

Here, Coffill et al. characterize Tp53, Tp63, and Tp73 in a jawless vertebrate, the Japanese lamprey, as well as the Mdm2 and Mdm4 genes using genome analysis. Functional analysis reveals conservation of p63 and p73 compared with p53, which shows substantial variability within the C-terminal and N-terminal domains, and that lamprey Mdm2 degrades human p53 with great efficiency; however, this interaction is not inhibited by currently available small molecule inhibitors of the human HDM2 protein.
Keywords: p53, Mdm2, Mdm4, evolution, lamprey
Abstract
The extant jawless vertebrates, represented by lampreys and hagfish, are the oldest group of vertebrates and provide an interesting genomic evolutionary pivot point between invertebrates and jawed vertebrates. Through genome analysis of one of these jawless vertebrates, the Japanese lamprey (Lethenteron japonicum), we identified all three members of the important p53 transcription factor family—Tp53, Tp63, and Tp73—as well as the Mdm2 and Mdm4 genes. These genes and their products are significant cellular regulators in human cancer, and further examination of their roles in this most distant vertebrate relative sheds light on their origin and coevolution. Their important role in response to DNA damage has been highlighted by the discovery of multiple copies of the Tp53 gene in elephants. Expression of lamprey p53, Mdm2, and Mdm4 proteins in mammalian cells reveals that the p53–Mdm2 interaction and the Mdm2/Mdm4 E3 ligase activity existed in the common ancestor of vertebrates and have been conserved for >500 million years of vertebrate evolution. Lamprey Mdm2 degrades human p53 with great efficiency, but this interaction is not blocked by currently available small molecule inhibitors of the human HDM2 protein, suggesting utility of lamprey Mdm2 in the study of the human p53 signaling pathway.
More than 50% of all human cancers contain mutations in the p53 tumor suppressor protein (Petitjean et al. 2007). From among the three family members p53, p63, and p73, the founding member p53 is known to be a highly inducible transcriptional regulator that can activate or repress the activity of a large number of genes, resulting in diverse cellular effects, including cell cycle arrest, apoptosis, DNA repair, or metabolism (Vousden and Lu 2002; Reinhardt and Schumacher 2012; Hock and Vousden 2014; Khoo et al. 2014; Meek 2015). The p53 key negative regulator Mdm2, which has a potent ubiquitin E3 ligase activity, acts in a heteromeric complex with its partner, Mdm4, to target p53 for degradation. The p53 response is activated in part through increased protein levels and decreased degradation when the p53/Mdm2/4 complex is disrupted; for example, in response to oncogene activation, nuclear stress, and DNA damage (Vousden and Lu 2002; Reinhardt and Schumacher 2012; Hock and Vousden 2014; Khoo et al. 2014; Meek 2015). The significant role of p53 in response to DNA damage is highlighted by the recent discovery of multiple functional copies of the Tp53 gene in elephants (Abegglen et al. 2015). The Tp63 gene also shows a response to DNA damage, acting as a major regulator of the response in female germ cells (Suh et al. 2006; Gonfloni et al. 2009; Levine et al. 2011). The Mdm component of the p53 pathway has been conserved in evolution (Lu et al. 2009), as it is found in all jawed vertebrates and some invertebrate species. Some of the invertebrate Mdm homologs identified can be found in urochordates (e.g., sea squirt, Ciona intestinalis), cephalochordates (Florida lancelet, Branchiostoma floridae), molluscs (owl limpet, Lottia gigantean; bay mussel, Mytilus trossulus), hemichordates (acorn worm, Saccoglossus kowalevskii), arothropoda (deer tick, Ixodes scapularis), and placozoa (Trichoplax adhaerens) (Momand et al. 2011). However, the Caenorhabditis elegans and Drosophila melanogaster genomes lack an Mdm gene, but, notably, their Tp53-like gene activity is still induced by DNA damage (Brodsky et al. 2004). The p53 family shares a common ancestor, and most conservation is seen in the DNA-binding domain. Tp53/Tp63/Tp73-like genes have been found in all animal species but are absent in plants and yeasts (Lu et al. 2009). In addition, multiple isoforms of the p53 family members are produced by use of alternate initiation codons and by alternate splicing. Understanding more about this vital human tumor repressor pathway is of high importance, as recent studies of p53 mutations in mice have shown that some p53 mutants lack substantial transcriptional activity yet retain some tumor suppressor functions (Brady et al. 2011; Li et al. 2012). Indeed, mice that lack what were thought to be vital p53 downstream response genes—p21, Puma, and Noxa—are as tumor-resistant as wild-type mice, yet p53-null mice show an extraordinary propensity to develop cancer, suggesting that other activities of the p53 system are required for tumor suppression (Valente et al. 2013). Biochemical studies of the evolutionary conservation of function of p53 family members and their regulators may help to identify the most conserved and essential functions of these complex and highly regulated proteins.
The extant vertebrates are divided into two major groups—the jawed vertebrates (gnathostomes) and the jawless vertebrates (cyclostomes)—that diverged ∼500 million years ago. While the jawed vertebrates include cartilaginous fish, ray-finned fish, lobe-finned fish, amphibians, reptiles, birds, and mammals, the cyclostomes are represented by only two lineages, the lampreys and hagfish, which constitute a monophyletic group. The p53 pathway members have been investigated extensively in representative jawed vertebrates, including a cartilaginous fish, the elephant shark (Lane et al. 2011), and thus it has been inferred that genes for all three Tp53 family members as well as Mdm2 and Mdm4 were present in the common ancestor of jawed vertebrates. However, it is unclear whether jawless vertebrate genomes code for all of the components of the jawed vertebrate p53 pathway and whether the p53 pathway proteins in jawless vertebrates are functionally equivalent to their counterparts in humans and other jawed vertebrates.
Jawless vertebrates are regarded as “primitive” vertebrates because of their relatively simple morphological and physiological traits compared with jawed vertebrates. For example, jawless vertebrates possess a single median dorsal nostril, in contrast to ventral nostrils in jawed vertebrates, and lack mineralized tissues, articulate jaws, paired appendages, the pancreas, and the spleen (Osorio and Retaux 2008; Shimeld and Donoghue 2012). The adaptive immune system of jawless vertebrates comprises variable lymphocyte receptors (VLRs) assembled from leucine-rich repeat modules (LRRs), in contrast to T-cell and B-cell receptors of jawed vertebrates that are generated from the immunoglobulin superfamily (Kasahara and Sutoh 2014). A unique feature of lampreys (and probably hagfish) is the programmed loss of ∼20% of germline DNA from somatic cells during early stages of embryonic development such that only the germline cells contain 100% of the genome (Smith et al. 2009). This programmed genome rearrangement has been proposed to serve as a biological strategy to restrict pluripotency functions to the germline (Smith et al. 2012).
To date, draft genome sequences of two lamprey species, the sea lamprey (Petromyzon marinus) (genome size 2.4 Gb) (Smith et al. 2013) and the Japanese lamprey (Lethenteron japonicum) (genome size 1.6 Gb) (Mehta et al. 2013), have been generated. While the sea lamprey genome sequence was generated using DNA from a somatic tissue, the Japanese lamprey genome was sequenced using DNA from the testis. In this study, we mined the genome assembly of the Japanese lamprey for members of the p53 pathway and obtained their complete coding sequences by a combination of RT–PCR and 5′/3′ RACE. To determine the extent of conservation of p53 pathway function during the evolution of vertebrates, we examined the biochemical function of the lamprey p53 (Lj-p53) and Mdm2 (Lj-Mdm2) proteins and their interactions with human p53 pathway proteins (Hs-p53 and HDM2). Lj-Mdm2 was remarkably active in degrading Hs-p53 when tested in a cellular background derived from mice lacking endogenous p53 and Mdm2, indicating that the p53-specific E3 ligase activity of Mdm2 already existed in the common ancestor of vertebrates.
Results
Lj-p53 family members
We searched the Japanese lamprey genome assembly (Japanese Lamprey Genome Project, http://jlampreygenome.imcb.a-star.edu.sg) for members of the p53 family using human and elephant shark p53, p63 and p73 protein sequences as the query (see the Materials and Methods). Our searches identified a complete gene on scaffold 216 with high similarity to Tp53. The protein encoded by this gene lacks a SAM domain, thereby providing further evidence that it is likely the p53 ortholog of lamprey. The Lj-p53 protein shows an overall identity of 38% with the Hs-p53 protein. However, the identity is higher in the DNA-binding domain (60%) and the oligomerization domain (39%) (Table 1; Supplemental Fig. S1). By performing 5′ RACE using different sets of primers, we were able to identify four isoforms (Lj-p53, Lj-Δ27p53, Lj-Δ30p53, and Lj-Δ108p53)) of the lamprey Tp53 gene (Fig. 1; Supplemental Fig. S2). The Lj-Δ27p53 and Lj-Δ30p53 isoforms are produced by the use of alternate methionine residues as initiation codons, creating N′-terminal deletions of 27 and 30 amino acids, respectively. Such ΔN′ isoforms are a common feature of the p53 family members found in jawed vertebrates. They are functionally highly significant, as their orthologs studied from other organisms have impaired transcription activation function and can therefore act as repressors rather than activators of gene expression (Marcel et al. 2011). They also lack the critical N′-terminal Mdm-binding peptide, making them more resistant to Mdm-mediated proteasomal degradation. This is likely the case for the lamprey ΔN′ isoforms, as the Mdm-binding peptide occurs at amino acids F15–W19 of Lj-p53. The third isoform found in this analysis is more complex, as it is generated by alternate splicing to produce a novel N′-terminal segment of 11 amino acids that is then fused in-frame to the main p53 sequence at amino acid 108. This truncated protein can be considered similar in general architecture to ΔN′ isoforms that lack the N′-terminal amino acids preceding the DNA-binding domain that have been found in other species. In zebrafish, an alternative Δ113 product is prominent (Guo et al. 2010), while the deer tick genome contains a separate gene that initiates at this intron/exon domain boundary (Lane et al. 2010). In humans and mice, Δ133 and Δ160 p53 protein species are also found (Marcel et al. 2011).
Table 1.
Amino acid similarity between human and lamprey protein sequences
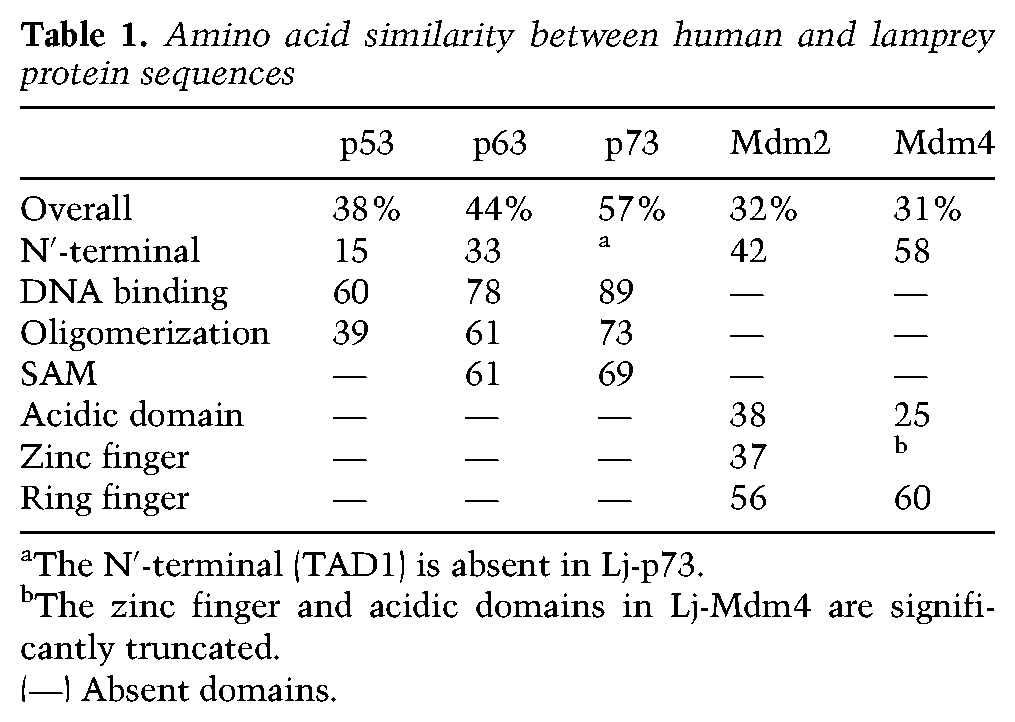
Figure 1.
Lamprey Tp53 gene structure and isoforms. By performing 5′ RACE using different sets of primers, we were able to identify four isoforms of the lamprey Tp53 gene. We designated these isoforms as Lj-p53, Lj-Δ27p53, Lj-Δ30p53, and Lj-Δ108p53. Coding exons are designated by open boxes, and noncoding exons are indicated by the purple boxes. The transcription start site is indicated by an arrow, and the sizes of the 5′ introns are labeled. Lj-Tp53 exons that encode the Mdm2-interacting region and the DNA-binding domain are labeled. The novel N′-terminal segment of 11 amino acids that is fused in-frame to the main p53 sequence at amino acid 108 in Lj-Δ108p53 is underlined.
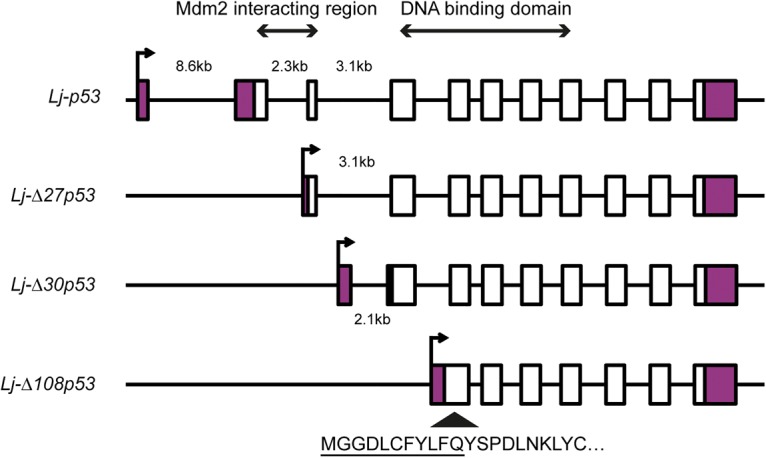
In addition to the Lj-Tp53 gene, two near-complete genes—one on scaffold_21 and another on scaffold_926—with similarity to both Tp63 and Tp73 were identified in the Japanese lamprey genome. The coding sequences of these genes were completed by sequencing 5′ RACE products, and the overall exon–intron structures were confirmed by RT–PCR. The proteins encoded by both genes contain a SAM domain, indicating that they are related to Tp63 and Tp73 genes of jawed vertebrates. In order to verify their identity, we carried out phylogenetic analysis (maximum likelihood) of all three lamprey proteins as well as p53, p63, and p73 proteins from selected jawed vertebrates. However, the phylogenetic analysis was unable to establish the identity of the lamprey proteins, as they were found to form independent branches with low bootstrap supports between the gnathostome p53 and p63 clades (Fig. 2), presumably due to the GC bias of lamprey genes (see Mehta et al. 2013). We then compared the synteny of genes at the lamprey gene loci with the synteny of genes at human and coelacanth Tp53, Tp63, and Tp73 gene loci (Supplemental Fig. S3). However, the synteny was also not informative, as the genes flanking the lamprey genes were not conserved in either human or coelacanth loci. We then used detailed protein alignments to designate the gene on scaffold_21 as Lj-Tp63 gene and the gene on scaffold_926 as Lj-Tp73 gene.
Figure 2.
Phylogenetic tree placement of Lj-p53, Lj-p63, and Lj-p73. The full-length protein sequences were aligned using ClustalW (default parameters) in MEGA6. Gaps in the alignment were removed, leaving a total of 252 positions in the final alignment. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test is shown next to the branches. (Eshark) Elephant shark. The accession numbers of protein sequences that were used in the phylogenetic analysis are in the Materials and Methods.
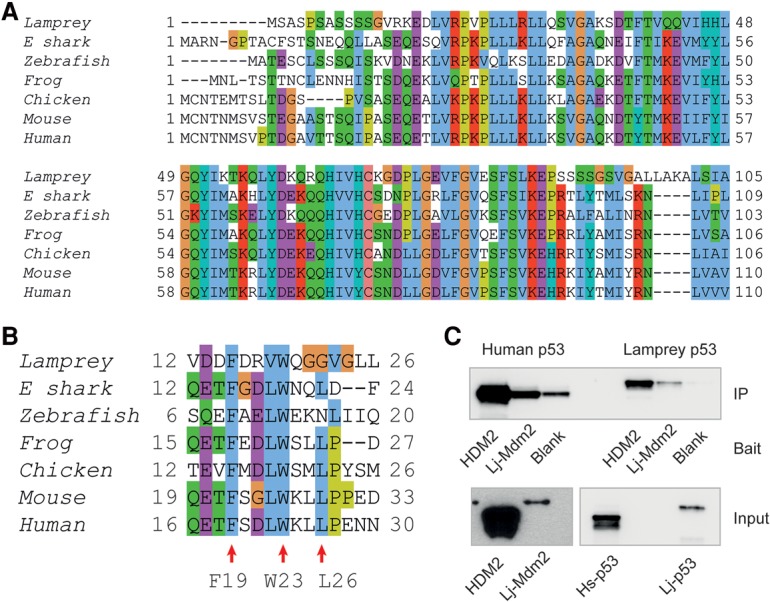
The three members of the Lj-Tp53 gene family exhibit very similar exon–intron organizations with highly conserved intron positions and phases (Supplemental Fig. S4), which supports the hypothesis that they are the result of duplications of an ancestral gene. The protein sequence alignment for Lj-p63 (Supplemental Fig. S5A,B) and Lj-p73 (Supplemental Fig. S6A,B) with human and other jawed vertebrate proteins revealed the overall amino acid similarity between Hs-p63 and Lj-p63 to be 44%, while the overall similarity for p73 for these two species was higher, at 57% (Table 1). Lj-p63 possesses an N′-terminal transactivation domain (TAD) with a 33% similarity; however, Lj-p73, which lacks this domain, is more similar to Hs-ΔNp73. The DNA-binding domain identities of Lj-p63 and Lj-p73 were 78% and 89%, respectively, to their human counterparts and were found to be significantly higher than that between Hs-p53 and Lj-p53 (60%). The SAM domains were found to have 61% and 69% identities between human and Lj-p63 and Lj-73, respectively. For each domain analyzed, Lj-p73 consistently displayed the closest similarity to the human protein, followed by Lj-p63 and then Lj-p53.
5′ RACE analysis identified three isoforms of the Lj-Tp63 gene, whereas no isoforms were identified for the Lj-Tp73 gene (Supplemental Fig. S7). All three isoforms of Lj-p63 have the same start site, which is 39 amino acids after the initiation of the longest Hs-p63 isoform, Hs-TAp63α (Supplemental Fig. S8A–C). The lamprey genome therefore does not appear to have an isoform equivalent to the well-described and important Hs-ΔNp63α, which is missing the first 94 amino acids that constitute a functional TAD. However, both zebrafish p63 and frog p63 (Supplemental Fig. S5A) have a translation start site similar to that of the Hs-ΔNp63 isoforms. Lj-p63_A is 774 amino acids and Lj-p63_B is 761 amino acids, but both terminate eight amino acids following the final amino acid of Hs-TAp63α based on the alignment of the full proteins. Lj-p63_C is 650 amino acids long and terminates at amino acid 613 of Hs-TAp63α but is longer than Hs-TAp63β or Hs-TAp63γ, which are missing 125 and 193 amino acids, respectively, at the C′ terminus of Hs-TAp63α. Therefore, in comparison with one another, the Lj-p63 isoforms lack amino acids in the central regions of the protein rather than at the N′-terminal or C′-terminal ends as found in human and other vertebrates.
Lj-Mdm2 and Lj-Mdm4 genes
Searches of the Japanese lamprey genome assembly identified several exons of an Mdm2 gene on scaffold_29. The coding sequence of this gene was completed by RT–PCR and 5′ RACE. The Lj-Mdm2 gene encodes a protein with 603 amino acids and includes all of the key functional domains such as the p53-binding domain, acidic region, zinc finger, and C′-terminal RING domain. Overall, it is 32% identical to human HDM2, with the RING finger domain exhibiting a higher identity of 56% (Table 1; Supplemental Fig. S9A,B). Consistent with other C′-terminal RING domain-containing proteins (Dolezelova et al. 2012), there are exactly 13 amino acids following the final cysteine residue, and, as with HDM2 (Fang et al. 2000), there are seven critical cysteine residues that are conserved for ubiquitin ligation activity and p53 degradation (Supplemental Fig. S10).
The Japanese lamprey genome also contains an Mdm4 gene (spread over two short scaffolds, 2349 and 14666) that encodes a 280-amino-acid protein. The full-length coding sequence of the Lj-Mdm4 was confirmed by RT–PCR. Alignment of this sequence with Mdm4 from a number of jawed vertebrate species shows that ∼200 amino acids that are present in the middle of jawed vertebrate proteins are missing in the Lj-Mdm4 in two regions (Supplemental Fig. S11A,B). In the human protein, an acidic domain is found from amino acid 215 to amino acid 255, and a zinc finger is located from amino acid 290 to amino acid 332, but these regions are either partially or completely absent in Lj-Mdm4 in addition to ∼50 amino acids located before the acidic domain. A caspase-3 cleavage site and a portion downstream from the zinc finger are also missing in Lj-Mdm4. Overall, the amino acid sequence similarity between Lj-Mdm4 and human HDM4 is 31%, with the highest identity in the RING domain at 60% (Table 1). As with Lj-Mdm2, there are exactly 13 amino acids following the final cysteine residue in the RING domain. However, in contrast to jawed vertebrate Mdm4 proteins, which have six cysteine residues, Lj-Mdm4 contains seven—the same number found in Mdm2 (Momand et al. 2011). This additional cysteine residue corresponds to C449 of human HDM2 but is absent in HDM4 and has been found to be important for p53 degradation and ubiquitination (Fang et al. 2000).
The synteny of genes flanking Lj-Mdm2 and Lj-Mdm4 and their orthologs in human and coelacanth are compared in Supplemental Figure S12. As with lamprey Tp53/Tp63/Tp73 loci, none of the genes flanking Lj-Mdm2 and Lj-Mdm4 is conserved in human and coelacanth loci. Interestingly, of the invertebrate genomes sequenced thus far and of those containing an Mdm gene, only one gene has been identified per organism, and this typically encodes a protein with six cysteine residues in its RING domain (Momand et al. 2011). How many cysteine residues were present in the Mdm gene of the common vertebrate ancestor, whether this gene is closer to jawed vertebrates Mdm2 or Mdm4 genes, and whether the Mdm proteins of invertebrates are functional in terms of p53 degradation are all questions worth exploring.
Conservation of the p53–Mdm2 interaction in lamprey
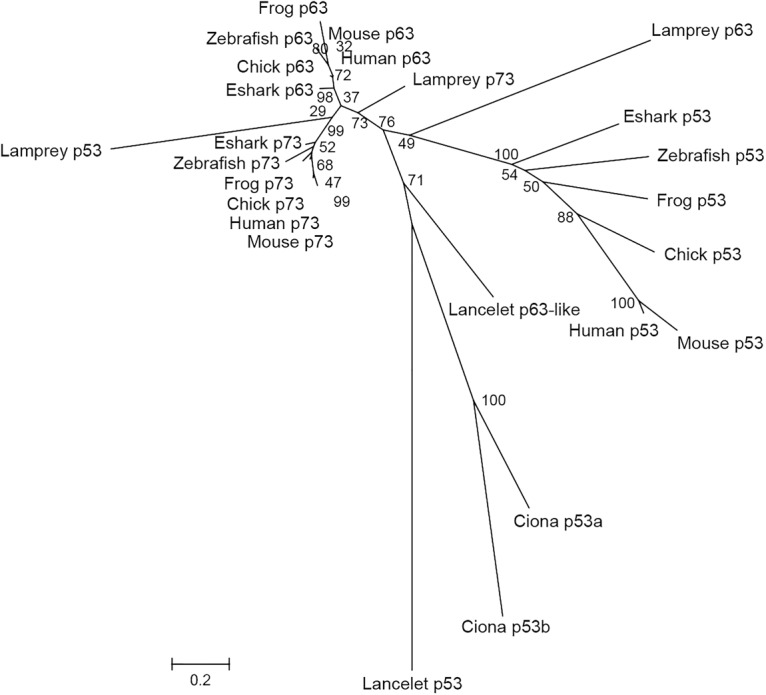
In jawed vertebrates, p53 protein levels are highly regulated, and the main negative regulator is Mdm2. In order to determine whether the p53 pathway function is conserved in lamprey, we first investigated the ability of Lj-Mdm2 to bind to Lj-p53 and Hs-p53. Detailed sequence analysis of the p53-binding domain within Lj-Mdm2 (Fig. 3A) and the N′-terminal Mdm2-interacting region of p53 (Fig. 3B) showed that Lj-p53 contains the key F19 and W23 motifs (human protein numbering), which figure prominently in the crystal structure of the complex between a peptide derived from the TAD of p53 and the N′-terminal region of Mdm2 (Kussie et al. 1996). Residue 26 is leucine in Hs-p53 and is one of three key residues required for binding to HDM2 (Hs-p53 F19, W23, and L26) (Bottger et al. 1997). In Lj-p53, this leucine is replaced by glycine (Figs. 3B, 4B), which incurs a complete loss of side chain interactions within the p53-binding cleft of HDM2. In vitro translation of Lj-p53, Lj-Mdm2, Hs-p53, and HDM2 in an Escherichia coli-based system showed that Lj-p53 is bound by both Lj-Mdm2 and HDM2. Furthermore, Lj-Mdm2 displayed binding to both Lj-p53 and Hs-p53 (Fig. 3C). This was clearly visible despite the lower level of expression of the lamprey proteins in this in vitro system. Molecular models show that, structurally, both p53 peptides can easily be accommodated by the binding site in the N′-terminal domain of HDM2 (Fig. 4A) and Lj-Mdm2 (Fig. 4B). However, fluorescent polarization (FP) analysis showed that an Lj-p53 peptide was unable to displace an Hs-p53-based peptide (carboxyfluorescein [FAM]-labeled p53-binding peptide 12.1 [FAM-12.1]) from the first 125 amino acids of HDM2 (HDM2Nterm) (Fig. 4C; Supplemental Tables S2, S3). The ability of the two proteins to interact may therefore depend more on other sites of interaction between Lj-Mdm2 and Lj-p53. In Hs-p53, the leucine at position 26 is also critical in the interaction with HDM2 (Kussie et al. 1996), so the corresponding glycine residue in Lj-p53 was mutated to generate the peptide Lj-p5312–25(G22L). By FP analysis, it was demonstrated that this peptide was able to displace FAM-12.1 from HDM2Nterm but with a lower affinity compared with wild-type Hs-p5316–29 (Supplemental Table S2).
Figure 3.
Partial conservation of the binding pocket between Mdm2 and p53 among vertebrate species. (A) Protein sequence alignment of the p53-binding region of Mdm2 from lampreys (this study), elephant sharks (Esharks, G9J1M1), zebrafish (Q561Z0), frogs (P56273), chickens (F1NGX6), mice (Q569X0), and humans (Q00987) (corresponding to human protein residues 1–110). (B) Protein sequence alignment of the Mdm2-interacting region of p53 sequences from lampreys (this report), elephant sharks (G9J1L8), zebrafish (P79734), frogs (P07193), chickens (P10360), mice (Q549C9), and humans (P04637) (corresponding to human protein residues 16–30). (C, top panel) Western blot showing in vitro translation and immunoprecipitation (IP) of Hs-p53 and Lj-p53 by either HDM2 or Lj-Mdm2, which was used as bait. Input levels can be seen in the bottom panel.
Figure 4.
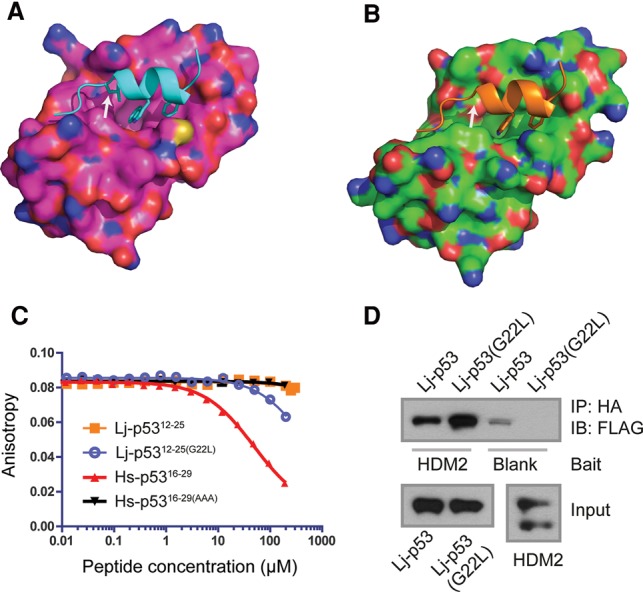
Binding of Lj-p53 to HDM2 is enhanced by Lj-p53(G22L) mutation. (A) A surface presentation (pink) of the human crystal structure of HDM2Nterm (residues 25–109) in complex with a fragment of Hs-p53 (cyan) showing the interaction of the three critical residues F19, W23, and L26. (B) A surface presentation (green) of a homology model of the p53-binding region of Lj-Mdm2Nterm showing the putative interaction with Lj-p53 (orange). Note that the equivalent of L26 in Hs-p53 is a glycine, G22, in Lj-p53 (arrow in both figures). (C) Competition titrations of Hs-p53 and Lj-p53 peptides against FAM-12.1 for binding to HDM2Nterm. KD values are in Supplemental Table S2. (D) Western blot of Lj-p53 and Lj-p53(G22L) immunoprecipitated (IP) by HDM2Nterm, which was used as bait. All proteins were expressed by in vitro translation. Input levels can be seen in the bottom panels.
In order to understand the mechanistic basis for this differential binding of the p53 peptides to HDM2Nterm, we carried out modeling studies using extensive molecular dynamics simulations. We investigated the propensities of the peptides to partition themselves from an aqueous environment to the hydrophobic environment on the surface of HDM2Nterm. Simulations of the complex state (peptide bound to HDM2Nterm) suggest that, unsurprisingly, residue number 26 (Leu in Hs-p53; Gly in Lj-p53) affords the largest discrimination between the human and lamprey peptides’ interactions with HDM2Nterm (Supplemental Table S1; Supplemental Fig. S13). However, the overall affinity as predicted by the modeling suggests that the Lj-p5312–25(G22L) peptide has a higher affinity for HDM2Nterm than does Hs-p5316–29 (Supplemental Table S1). To understand this discordance with respect to the above experimental observations, we examined the behavior of the peptides in the aqueous environment (Supplemental Fig. S14), as the p53 peptide is known to bind as an α helix to HDM2Nterm (Kussie et al. 1996). It is clear that the Hs-p5316–29 peptide (Supplemental Fig. S14, column A) can exist in dominant helical conformations (>90% of the conformations sampled), particularly within the region F19–L26 (the region essential for binding to HDM2), and is in good agreement with nuclear magnetic resonance (NMR) studies (Lee et al. 2000) and other simulation studies (Lee et al. 2007; Dastidar et al. 2008; Mavinahalli et al. 2010). In contrast, Lj-p5312–25 shows poor helicity (Supplemental Fig. S14, column B). In Lj-p5312–25, the C′-terminal region consists of three glycine residues that are known to be helix breakers (Li and Deber 1992; Forood et al. 1993). The G22L mutation in the Lj-p53 peptide (Supplemental Fig. S14, column C) leads to an increase in helicity relative to Lj-p5312–25, although the population of helical states is still lower than that observed by Hs-p5316–29. This suggests that it is easier for the Hs-p53 peptide to partition from the aqueous environment to the hydrophobic surface of HDM2Nterm in an α-helical mode compared with both the Lj-p5312–25 and Lj-p5312–25(G22L) peptides, thus explaining the binding affinity trend observed in the experiments.
In addition, the residues T18 and S20 in Hs-p53 are replaced by D14 and D16, respectively in lampreys. In humans, kinase-modulated phosphorylation of T18 or S20 (or replacement by phosphomimetic Asp/Glu) leads to an attenuation of the p53–HDM2 interaction and a concomitant stabilization of p53 (Meek 1999; Schon et al. 2002). The underlying mechanism engages an acidic patch on the surface of HDM2 (Lee et al. 2007). The residues corresponding to T18 and S20 of Lj-p53 are both aspartic acids and would further contribute to the loss of affinity for HDM2Nterm. Thus, it is clear that the differences in the conformational landscapes of the Hs-p53 and Lj-p53 peptides and the physicochemical character of the HDM2 surface together modulate the differential binding of these peptides to HDM2Nterm.
To further explore the affinity of Lj-p53 with HDM2, we generated a full-length Lj-p53(G22L) expression construct to test its ability to bind to full-length HDM2 in an in vitro system. Figure 4D shows that, as predicted, the mutation Lj-p53(G22L) enhanced the ability of Lj-p53 to bind to HDM2. Conversely, this mutation displayed reduced ability to bind Lj-Mdm2 (Supplemental Fig. S15A). These studies reflect on the interesting topic of the coevolution of proteins that must interact with each other for regulated function.
Conservation of the DNA binding of Lj-p53
The DNA-binding domain of Lj-p53 shows 60% homology with Hs-p53 (Table 1), and Figure 5A shows the alignment of the p53 DNA-binding domain from seven vertebrate species. The sequence-specific DNA-binding function of Lj-p53 could be demonstrated in an in vitro DNA-binding assay using in vitro synthesized proteins. Lj-p53 was able to bind to an optimized p53 consensus DNA-binding element, “ConA” (Fig. 5B; Goh et al. 2010), in this immunoprecipitation- and PCR-based assay but was only able to bind weakly to the Hs-p21 and Hs-PUMA p53-binding sites, reflecting a DNA-binding specificity subtly different from that of the human protein. This is consistent with Lj-p53 retaining certain critical contact residues, including K120, S241, R248, A276, C277, and R280 (Hs-p53 protein numbering), deduced from the X-ray structure analysis of the Hs-p53 and DNA interaction sites (Fig. 5A; Cho et al. 1994). Lj-p53 was not able to induce a transcription response in a cell-based assay using a reporter gene system comprising the ConA response element (data not shown). The lack of transcriptional activity could be due to the inability of Lj-p53 to recruit appropriate cotranscription factors in a human cell line, especially given the low sequence homology between the N′-terminal domains (Table 1; Supplemental Fig. S1). In addition, a comparison of the sequences of the tetramerization domains of Hsp53 (EYFTLQIRGRERFEMFRELNEALELKDAQAG) and Ljp53 (ELFLIPVRGRENYELLLHLKESLEMKQLVPQ) (39% of residues are identical and shown in bold, and 58% of residues are similar in character and shown underlined) suggests that the packing of the tetramers in humans is mediated by larger side chains, thus suggesting that the human tetramer is likely more stable and hence may contribute to more effective transcription.
Figure 5.

The DNA-binding region of Lj-p53 is conserved. (A) Protein sequence alignment of the DNA-binding domain of p53 from lampreys, elephant sharks (Eshark), zebrafish, frogs, chickens, mice, and humans. Accession numbers are in the legend for Figure 3B. Amino acids important for DNA binding and/or human cancer hot spot mutations (shown in red) are indicated by an asterisk. (B) Nucleotides representing the Hs-p53-binding consensus sequence ConA, which was used for DNA-binding and activation studies. Binding of in vitro translated Hs-p53Δ and Lj-p53Δ to the p21-2, PUMA-2, and ConA response elements. DNA binding corresponds to fold difference over nonspecific DNA binding between the respective p53 and equal molar concentrations of “no response element” control DNA. Error bars represent mean ± SEM of three independent binding experiments.
Conservation of E3 ligase activity in Lj-Mdm2
An analysis of the functional properties of the Lj-Mdm2 and Lj-Mdm4 proteins exploited key features of the known properties of the human homolog. HDM2 acts to promote the degradation of p53 through its action as an E3 ligase. Mutational studies have determined that the addition of amino acids to the C′ terminus but not the N′ terminus of HDM2 inhibits its enzymatic function, as do point mutations in the RING finger domain (Dolezelova et al. 2012). Figure 6A shows an alignment of the RING finger domain and the C′-terminal sequence of a selection of vertebrate Mdm2 proteins. All proteins show a high degree of conservation, with all seven cysteine residues completely conserved. Mutation of cysteine residue 464 to an alanine residue inhibits the human protein (Fang et al. 2000), so the equivalent point mutation was made in the lamprey protein. The lamprey and all other sequenced Mdm2s have exactly 13 amino acids C′-terminal to the last cysteine residue in the RING finger. Addition of amino acids at this C′ terminus also inactivates the HDM2 protein (Dolezelova et al. 2012), so an HA tag was added to Lj-Mdm2 at this position as well as at the N′ terminus of the protein in an alternative construct. The N′-terminally or C′-terminally tagged Lj-Mdm2 proteins were then tested for function by transfecting them along with Lj-p53 into double-knockout cells, which were fibroblasts derived from mice in which the endogenous Trp53 and Mdm2 genes had been deleted (de Rozieres et al. 2000). The results are remarkably clear (Fig. 6B). The N′-terminally HA-tagged Lj-Mdm2 protein was able to reduce Lj-p53 levels (Fig. 6B, lane 3), and this was partially blocked (Fig. 6B, lane 4) by the proteasome inhibitor MG132. In contrast, the C′-terminally tagged Lj-Mdm2 protein was unable to reduce the levels of Lj-p53 Fig. 6B, (Fig. 6B, lane 5). These results demonstrate that Lj-p53 is a target for E3 ligases and that the Lj-Mdm2 protein was able to function effectively within a mammalian cell, indicating the functional conservation of the ubiquitin–proteasome pathway within vertebrates. This implies specifically that the E2 ubiquitin complex protein of mouse cells was able to interact with the Lj-Mdm2 protein. Further evidence of this conservation comes from the study of the C464A mutation (human protein numbering) in Lj-Mdm2 (Fig. 6B, lane 7). This point mutation acts just as has been reported for the human and mouse proteins (Fang et al. 2000). It abolishes the E3 ligase activity of Lj-Mdm2 (Fig. 6B, cf. lanes 7 and 3).
Figure 6.
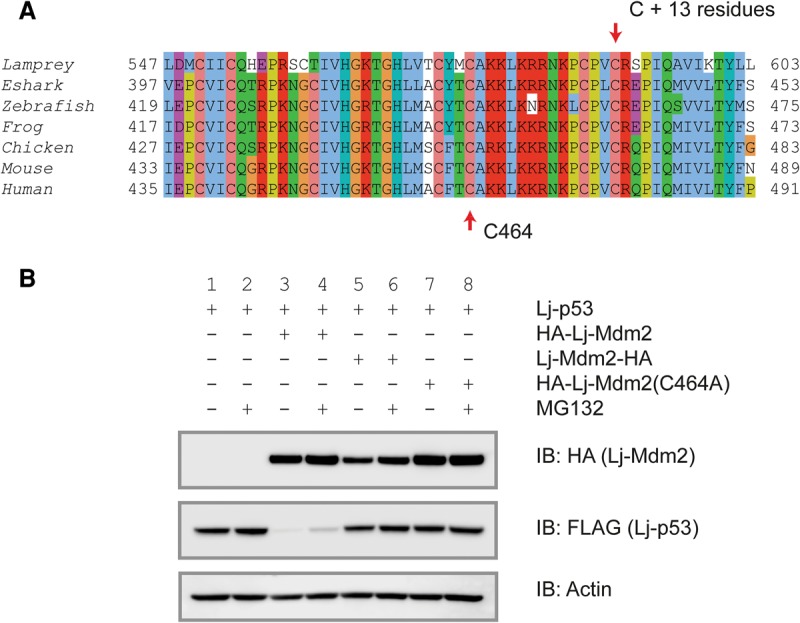
p53 degradation by Mdm2 is conserved in lampreys. (A) Protein sequence alignment of the RING domain of Mdm2 from lampreys, elephant sharks (Eshark), zebrafish, frogs, chickens, mice, and humans. Accession numbers are in the legend for Figure 3A. The top arrow indicates that all Mdm2 RING domains terminate exactly 13 amino acids residues following the last cysteine. The bottom arrow marks the cysteine residue mutated to generate a nonfunctional Lj-Mdm2 that is unable to degrade Lj-p53. (B) Western blot of Lj-p53 levels following cotransfection with various Lj-Mdm2-expressing constructs. (Lane 1) Lj-p53. (Lane 2) Lj-p53 with MG132. (Lane 3) Lj-p53 + HA-Lj-Mdm2. (Lane 4) Lj-p53 + HA-Lj-Mdm2 with MG132. (Lane 5) Lj-p53 + Lj-Mdm2-HA. (Lane 6) Lj-p53 + Lj-Mdm2-HA with MG132. (Lane 7) Lj-p53 + HA-Lj-Mdm2(C464A). (Lane 8) Lj-p53 + HA-Lj-Mdm2(C464A) with MG132. Lj-Mdm2 levels are shown in the top panel, and the loading control is shown in the bottom panel.
Western blot analysis of the function of Lj-Mdm4 (Supplemental Fig. S15B) showed that expression levels of both Lj-p53 and Lj-Mdm2 were decreased in the presence of very low levels of Lj-Mdm4 (we were unable to detect expression of the myc-tagged Lj-Mdm4 construct in the cell line used). The ability of Lj-Mdm4 to degrade Lj-p53 (in the absence of Lj-Mdm2) (Supplemental Fig. S15B, cf. lanes 9 and 1) can be noted. This function is possibly due to the extra cysteine in the RING domain of Lj-Mdm4 (see above), but further mutational studies are needed to confirm this.
The disruption of the p53 Mdm2/Mdm4 interaction can activate the p53 response in the absence of DNA damage and, as such, is being actively pursued as a drug target. Currently, two classes of molecules are in clinical trial (Khoo et al. 2014). The first class, represented by the small molecule “Nutlin,” consists of conventional low-molecular-weight drugs, while the second class consists of p53-binding peptides that have been stabilized by cross-linking agents, represented here by the stapled peptide “Staplin.” It was of great interest to test these molecules for their binding ability in the lamprey system. Again, using in vitro translated proteins, it could be shown that, as expected, both Nutlin and Staplin could block the binding of Hs-p53 to HDM2Nterm (Fig. 7A, lanes 1–3). Interestingly, when this experiment was repeated using Lj-Mdm2Nterm, binding to Hs-p53 was blocked only by Staplin but not by Nutlin (Fig. 7A, lanes 4–6). In this regard, Lj-Mdm2Nterm mimics the recently described M62A mutant of human HDM2 (Chee et al. 2014), but the structural basis is in fact distinct. The pocket of HDM2Nterm/Lj-Mdm2Nterm that nestles the bromobenzoyl moiety of Nutlin (orange patch in Supplemental Fig. S16) is made up of residues Leu54/Ile37, His96/Pro79, Ile99/Ser82, and Tyr100/Val86. It is very clear that the pocket in Lj-Mdm2Nterm is much more open and would lead to ineffective packing of the bromobenzoyl moiety; molecular dynamics simulations (data not shown) show that Nutlin remains stably bound only in HDM2Nterm.
Figure 7.
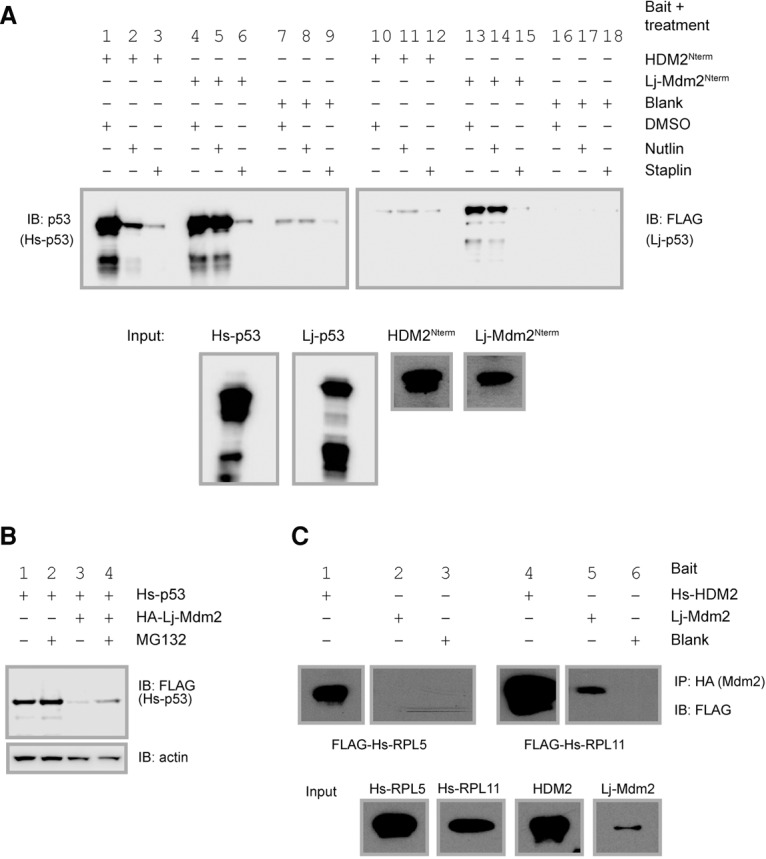
Lj-Mdm2 function and binding partners are conserved across species. (A) In vitro pull-down assay of Hs-p53 or Lj-p53 by HDM2Nterm or Lj-Mdm2Nterm in the presence of Nutlin or Staplin. (D) DMSO; (N) Nutlin; (S) Staplin. The top panel shows immunoprecipitated p53, and the bottom panel shows input levels. Hs-p53 was detected using DO1 antibody, and Lj-p53 was detected using anti-Flag antibody. (B) Western blot of Hs-p53 levels following cotransfection with an Lj-Mdm2-expressing construct. (Lane 1) Flag-Hs-p53. (Lane 2) Flag-Hs-p53 with MG132. (Lane 3) Flag-Hs-p53 + HA-Lj-Mdm2. (Lane 4) Flag-Hs-p53 + HA-Lj-Mdm2 with MG132. The loading control is shown in the bottom panel. (C, top panel) Western blot of in vitro translation and immunoprecipitation (IP) of Hs-RPL5 or Hs-RPL11 by either HDM2 or Lj-Mdm2, which was used as bait. (Lane 1) Hs-RPL5 with HDM2. (Lane 2) Hs-RPL5 with Lj-Mdm2. (Lane 3) Hs-RPL5 with blank. (Lane 4) Hs-RPL11 with HDM2. (Lane 5) Hs-RPL11 with Lj-Mdm2. (Lane 6) Hs-RPL11 with blank. Input levels are shown in the bottom panel.
In this in vitro system that uses only the N′-terminal domain fragments of Lj-Mdm2 and HDM2, we were not able to detect significant binding between HDM2Nterm and Lj-p53 (Fig. 7A, lanes 10–12), but, as shown earlier, we demonstrated such an interaction when the full-length proteins were examined in vitro (Fig. 3C). In contrast and as expected, we showed a strong interaction between Lj-Mdm2Nterm and Lj-p53. This was in turn blocked by Staplin but not by Nutlin, confirming that the p53-binding pocket in Lj-Mdm2Nterm interacts poorly, if at all, with Nutlin while binding much more strongly to Staplin, as has been previously demonstrated with HDM2Nterm (Wei et al. 2013a,b); this was further confirmed by molecular dynamics simulations (data not shown).
Given the strong interaction seen between full-length Lj-Mdm2 or Lj-Mdm2Nterm and Hs-p53, the ability of Lj-Mdm2 to degrade Hs-p53 was examined in double-knockout cells. Consistent with the binding data, Lj-Mdm2 acted as a very efficient enzyme in promoting the degradation of Hs-p53, and this degradation process was only partially blocked by MG132. The lamprey protein may therefore prove very useful in further analyses of the regulation of Mdm2 proteins. One aspect of the regulation of the pathway in mammalian cells is the induction of the p53 response associated with nucleolar disruption (Rubbi and Milner 2003) and the release of free ribosomal proteins. This interaction appears to be highly conserved as well, as the Hs-RPL11 protein, when examined in vitro and in tissue culture-based models, was found to bind Lj-Mdm2; however, no interaction was detected with Hs-RPL5 (Fig. 7C).
Discussion
The evolutionary conservation of the p53 pathway is remarkable and poses an intellectual challenge, as organisms in which the gene is deleted or lost are completely viable and fertile (Lozano 2010). One attractive explanation of this apparent dilemma is the concept of robustness, where p53, as a highly inducible transcription factor, acts to ensure the genetic stability required for the long-term competitive success of a species (Lane 1992). Cyclostomes, comprising jawless vertebrates such as lampreys and hagfish, are the sister group of the living jawed vertebrates (gnathostomes) and thus are an important group for studying vertebrate evolution. Here we show that homologs of all members of the p53 family as well as the negative regulators Mdm2 and Mdm4 are present in the Japanese lamprey. Functional studies showed that sequence conservation was reflected in both biochemical activity and the ability of Lj-Mdm2 protein to function as a very effective E3 ligase in mouse cells. The coevolution of proteins that interact with and regulate each other is of great interest, and here the p53–Mdm2 interaction is a very clear example. Detailed biochemical and molecular dynamics simulations act to reinforce our understanding of this key molecular interaction, and the study of evolutionary variants of interacting partners that are targets of clinically relevant drugs is of marked interest. Thus, our finding that Staplin but not Nutlin can block the interaction of Lj-p53 or Hs-p53 with Lj-Mdm2Nterm is helpful in the design of more potent and specific inhibitors of HDM2 and may provide a useful genetic tool for the study of p53 regulation. Further elucidation of Lj-Mdm2 function and its response to these and other types of inhibitors in cellular systems may help in the dissection of critical post-translational modifications that regulate the p53 pathway, offering insights into the basis of tumor suppression by the human TP53 gene, given its high mutation rate in cancer.
Materials and methods
Identification of Tp53 family members and Mdm2 and Mdm4 genes in the lamprey genome
The Japanese lamprey genome assembly at the Japanese Lamprey Genome Project (http://jlampreygenome.imcb.a-star.edu.sg) was searched for members of the Tp53 family and Mdm2 and Mdm4 genes by TBlastN using human and elephant shark p53, p63, p73, Mdm2, and Mdm4 protein sequences as queries. The genomic regions showing similarity to human or elephant shark protein sequences were searched against the NCBI nonredundant protein database using Blastx to confirm the identity of the gene. Any exons missing in the genome assembly were identified by sequencing RT–PCR or 5′/3′ RACE products. 5′ RACE-ready single-strand cDNA was used as a template for RT–PCR. Total RNA was extracted from the gills, kidney, muscle, and intestine using Trizol reagent (GIBCO-BRL) according to manufacturer's protocol. Single-strand cDNA was prepared from total RNA using the SMART RACE cDNA amplification kit (Clontech). Primers were designed for representative exons identified in the Japanese lamprey genome assembly and used for RT–PCR and 5′/3′ RACE (primer sequences available on request). RT–PCR and RACE products were sequenced directly or cloned into the pGEM-T Easy Vector (Promega) and sequenced completely using the BigDye Terminator Cycle sequencing kit (Applied Biosystems) on an ABI 3730xl capillary sequencer (Applied Biosystems). Sequences of the Japanese lamprey genes characterized in this study have been deposited in GenBank (Lj-p53, KT960978; Lj-p63, KT960979; Lj-p73, KT960980; Lj-Mdm2, KT960981; and Lj-Mdm4, KT960982).
Phylogenetic analysis of Lj-p53, Lj-p63, and Lj-p73 sequences
The full-length protein sequences were aligned using ClustalW (default parameters) in MEGA6. Gaps in the alignment were removed completely, leaving a total of 252 positions in the final alignment. A maximum likelihood (JTT+G substitution model) bootstrap consensus tree was inferred from 100 replicates. The consensus tree is shown in Figure 2. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test is shown next to the branches. The accession numbers of protein sequences that were used in phylogenetic analysis are Chick_p53 P53_CHICK, Chick_p63 F1N8Z7_CHICK, Chick_p73 XP_417545.3, Ciona_p53a NP_001122370.1, Ciona_p53b NP_001071796.1, Eshark_p53 AEW46988.1, Eshark_p63 AEW46989.1, Eshark_p73 AEW46990.1, Frog_p53 F7A9U0_XENTR, Frog_p63 F6ZGN7_XENTR, Frog_p73 F6TKT0_XENTR, Human_p53 P53_HUMAN, Human_p63 P63_HUMAN, Human_p73 P73_HUMAN, Lancelet_p53 XP_002598770.1, Lancelet_p63like XP_002613954.1, Mouse_p53 P53_MOUSE, Mouse_p63 P63_MOUSE, Mouse_p73 P73_MOUSE, Zebrafish_p53 P53_DANRE, Zebrafish_p63 A7YYJ7_DANRE, and Zebrafish_p73 B0S576_DANRE.
Plasmids
For details about plasmids, see Supplemental Table S4.
Cell culture, immunoprecipitation, and Western blot analysis
Mouse embryonic fibroblast p53/Mdm2 double-knockout cells (a kind gift from Guillermina Lozano) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% (v/v) foetal calf serum (FCS) and 1% (v/v) penicillin/streptomycin. To ensure a high transfection efficiency to reliably measure p53 protein levels, double-knockout cells were cotransfected with various combinations of Hs-p53, Lj-p53, Lj-Mdm2, Lj-Mdm4, and vector plasmids using Nucleofector II (Lonza) according to the manufacturer's instructions for mouse embryonic fibroblasts with MEF 2 Nucleofector solution and program A-023. In all cases, the total amount of plasmid DNA transfected per well was equalized by addition of parental vector. Four hours prior to harvesting, cells were treated with proteasome inhibitor MG132 (Selleckchem). Double-knockout cells were harvested 24 h after transfection and lysed with NP40 buffer (50 mM Tris-HCl at pH 7.4–8.0, 150 mM NaCl, 1% NP-40) supplemented with protease inhibitors. Following protein concentration determination by BCA protein assay (Thermo Scientific Pierce), equal protein amounts of sample were added to SDS-PAGE loading buffer and incubated for 5 min at 95°C. The proteins were then separated by SDS-PAGE and transferred to nitrocellulose. Immunoblotting was carried out with the relevant antibodies.
Immunoprecipitation and Western blot analysis
Protein G beads (Invitrogen) were incubated with 1 µg of anti-HA per 10 µL of beads for 1 h in PBST–3% BSA and subsequently washed twice in PBST–0.1% BSA. In vitro translation-expressed HDM2 or Lj-Mdm2 was incubated with the beads on a rotator for 30 min. Nutlin or stapled peptides were added at required concentrations and incubated for 30 min. In vitro translation-expressed Hs-p53, Lj-p53, Hs-RPL5, or Hs-RPL11 was added to the mixture and incubated for 1 h. Beads were finally washed three times in PBST-0.1%BSA and three times with PBS, and bound proteins were eluted by resuspension in 20 µL of LDS-PAGE loading buffer and incubated for 5 min at 95°C. Both the eluates and inputs were subjected to electrophoresis, transferred to nitrocellulose membranes, and probed for Hs-p53 with horseradish peroxidase-conjugated DO1 antibody (Santa Cruz Biotechnology); for Lj-p53, Hs-RPL5, or Hs-RPL11 with horseradish peroxidase-conjugated Flag-M2 antibody (Sigma); and/or for HDM2 or Lj-Mdm2 with anti-HA antibody (Santa Cruz Biotechnology) followed by rabbit anti-mouse immunoglobulin (Dako Cytomation).
DNA-binding assay
Protein G beads were incubated with 1 µg of anti-HA antibody per 5 µL of beads for 1 h in PBST–3% BSA and subsequently washed three times in PBST–0.1% BSA. In vitro translated Hs-p53 or Lj-p53 was incubated with the beads on a rotator for 30 min. PUMA, p21, 2ConA DNA template, or negative control DNA (20 ng) was added to the mixture and incubated for 1 hr. The negative control DNA was added as a measurement of nonspecific binding by p53. The beads were finally washed three times in PBST–0.1% BSA and three times with PBS, and bound DNA was eluted by resuspension in 20 µL of nuclease-free water and incubated for 5 min at 95°C. Real-time PCR quantification of the eluates was performed using primers petF3 and WpetR1 at 250 nM each with SsoAdvanced Universal SYBR Green super mix (Bio-Rad Laboratories) and quantified via a CFX96 real-time system (Bio-Rad Laboratories). Data were interpreted as fold differences (calculated based on cycle threshold differences) over nonspecific DNA-binding control (neutral DNA).
Fluorescence anisotropy
Fluorescence anisotropy titrations of purified wild-type HDM2Nterm (both 1–125 and 6–125) with 50 nM FAM-12.1 (FAM-RFMDYWEGL-NH2) were performed as previously described (Brown et al. 2013; Chee et al. 2014) to first determine the dissociation constants for the peptide–protein interaction. Subsequent determination of apparent KDs of Nutlin, human, and lamprey peptides was performed by competitive fluorescence anisotropy in which titrations of the peptides were carried out with a constant concentration of HDM2 and FAM-labeled peptide (Brown et al. 2013; Chee et al. 2014). Readings were carried out using the Envision multilabel reader (PerkinElmer). All experiments were carried out in PBS (2.7 mM KCl, 137 mM NaCl, 10 mM Na2HPO4, 2 mM KH2PO4 at pH 7.4), 3% DMSO, and 0.1% Tween-20 buffer. All titrations were carried out in replicates (n = 2–6). Curve fitting was carried out using Prism 4.0 (GraphPad).
Supplementary Material
Acknowledgments
This work was supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology, and Research), Singapore.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.274118.115.
Freely available online through the Genes & Development Open Access option.
References
- Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, Kiso WK, Schmitt DL, Waddell PJ, Bhaskara S, et al. 2015. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA 314: 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottger A, Bottger V, Garcia-Echeverria C, Chene P, Hochkeppel HK, Sampson W, Ang K, Howard SF, Picksley SM, Lane DP. 1997. Molecular characterization of the hdm2–p53 interaction. J Mol Biol 269: 744–756. [DOI] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. 2011. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145: 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, Golic KG, Rio DC, Rubin GM. 2004. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol 24: 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, Khoo KH, Choong ML, Lee MA, Yurlova L, Zolghadr K, et al. 2013. Stapled peptides with improved potency and specificity that activate p53. ACS Chem Biol 8: 506–512. [DOI] [PubMed] [Google Scholar]
- Chee SM, Wongsantichon J, Soo Tng Q, Robinson R, Joseph TL, Verma C, Lane DP, Brown CJ, Ghadessy FJ. 2014. Structure of a stapled peptide antagonist bound to nutlin-resistant Mdm2. PLoS One 9: e104914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. 1994. Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science 265: 346–355. [DOI] [PubMed] [Google Scholar]
- Dastidar SG, Lane DP, Verma CS. 2008. Multiple peptide conformations give rise to similar binding affinities: molecular simulations of p53–MDM2. J Am Chem Soc 130: 13514–13515. [DOI] [PubMed] [Google Scholar]
- de Rozieres S, Maya R, Oren M, Lozano G. 2000. The loss of mdm2 induces p53-mediated apoptosis. Oncogene 19: 1691–1697. [DOI] [PubMed] [Google Scholar]
- Dolezelova P, Cetkovska K, Vousden KH, Uldrijan S. 2012. Mutational analysis of Mdm2 C-terminal tail suggests an evolutionarily conserved role of its length in Mdm2 activity toward p53 and indicates structural differences between Mdm2 homodimers and Mdm2/MdmX heterodimers. Cell Cycle 11: 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. 2000. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275: 8945–8951. [DOI] [PubMed] [Google Scholar]
- Forood B, Feliciano EJ, Nambiar KP. 1993. Stabilization of α-helical structures in short peptides via end capping. Proc Natl Acad Sci 90: 838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh W, Lane D, Ghadessy F. 2010. Development of a novel multiplex in vitro binding assay to profile p53–DNA interactions. Cell Cycle 9: 3030–3038. [DOI] [PubMed] [Google Scholar]
- Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, Mattei M, Candi E, De Felici M, Melino G, et al. 2009. Inhibition of the c-Abl–TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 15: 1179–1185. [DOI] [PubMed] [Google Scholar]
- Guo L, Chua J, Vijayakumar D, Lee KC, Lim K, Eng H, Ghadessy F, Coomber D, Lane DP. 2010. Detection of the 113p53 protein isoform: a p53-induced protein that feeds back on the p53 pathway to modulate the p53 response in zebrafish. Cell Cycle 9: 1998–2007. [DOI] [PubMed] [Google Scholar]
- Hock AK, Vousden KH. 2014. The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta 1843: 137–149. [DOI] [PubMed] [Google Scholar]
- Kasahara M, Sutoh Y. 2014. Two forms of adaptive immunity in vertebrates: similarities and differences. Adv Immunol 122: 59–90. [DOI] [PubMed] [Google Scholar]
- Khoo KH, Verma CS, Lane DP. 2014. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 13: 217–236. [DOI] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. 1996. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274: 948–953. [DOI] [PubMed] [Google Scholar]
- Lane DP. 1992. Cancer. p53, guardian of the genome. Nature 358: 15–16. [DOI] [PubMed] [Google Scholar]
- Lane DP, Cheok CF, Brown CJ, Madhumalar A, Ghadessy FJ, Verma C. 2010. The Mdm2 and p53 genes are conserved in the Arachnids. Cell Cycle 9: 748–754. [DOI] [PubMed] [Google Scholar]
- Lane DP, Madhumalar A, Lee AP, Tay BH, Verma C, Brenner S, Venkatesh B. 2011. Conservation of all three p53 family members and Mdm2 and Mdm4 in the cartilaginous fish. Cell Cycle 10: 4272–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Mok KH, Muhandiram R, Park K-H, Suk J-E, Kim D-H, Chang J, Sung YC, Choi KY, Han K-H. 2000. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem 275: 29426–29432. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Srinivasan D, Coomber D, Lane DP, Verma CS. 2007. Modulation of the p53–MDM2 interaction by phosphorylation of Thr18: a computational study. Cell Cycle 6: 2604–2611. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. 2011. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 12: 259–265. [DOI] [PubMed] [Google Scholar]
- Li SC, Deber CM. 1992. Glycine and β-branched residues support and modulate peptide helicity in membrane environments. FEBS Lett 311: 217–220. [DOI] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149: 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano G. 2010. Mouse models of p53 functions. Cold Spring Harb Perspect Biol 2: a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WJ, Amatruda JF, Abrams JM. 2009. p53 ancestry: gazing through an evolutionary lens. Nat Rev Cancer 9: 758–762. [DOI] [PubMed] [Google Scholar]
- Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz-Cuaran S, Olivier M, Hall J, Mollereau B, Hainaut P, et al. 2011. Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ 18: 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavinahalli JN, Madhumalar A, Beuerman RW, Lane DP, Verma C. 2010. Differences in the transactivation domains of p53 family members: a computational study. BMC Genomics 11: S5–S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW. 1999. Mechanisms of switching on p53: a role for covalent modification? Oncogene 18: 7666–7675. [DOI] [PubMed] [Google Scholar]
- Meek DW. 2015. Regulation of the p53 response and its relationship to cancer. Biochem J 469: 325–346. [DOI] [PubMed] [Google Scholar]
- Mehta TK, Ravi V, Yamasaki S, Lee AP, Lian MM, Tay BH, Tohari S, Yanai S, Tay A, Brenner S, et al. 2013. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc Natl Acad Sci 110: 16044–16049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Villegas A, Belyi VA. 2011. The evolution of MDM2 family genes. Gene 486: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio J, Retaux S. 2008. The lamprey in evolutionary studies. Dev Genes Evol 218: 221–235. [DOI] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Schumacher B. 2012. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet 28: 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi CP, Milner J. 2003. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22: 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon O, Friedler A, Bycroft M, Freund SM, Fersht AR. 2002. Molecular mechanism of the interaction between MDM2 and p53. J Mol Biol 323: 491–501. [DOI] [PubMed] [Google Scholar]
- Shimeld SM, Donoghue PC. 2012. Evolutionary crossroads in developmental biology: cyclostomes (lamprey and hagfish). Development 139: 2091–2099. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Antonacci F, Eichler EE, Amemiya CT. 2009. Programmed loss of millions of base pairs from a vertebrate genome. Proc Natl Acad Sci 106: 11212–11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Baker C, Eichler EE, Amemiya CT. 2012. Genetic consequences of programmed genome rearrangement. Curr Biol 22: 1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, Campbell MS, Yandell MD, Manousaki T, Meyer A, Bloom OE, et al. 2013. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat Genet 45: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh EK, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, Elvin JA, Bronson RT, Crum CP, McKeon F. 2006. p63 protects the female germ line during meiotic arrest. Nature 444: 624–628. [DOI] [PubMed] [Google Scholar]
- Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3: 1339–1345. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. 2002. Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604. [DOI] [PubMed] [Google Scholar]
- Wei SJ, Joseph T, Chee S, Li L, Yurlova L, Zolghadr K, Brown C, Lane D, Verma C, Ghadessy F. 2013a. Inhibition of nutlin-resistant HDM2 mutants by stapled peptides. PLoS One 8: e81068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SJ, Joseph T, Sim AY, Yurlova L, Zolghadr K, Lane D, Verma C, Ghadessy F. 2013b. In vitro selection of mutant HDM2 resistant to Nutlin inhibition. PLoS One 8: e62564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.