 2D,G,M) of ZnF8, ZnF9, and ZnF11. The terminal Nη1 and Nη2 groups of Arg736, Arg764, and Arg820 donate H bonds to the guanine O6 and N7 atoms (interatomic distances 2.7–3.0 Å), a pattern specific to Gua. Many sequence-specific proteins recognize Gua in this same way as, for example, the SfiI endonuclease (recognition sequence: GGCCN5GGCC), where three of the four guanines in each half-site form identical H bonds with Arg. (The fourth Gua H-bonds with lysine in almost the same manner [Vanamee et al. 2005]). Depending on side chain rotomer conformation, the Nε2 group of His733, His761, and His817 donate one H bond to either guanine O6 or guanine N7 (2.6–2.9 Å), and the adjacent ring Cε1 atom donates a C–H…N or C–H…O-type bond (3.0–3.2 Å) to the other (Horowitz and Trievel 2012). Due to the locations of the imidazole side chains of these histidines, only guanine can occupy these positions in the consensus sequence without encountering a steric or electrostatic obstruction, a feature that likely contributes to specificity at these positions.
In addition to these interactions, the last base pair of each triplet also H-bonds with a residue in the next finger. Thus, S760 (position −5) of ZnF9 accepts a weak H bond (3.4 Å) from the N4 group of Cyt5 in the preceding triplet, and D786 at position −7 of ZnF10 accepts a similar H bond (3.1 Å) from Cyt8 (Fig. 2D,G). These C:G base pairs each engage in three H bonds, therefore saturating their major groove H-bonding capacities. Cyt14 of triplet 4 might interact in a similar way with N842 or S844 of ZnF12, but the absence of this finger in the structure prevents us from visualizing this juxtaposition (Fig. 2, cf. M and D or G).
Triplet 3 of the consensus sequence (NTN) includes an invariant T:A base pair. In place of His and Arg at positions −4 and −1, with which ZnF8, ZnF9, and ZnF11 specify C:G base pairs, ZnF10 contains Asn (N789 at −4) and Ser (S792 at -1). The side chain of N789 donates one H bond to adenine N7 and accepts one from adenine N6 (Fig. 2I). The O4 atom of the partner thymine interacts nonspecifically with water molecules (Fig. 2I). Juxtaposition of Asn with adenine is a common mechanism for A:T base pair recognition (Luscombe et al. 2001), as occurs, for example, with Asn117 of EcoRI (Kim et al. 1990), Asn185 of EcoRV (Winkler et al. 1993), and Asn140 of PvuII (Cheng et al. 1994).
Due to its short side chain, S792 cannot interact directly with the last base pair of triplet 3 (A:T in our structure). As a result, this base pair is variable, and S792 interacts with Thy11 ambiguously via intermediate water molecules (Fig. 2J). Like the last base pairs of triplets 1 and 2, its adenine partner also interacts with a residue in the next finger, S816 at position −5 of ZnF11 (Fig. 2J). Despite complete conservation of Ser at this position in each ZnF, the way in which it interacts with DNA differs from triplet to triplet. Thus, S732 (ZnF8) interacts with guanine, S760 (ZnF9) interacts with cytosine, S788 (ZnF10) interacts with a phosphate, and S816 (ZnF11) interacts with the adenine (Supplemental Fig. S3d). This “adaptability” stems in part from the ability of serine to act as either an H-bond donor or acceptor or both at the same time.
Nonspecific interactions
H bonds are present in our structure between ZnF8–11 and many of the variable base pairs of the consensus sequence, implying adaptability to sequence differences. The nonspecific first base pair of consensus triplet 1 (G:C in our structure) engages in two H bonds, with Asn730 (3.0 Å) and Ser732 (2.9 Å) at positions −7 and −5 of ZnF8 (Fig. 2B). Like serine, asparagine can also act as an H-bond donor or acceptor by rotation of its side chain, explaining how it, too, might accommodate alternative base pairs. The first base pair of triplet 2 (a T:A in our structure) also engages in an H bond, with Asp758 (3.1 Å) at position −7 of ZnF9 (Fig. 2E). Each of the two nonspecific base pairs (the first and the third) of triplet 3 engages in two H bonds: the first (a C:G) with Arg785 (2 × 2.9 Å) at position −8 of ZnF10 and the third (an A:T) with Ser816 (3.0 Å and 3.2 Å) at position −5 of the following finger (Fig. 2H,J). The former interaction, with Arg785, is remarkably similar to those between the conserved arginines at position −1 of ZnF8, ZnF9, and ZnF11 that specify the invariant C:G base pairs (see the Discussion). Finally, the first base pair of triplet 4 (a G:C) engages in a single H bond with Asn814 (3.2 Å) at position −7 of ZnF11 (Fig. 2K).
The missing ZnF12
The peptide ZnF8–12, comprising five fingers, was used for crystallization, but only fingers 8–11 were visible in our structure. The cocrystallization oligo included additional downstream triplets, -ACGTGG-, from the THE1B hot spot with which ZnF12 could have interacted, and these base pairs are clearly visible in the structure (Fig. 1C). We confirmed that the protein had not degraded during crystallization (Supplemental Fig. S2d) and then measured the binding affinities of ZnF8–11, ZnF8–12, and ZnF8–13 against the THE1B hot spot sequence (Fig. 2O). The binding affinities of ZnF8–12 and ZnF8–13 were found to be similar, suggesting that ZnF13 does not provide extra binding. In contrast, eliminating ZnF12 caused affinity to drop by a factor of ∼12 (Fig. 2O), indicating that ZnF12 interacts favorably with the hot spot sequence and presumably with triplet 5, -ACG. Within triplet 5 is a CpG dinucleotide (Fig. 2A), the canonical site for cytosine methylation in eukaryotic DNA. We repeated the binding assay with an oligo containing 5-methylcytosine in place of cytosine at position 17 in the bottom strand. Binding affinity remained the same, indicating that it is not affected by such methylation (Fig. 2P). The effect of methylation of the cytosine in the top strand at position 16 was not investigated, since the ZnF12 likely contacts only the bottom strand guanine of this base pair, in much the same way as ZnF8, ZnF9, and ZnF11.
ZnF12 has Arg at position −1, which in ZnF8, ZnF9, and ZnF11, interacts specifically with the guanine of invariant C:G base pairs. Triplet 5 has a G:C base pair at this position instead of C:G, suggesting that binding by ZnF12 might unfavorably juxtapose arginine with cytosine. To investigate this, we replaced G:C base pair 17 with a C:G base pair, in essence to mimic triplets 1, 2, and 4. We measured affinity by binding ZnF8–12 to the 6-carboxyfluorescein (FAM)-labeled THE1B oligo and competitively displacing it with either the unlabeled THE1B (G:C, control) oligo or the reversed (C:G, test) oligo. As anticipated, the latter competed approximately threefold more effectively, indicating that ZnF12 has higher affinity when triplet 5 is ACC than ACG (Supplemental Fig. S2e). Suspecting that the inability to observe ZnF12 in our structure resulted from motion due to the unfavorable proximity of Arg848 and Cyt17, we attempted to cocrystallize ZnF8–12 with the higher-affinity ACC oligo. To our surprise, we were unable to produce crystals with these (Supplemental Fig. S5).
Allelic variants of hPRDM9
The preceding analysis relates to hPRDM9A, the most common European allele and the predominant one in Africa. Allele B differs from A by a serine-to-threonine change, S680T, at position −1 of ZnF6 (Baudat et al. 2010). This does not affect ZnF8–12, which are responsible for hot spot recognition (Fig. 3A). Eighty-eight percent of hot spots in a heterozygous A/B individual overlapped those in two A/A individuals, which themselves overlapped by 89%, suggesting that PRDM9B does not specify a distinct set of hot spots (Pratto et al. 2014).
Figure 3.

The C allele of hPRMD9. (A) hPRDM9 contains a C-terminal tandem ZnF DNA-binding array comprising 13 (alleles A and B) or 14 (allele C) fingers. A single amino acid substitution at ZnF6 resulted in allele B. (B) The allele A-specific sequence (THE1B) (Myers et al. 2008) and allele C-specific sequence (Motif-1) (Hinch et al. 2011) are aligned with each ZnF with amino acids at the −8, −7, −4, and −1 positions. (C,D) Comparison of DNA-binding affinities by PRDM9A (ZnF8–12) and PRDM9C (ZnF8–13) against allele A-specific and allele C-specific sequences. Because PRDM9C binds too tightly against Motif-1 (KD being lower than probe concentration of 5 nM), we increased NaCl concentration in the binding assays from 300 mM (C) to 350 mM (D).
PRDM9 allele C is the second most common allele in African populations, with a frequency of 12.8% (Berg et al. 2010, 2011). In A/C heterozygotes, PRDM9C-specific hot spots are more frequent (56% vs. 44%) and more active than PRDM9A-specific hot spots (Pratto et al. 2014), suggesting partial dominance of allele C, as was observed in mice (Brick et al. 2012). Allele C differs from A by an arginine-to-serine change, R764S, at position −1 of ZnF9 and by a substitution that replaces ZnF11 with two other fingers, resulting in an extra finger (Fig. 3A; Baudat et al. 2010; Berg et al. 2010; Jeffreys et al. 2013). We expressed and purified ZnF8–13 of PRDM9C and measured its affinity for oligos containing the C consensus sequence (Motif 1 in Hinch et al. 2011) or the THE1B hot spot (Fig. 3B). We found that its affinity for the C sequence was >10× higher than the affinity of PRDM9A for the THE1B sequence (Fig. 3C), consistent with the observation that PRDM9C is partially dominant over PRDM9A (Pratto et al. 2014). PRDM9C, moreover, barely bound the THE1B sequence (Kd > 600× higher), consistent with PRDM9A and PRDM9C acting at entirely different hot spots (Pratto et al. 2014). PRDM9A also discriminates between the THE1B and C sequences, albeit to a smaller extent (>10×).
Allele L20 differs from A by an asparagine-to-histidine change, N789H, at position −4 of ZnF10 (Fig. 4A; Berg et al. 2010). N789 recognizes the T:A base pair in consensus triplet 3 (Fig. 2I). We found that the N789H variant of ZnF8–12 reduced affinity for the THE1B sequence approximately sixfold (Fig. 4B). Since histidine at −4 of ZnF8, ZnF9, and ZnF11 specifically interacts with guanine (Fig. 2C,F,L), we substituted C:G for T:A in triplet 3 of the THE1B sequence. This change increased affinity greater than twofold for the N789H variant and reduced affinity approximately fivefold for wild-type ZnF8–12 (Fig. 4C), revealing a marked difference in sequence preference between hPRDML20 and hPRDM9A.
Figure 4.

The non-A alleles of hPRDM9. (A) From hPRDM9A, a single amino acid substitution at ZnF10 and ZnF11 resulted in alleles L9/L24, L20, or L13, respectively. (B) Comparison of DNA-binding affinities by different alleles (in the context of ZnF8–12) with the oligo containing the hot spot THE1B sequence. (C) A single base pair change at THE1B (from T:A to C:G) resulted in stronger binding by the N789H variant (allele L20). (D,E) Comparison of DNA-binding affinities by different alleles (in the context of ZnF8–12) with oligos containing hot spot sequences (MSTM1a and MSTM1b).
PRDM9L20 enhances recombination at MSTM1b (Berg et al. 2010), a hot spot on chromosome 1q42.3 (Neumann and Jeffreys 2006). A candidate recognition motif for hPRDM9L20 (“MSTM1b-0”) occurs ∼400 base pairs (bp) upstream of the center of crossover (Neumann and Jeffreys 2006). We identified additional motifs ∼300 bp upstream of (MSTM1b-1), and ∼400 bp and ∼500 bp downstream from (MSTM1b-2 and MSTM1b-3) the crossover center (Supplemental Fig. S6a,b). We compared the affinities of ZnF8–12 N789 (=hPRDM9A) and H789 (=hPRDM9L20) for all four sequences. The two peptides behaved alike, and we found that affinity for the MSTM1b-1 was very much higher than for the other sequences (>25-fold, ∼10-fold, and approximately eightfold, respectively, for MSTM1b-0, MSTM1b-2, and MSTM1b-3) (Supplemental Fig. S6c–f). Our finding that PRDM9A and PRDM9L20 bind these targets with similar affinities sheds little light on how L20 enhances recombination at MSTM1b in vivo or why A/L20 individuals have the highest crossover frequency at the MSTM1b hot spot of all variants examined, including A/A homozygotes (Neumann and Jeffreys 2006; Berg et al. 2010). We speculate that PRDM9 function might be dosage-sensitive (Baker et al. 2015) and that PRDM9L20 binds MSTM1b best, thereby raising the local concentration, whereas PRDM9A occupies many hot spots and is relatively dilute at this site. However, other allele combinations with reduced DNA-binding affinity, such as A/L9 and A/L24, do have lower crossover activity at MSTM1b, as expected (Berg et al. 2010).
Alleles L9 and L24 differ from allele A by a lysine-to-glutamate (K787E) change at position −6 of ZnF10 (Fig. 4A). L9 and 24 have different, but synonymous, base substitutions and code for the same ZnF array (Berg et al. 2010). K787 juxtaposes the phosphate groups of Cyt7 and Cyt8 in triplet 2 of the hot spot sequence (Supplemental Fig. S3a,d). Changing K787 to a negatively charged residue such as Glu changes an electrostatic attraction into repulsion, and this is expected to reduce binding affinity. We measured the affinity of ZnF8–12 K787E (=hPRDM9L9/L24) for a variety of target sequences. Affinity varied from sequence to sequence, but, in every case, K787E had the lowest of all of the alleles examined, confirming that this mutation is indeed deleterious (Fig. 4B–E; Supplemental Fig. S6c–f). Of the sequences examined, K787E exhibited the highest affinity for MSTM1b-1 (Fig. 4D).
MSTM1a, a recombination hot spot neighboring MSTM1b, was reported to be active only in A/L9 and A/L24 individuals (who have similar level of activity at the MSTM1b) (Berg et al. 2010) and not in those carrying, for example, A/L20 or A/A. Contrary to our expectation, K787E (=hPRDM9L9/L24) bound the putative MSTM1a hot spot with fivefold to 10-fold lower affinity than ZnF8–12 N789 (=hPRDM9A) and H789 (=hPRDM9L20) (Fig. 4E), again shedding little light on the in vivo situation (Berg et al. 2010). It is possible that K787E localizes preferentially to MSTM1b, resulting in spillover to MSTM1a, a phenomenon observed between adjacent hot spots (Fan et al. 1997; Tiemann-Boege et al. 2006). It has been suggested that MSTM1a is a “young” hot spot that is yet to leave a significant mark on haplotype diversity (Jeffreys et al. 2005).
The allele L13 differs from allele A by a serine-to-arginine (S813R) change at position −8 of ZnF11 (Fig. 4A). S813 makes a weak contact (3.5 Å) with a backbone phosphate group in triplet 4 (Supplemental Fig. S3d,f). Changing S813 to a positively charged residue such as arginine is expected to increase binding affinity and was found to do so modestly for all of the sequences tested (Supplemental Fig. S7).
Discussion
Our results confirm that the ZnF array 8–12 of PRDM9A, the major allelic variant in European and African populations, recognizes the consensus motif 5′-NCCNCCNTNNCCNCN-3′. The C:G base pairs in this consensus are recognized by conserved histidine residues at position −4 or conserved arginine residues at position −1 of ZnF8, ZnF9, ZnF11, and probably ZnF12. The T:A base pair is recognized by an asparagine at position −4 of ZnF10 that replaces the histidine conserved in the other fingers. These specific Gua–Arg, Gua–His, and Ade–Asn interactions involve bidentate contacts.
Of interest is our finding that, like the invariant bases, many of the variable bases in the THE1B sequence—mainly those at the first position of each triplet—also form H bonds with amino acids. These H bonds are “versatile” in the sense they can arise with some bases but not with others. This implies that the participating amino acids can alter conformation to suit the substrate and, in this way, intimately fit the ZnF array to a variety of different sequences. The Arg–Asp (RD) dipeptide at positions −8 and −7 of ZnF9 and ZnF10 are examples of such adaptability. In ZnF9, D758 conforms to T:A as the first base pair of its triplet (TCC) and H-bonds (3.1 Å) with the adenine in the bottom strand (Fig. 2E), while R757 H-bonds (2.5 Å) with a backbone phosphate group (Supplemental Fig. S3d). In ZnF10, these same amino acids adopt different conformations and partners (Fig. 2N). D786 conforms to the last base pair of the previous triplet (Fig. 2G) and, by doing so, makes space for R785 instead to conform to the first base pair of its triplet (CTN) (Fig. 2H). D786 H-bonds (3.1 Å) with the cytosine in the top strand of triplet 2 (TCC), while R785 forms two H bonds (2× 2.9 Å) with the guanine in the bottom strand of triplet 3 (Fig. 2H). Other examples of adaptability are also evident in our structure, such as N730 and N814 (Fig. 2B,K) and the four conserved serine residues at position −5 discussed earlier.
Another interesting result concerns the ability of ZnF8–12 (and perhaps other ZnF combinations) to bind to sequences that differ from the consensus at one or more positions without loss of affinity. For example, PRDM9A binds with similar affinity to the THE1B and MSTM1b-1 sequences even though these differ at nine out of 15 base pair positions (five triplets), including the entire triplet 3 (from CTA to TCT) (Fig. 4D). Except for the two changes in the conserved positions (T-to-C change in the middle of triplet 3 and C-to-A change in the last base pair of triplet 4), all other seven changes are in the variable “N” positions. This might account for the observation that 88% of the variable hot spot sequences differ in at least one position between the genomes of two A/A individuals (Pratto et al. 2014). Notwithstanding, a single amino acid change at position −4 of ZnF10, from Asn (PRDM9A) to His (PRDM9L20), resulted in a switch in DNA sequence preference at the middle base pair of triplet 3 from T:A to C:G (Fig. 4B,C).
Genome-wide recombination initiation maps of individual human males suggest that sequence changes at PRDM9-binding sites explain less than half of the variation in hot spot intensity (Pratto et al. 2014). Evidently, factors other than DNA sequence affinity must influence crossover activity (Neumann and Jeffreys 2006), factors such as accessibility of binding sites within chromatin, recruitment of additional proteins via the N-terminal putative KRAB domain, or dimerization (Supplemental Fig. S8; Baker et al. 2015). Further studies of PRDM9 protein–protein interactions (via the KRAB domain), histone methylation at H3K4 (via the PR-SET domain and the yet to be identified reader domain), and sequence-specific DNA binding (via the ZnF array) could lead to a more complete understanding of meiotic recombination and mammalian genome evolution.
Materials and methods
We designed synthetic hPRDM9A ZnF8–13 by optimizing the codon set for Escherichia coli. We generated a GST-tagged construct containing residues 716–857 (pXC1378). The tag was cleaved, and the ZnF8–12 was crystallized with 20 + 1-bp DNA containing THE1B hot spot sequence. The structure was determined by single anomalous diffraction at a wavelength near the zinc absorption edge. The DNA-binding activities of allele A (wild type), allele L20 (N789H), allele L9/L20 (K787E), allele L13 (S813R), and allele C were assayed by fluorescence polarization.
Protein expression and purification
The cDNA fragments encompassing the C-terminal array of ZnF8–13 (residues 716–893; pXC1377) from hPRDM9A (accession no. Q9NQV7) and ZnF8–14 (residues 716–919; pXC1504) from hPRDM9C were synthesized (Genewitz) and subcloned into pGEX-6p1 vector. PRDM9A ZnF8–12 (residues 716–857; pXC1378) and ZnF8–11 (residues 716–829; pXC1381) were generated by PCR from ZnF8–13 (pXC1377) plasmid DNA. In addition, mutants N789H (pXC1379), K787E (pXC1380), and S813R (pXC1439) of ZnF8–12 were generated using site-directed mutagenesis protocol; mutations were confirmed by sequencing. PRDM9C ZnF8–13 (residues 716–885; pXC1505) was generated by PCR from ZnF8–14 (pXC1504) plasmid DNA. All ZnF proteins were expressed as glutathione S-transferase (GST)-tagged fusion proteins in E. coli BL21 (DE3) codon plus RIL and purified using the same protocol.
Cells were grown in LB medium at 37°C until the OD600 reached 0.5, when the temperature was lowered to 16°C. The culture was supplemented with 100 µM ZnCl2 and induced by 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight. Cells were harvested by centrifugation and resuspended into lysis buffer containing 20 mM Tris (pH 7.5), 700 mM NaCl, 5% glycerol, 25 µM ZnCl2, 0.5 mM tris(2-carboxyethyl) phosphine (TCEP), and 0.1 mM phenylmethylsulphoyl fluoride (PMSF). Cells were lysed by sonication for 8 min with 1 sec on and 2 sec off. Lysate was treated with neutralized polyethylenimine (Sigma) to a final concentration of 0.1% and clarified by centrifugation. Clear lysate was loaded onto a glutathione Sepharose 4B column (GE Healthcare), and the GST-tagged protein was eluted from the column with buffer containing 100 mM Tris (pH 8.0), 500 mM NaCl, 5% glycerol, 25 µM ZnCl2, 0.5 mM TCEP, and 20 mM reduced glutathione. The GST tag was removed by treating the eluted protein with ∼100 µg of Precission protease (purified in-house) overnight at 4°C. Cleaved protein was further purified to homogeneity by ion exchange chromatography on tandem Hitrap Q-SP columns (GE Healthcare). Most of the protein flowed through the Q column onto the SP column, from which it was eluted as a single peak at ∼0.8 M NaCl using a linear gradient of NaCl from 0.5 M to 1 M. Finally, the protein was eluted out as a single peak on a Superdex-200 (16/60) column with the same lysis buffer except at 500 mM NaCl.
Fluorescence-based DNA-binding assay
DNA binding was performed using a fluorescence polarization assay. Various amounts of ZnF protein was incubated with 10 or 5 nM FAM-labeled dsDNA probe in buffer containing 20 mM Tris (pH 7.5), 300 mM NaCl, 5% glycerol, 25 µM ZnCl2, and 0.5 mM TCEP, with a final volume of 50 µL for 30 min at room temperature. The fluorescence polarization was measured using a Synergy 4 microplate reader (BioTek). Curves were fit individually using the equation [mP] = [maximum mP] × [C]/(KD + [C]) + [baseline mP], where mP is millipolarization, and [C] is protein concentration. KD values were derived (from two experimental replicates) by fitting the experimental data to the equation in Graphpad prim software (version 6.0).
Crystallography
Purified ZnF8–12 was incubated with the dsDNA (Supplemental Fig. S5) at an equimolar ratio to a final concentration of 25 µM on ice in 20 mM Tris (pH 7.5), 500 mM NaCl, 5% glycerol, 25 µM ZnCl2, and 0.5 mM TCEP. The protein–DNA complex was formed by dialysis against the same buffer components with 250 mM NaCl but without ZnCl2. The complex was further concentrated up to ∼0.6 mM prior to crystallization. An aliquot of protein–DNA complex (0.2 µL) was mixed with an equal volume of mother liquor by Phoenix (Art Robbins Instruments). Good diffracting crystals grew overnight at 16°C with three different DNAs (Supplemental Table S1). The best diffracting crystals used for data collection were obtained from mother liquor containing 58 µM Bis-Tris propane, 42 µM citric acid (pH 6.0), and 25% (w/v) polyethylene glycol 3350 using hanging drop vapor diffusion method. The crystals were flash-frozen under liquid nitrogen using 20% glycerol as a cryoprotectant.
X-ray diffraction data were collected at the SER-CAT 22-ID beamline of the Advanced Photon Source (Argonne National Laboratory). First, a data set at 2.4 Å resolution was collected and processed by HKL2000 (Otwinowski et al. 2003) from a single crystal at wavelength 1.28149 Å (∼20 eV above the zinc absorption edge). A total of 794 frames was collected with 1° oscillation, resulting in 98.8% reflections with Bijvoet pairs. The AutoSolve module of PHENIX (Adams et al. 2010) was used for initial crystallographic phasing calculation by single-wavelength anomalous dispersion of zinc signals. The initial electron density revealed clearly visible DNA molecules, and a B-DNA model made by the “make-na server” (http://structure.usc.edu/make-na/server.html) was placed into the density. The AutoBuild module of PHENIX built ZnF molecules into the electron density. A higher-resolution data set at 1.92 Å was collected and used for structural refinement. A total of 3568 frames collected from four different crystals, at the wavelength of 1.0 Å with 0.5° oscillation, was scaled and merged using HKL2000. Model refinements were performed using PHENIX, and the model was manually adjusted by COOT (Emsley and Cowtan 2004). ZnF8–12 was also crystallized in complex with two additional DNA sequences (Supplemental Table S1), and data sets were collected at 1.97 and 2.05 Å, respectively, and used for structural refinement. Structure quality was analyzed and validated by the Protein Data Bank (PDB) validation server (Read et al. 2011). Molecular graphics were generated using PyMol (DeLano Scientific, LLC).
Accession numbers
The X-ray structures (coordinates and structure factor files) of hPRDM9A ZnF8–12 with bound DNA have been submitted to PDB under accession numbers 5EGB (P21 space group), 5EH2, and 5EI9 (P1 space group).
Supplementary Material
Acknowledgments
We thank Dr. F. Baudat and B. de Massy for sending us the initial hPRDM9 construct, B. Baker of New England Biolabs for synthesizing the oligos, and Yusuf Olatunde Olanrewaju for providing purified proteins of ZFP57 and KAP1. The Department of Biochemistry of Emory University School of Medicine supported the use of SER-CAT beamlines. This work was supported by a grant from the National Institutes of Health (GM049245-22) to X.C. (who is a Georgia Research Alliance Eminent Scholar). A.P. performed all experiments. J.R.H. helped with crystallographic experiments. G.G.W. performed structural analysis and assisted in preparing the manuscript. X.Z. and X.C. organized and designed the scope of the study. All authors were involved in analyzing data and preparing the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.274928.115.
Freely available online through the Genes & Development Open Access option.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Petkova P, Walker M, Flachs P, Mihola O, Trachtulec Z, Petkov PM, Paigen K. 2015. Multimer formation explains allelic suppression of PRDM9 recombination hotspots. PLoS Genet 11: e1005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Lam KW, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ. 2010. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet 42: 859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Sarbajna S, Odenthal-Hesse L, Butler NJ, Jeffreys AJ. 2011. Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. Proc Natl Acad Sci 108: 12378–12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. 2012. Genetic recombination is directed away from functional genomic elements in mice. Nature 485: 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Balendiran K, Schildkraut I, Anderson JE. 1994. Structure of PvuII endonuclease with cognate DNA. EMBO J 13: 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Eram MS, Bustos SP, Lima-Fernandes E, Siarheyeva A, Senisterra G, Hajian T, Chau I, Duan S, Wu H, Dombrovski L, et al. 2014. Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J Biol Chem 289: 12177–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QQ, Xu F, White MA, Petes TD. 1997. Competition between adjacent meiotic recombination hotspots in the yeast Saccharomyces cerevisiae. Genetics 145: 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneveld LF, Atencia R, Garriga RM, Vigilant L. 2012. High diversity at PRDM9 in chimpanzees and bonobos. PLoS One 7: e39064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Christensen RG, Bell HA, Goodwin M, Patel RY, Pandey M, Enuameh MS, Rayla AL, Zhu C, Thibodeau-Beganny S, et al. 2014. An improved predictive recognition model for Cys2–His2 zinc finger proteins. Nucleic Acids Res 42: 4800–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. 2005. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438: 374–378. [DOI] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, Palmer CD, Chen GK, Wang K, Buxbaum SG, Akylbekova EL, et al. 2011. The landscape of recombination in African Americans. Nature 476: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S, Trievel RC. 2012. Carbon-oxygen hydrogen bonding in biological structure and function. J Biol Chem 287: 41576–41582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. 2005. A haplotype map of the human genome. Nature 437: 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R, Panayi M, Myers S, Donnelly P. 2005. Human recombination hot spots hidden in regions of strong marker association. Nat Genet 37: 601–606. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Cotton VE, Neumann R, Lam KW. 2013. Recombination regulator PRDM9 influences the instability of its own coding sequence in humans. Proc Natl Acad Sci 110: 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Grable JC, Love R, Greene PJ, Rosenberg JM. 1990. Refinement of Eco RI endonuclease crystal structure: a revised protein chain tracing. Science 249: 1307–1309. [DOI] [PubMed] [Google Scholar]
- Klug A. 2010. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79: 213–231. [DOI] [PubMed] [Google Scholar]
- Koh-Stenta X, Joy J, Poulsen A, Li R, Tan Y, Shim Y, Min JH, Wu L, Ngo A, Peng J, et al. 2014. Characterization of the histone methyltransferase PRDM9 using biochemical, biophysical and chemical biology techniques. Biochem J 461: 323–334. [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Gylfason A, Kristinsson KT, Gudjonsson SA, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103. [DOI] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM. 2001. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res 29: 2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G. 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129. [DOI] [PubMed] [Google Scholar]
- Nachman MW. 2001. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet 17: 481–485. [DOI] [PubMed] [Google Scholar]
- Neumann R, Jeffreys AJ. 2006. Polymorphism in the activity of human crossover hotspots independent of local DNA sequence variation. Hum Mol Genet 15: 1401–1411. [DOI] [PubMed] [Google Scholar]
- Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, Beatson SA, Lunter G, Malik HS, Ponting CP. 2009. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet 5: e1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. 2003. Multiparametric scaling of diffraction intensities. Acta Crystallogr A 59: 228–234. [DOI] [PubMed] [Google Scholar]
- Persikov AV, Singh M. 2014. De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res 42: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratto F, Brick K, Khil P, Smagulova F, Petukhova GV, Camerini-Otero RD. 2014. DNA recombination. Recombination initiation maps of individual human genomes. Science 346: 1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RJ, Adams PD, Arendall WB III, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, et al. 2011. A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19: 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurel L, Leffler EM, Przeworski M. 2011. The case of the fickle fingers: how the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. PLoS Biol 9: e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH, Emerson RO, Shendure J. 2009. Extraordinary molecular evolution in the PRDM9 fertility gene. PLoS One 4: e8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemann-Boege I, Calabrese P, Cochran DM, Sokol R, Arnheim N. 2006. High-resolution recombination patterns in a region of human chromosome 21 measured by sperm typing. PLoS Genet 2: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanamee ES, Viadiu H, Kucera R, Dorner L, Picone S, Schildkraut I, Aggarwal AK. 2005. A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. EMBO J 24: 4198–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler FK, Banner DW, Oefner C, Tsernoglou D, Brown RS, Heathman SP, Bryan RK, Martin PD, Petratos K, Wilson KS. 1993. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J 12: 1781–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO. 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 29: 183–212. [DOI] [PubMed] [Google Scholar]
- Wu H, Mathioudakis N, Diagouraga B, Dong A, Dombrovski L, Baudat F, Cusack S, de Massy B, Kadlec J. 2013. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep 5: 13–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.