

The heart consumes a significant amount of ATP to maintain its contractile performance, and fatty acid oxidation (FAO) serves as a primary source of energy in the adult heart. Research in the past several decades has demonstrated that the nuclear receptor family, in particular peroxisome proliferator-activated receptor α (PPARα), is a major transcriptional mechanism regulating expression of proteins involved in fatty acid metabolism. Although much progress has been made in understanding the functional role of PPARα in cardiac physiology and disease, less is known about the regulation of PPARα itself1. PPARα-mediated transcriptional cascades are determined by the level of PPARα expression as well as its activity. Long chain fatty acids and synthetic ligands, such as fibrates, increase the transcriptional activity of PPARα in multiple cell types including cardiac myocytes. The endogenous ligands of PPARα are still debated but evidence indicates they may be associated with triglyceride lipolysis2–4. Expression of PPARα in the heart changes during the development, and under multiple pathological conditions including heart failure and diabetes. However, the mechanism(s) underlying these changes are poorly understood. In this issue of Circulation Research, Drosatos et al.5 report that Krüppel-like factor 5 (KLF5) regulates the transcription of PPARα in the heart (Figure).
FIGURE.
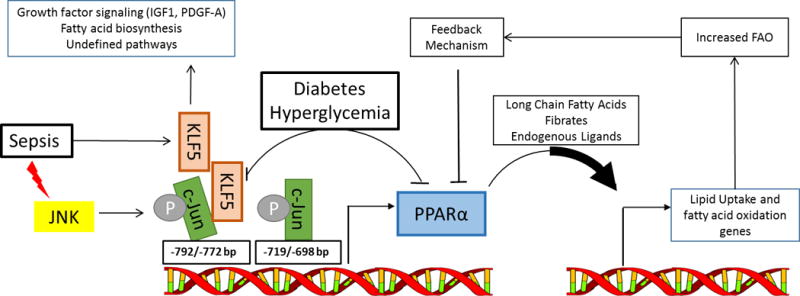
PPARα expression and fatty acid oxidation in the heart: proposed regulation by KLF5, sepsis and diabetes. PPARα level and the availability of its ligands regulate the expression of key proteins for fatty acids transport and oxidation (FAO) in the heart. Alterations in FAO act through feedback mechanisms to regulate PPARα expression and restore FAO to optimal levels. Deletion of KLF5 in the heart resulted in lower PPARα expression and decreased FAO. PPARα transcript levels are reduced in parallel to reduced KLF5 expression in early stage of diabetes, while in sepsis the activation of c-Jun N-terminal Kinase (JNK) phosphorylates c-Jun which binds to the PPARα promoter and prevents transcription of PPARα by KLF5. KLF5 has non-metabolic actions on the heart as well including effects on growth factor signaling and fatty acid biosynthesis in addition to other pathways suggested by microarray analysis of mouse hearts with cardiac-specific deletion of Klf5.
Krüppel-like transcription factors (KLFs) are a family of zinc-finger DNA-binding proteins known to be heavily involved in gene expression during development, but their role in adult organ/tissue is just begun to be revealed. KLF5 has been shown to regulate lipid metabolism in non-cardiac tissue such as lung development and adipogenesis6–8. In the heart, KLF5 was found to promote cardiac hypertrophy by driving platelet-derived growth factor-A (PDGF-A) expression and transactivating Igf1 in fibroblasts9, 10. Using both gain- and loss-of-function approaches, Drosatos et al. in the present study showed that KLF5 regulates the expression of PPARα in the heart. A binding site of KLF5 was mapped to the promoter region of PPARα. ChIP assay demonstrated the binding of KLF5 to the site in the cardiac-like HL-1 cell line during adenovirus mediated overexpression of KLF5, which is associated with increased PPARα expression (Figure). Furthermore, cardiac-specific deletion of KLF5 in mice led to decreased expression of PPARα and its target genes with concomitant decrease of FAO. The evidence collectively identifies KLF5 as a positive regulator of FAO in the heart via the transcription of PPARα.
Does the regulation of PPARα by KLF5 play a role in heart disease? The study examined two pathological conditions in which PPARα was downregulated in the heart, sepsis and diabetes. In mouse models of both type I (Streptozotocin injection) and type II (ob/ob and db/db mice) diabetes, the study found a nice parallel change of KLF5 and PPARα expression in the heart. It also showed that the down regulation of KLF5-PPARα in the early stage of diabetes could be restored by normalizing blood glucose levels (Figure). The relationship of hyperglycemia and KLF5 expression, however, appeared to be more complex in these models since a rebound of KLF5- PPARα level occurred at late stage of diabetes despite persistent hyperglycemia. Future studies determining the causal relationship and the molecular mechanisms are clearly warranted. Nevertheless, these observations provide an interesting new direction to dissect the regulation of PPARα in the diabetic heart. Although increased FAO has been consistently observed in diabetic hearts, the level of PPARα varies significantly depending on the model and the severity of disease11. It is worth testing whether the biphasic change of KLF5-PPARα is another regulator of fatty acid oxidation in the diabetic heart and hence a new target for modulating cardiac metabolism in diabetes.
PPARα expression is downregulated in the heart in sepsis, which however, was found negatively correlated with KLF5 level. A previous study by the same group showed that c-Jun N-terminal kinase (JNK) activation during sepsis was responsible for decreases in fatty acid oxidation through PPARα downregulation12. Here they further demonstrated in HL-1 cells treated with lipopolysaccharide (LPS) that activated c-Jun bound to the same promoter region as KLF5 of the Ppara gene, with c-Jun outcompeting KLF5 leading to suppression of PPARα (Figure). This is an excellent example illustrating the complexity of a very delicate molecular dance in the transcriptional regulation. The relative role of each specific regulatory mechanism is dependent on the contribution of other mechanisms, which are likely disease specific. In the clinical setting, sepsis has historically been considered primarily a disorder of acute inflammation, but this paradigm is under reconsideration after several anti-inflammatory interventions failed to improve patient outcomes. The role of metabolic failure in sepsis and cardiac metabolism specifically have received renewed attention recently13, 14. It has not been determined whether effective anti-inflammatory therapy removes the suppression of PPARα expression by c-Jun. Furthermore, it is unknown whether downregulation of PPARα in sepsis is adaptive or maladaptive. Additional studies in a more clinically relevant model of sepsis such as cecal ligation puncture (CLP), which better recapitulates the progressive organ system failure often observed in humans, will be necessary to elucidate fundamental mechanisms with translational therapeutic potential.
The study also addressed the functional significance of KLF5 in the heart using cardiac specific KLF5 deletion mouse model (cKO). Despite the decrease in FAO in cardiac muscle, cKO mice showed normal cardiac function at 2–3 months. This is consistent with a prior report by another group showing normal cardiac function and unaltered response to a moderate pressure overload in cKO15. However, the present study found that cKO developed contractile dysfunction after 6 months and progressively at 12 months where they displayed myocardial lipid accumulation. These changes resemble the observations made in hearts of PPARα-null mice suggesting that maintaining FAO is critical for normal cardiac function in the long term16. An important caveat of the experiment is that at 9 months the PPARα level is normalized and at 12 months the PPARα level is elevated in the cKO (supplemental figure VIII). As PPARα expression is under the control of multiple transcription and feedback mechanisms it is likely that compensatory mechanisms yet undefined take over and restore PPARα levels in cKO at older age. Other regulators of PPARα expression have been proposed and even another KLF, KLF15, has been shown to regulate PPARα expression and cardiac metabolism17. However, the researcher reported decreased ATP content and accumulation of lipids in the myocardium of cKO at older age suggesting defective energy metabolism despite the normalization of PPARα and its target gene expression. Direct measurements of metabolic fluxes are thus necessary to further dissect the metabolic mechanisms responsible for contractile dysfunction in cKO.
This work adds a much needed insight into how PPARα gene expression is regulated in the heart, but also raises many additional questions. The authors showed clearly that KLF5 and c-Jun have opposing regulatory actions on PPARα expression, however, the authors did not address what other factors would be involved in the compensatory PPARα expression in cKO hearts. This is especially important due to the fact that PPARα and downstream gene expression returns to normal levels after 9 months of age in the cKO hearts when reduced ATP content and increased triglyceride deposition occur; pointing to potential consequences of KLF5 loss outside of PPARα downregulation. The authors did indeed identify significant gene expression changes of many pathways in cKO hearts. Microarray data showed that deletion of KLF5 led to increase expression of 228 genes and decrease expression of 79 genes with the compliment and coagulation cascade pathway being the most affected, suggesting a much broader effect of KLF5 deficiency outside of PPARα regulation and opened up a swath of possibilities by which KLF5 deletion would impact cardiac function.
As with all good science, this line of investigation has opened the door for additional studies. Future experiments will be needed to fully elucidate the mechanism of cardiac dysfunction caused by KLF deficiency, as well as the significance of KLF5 mediated control of PPARα expression in diabetic cardiomyopathy and heart failure. It is exciting to speculate that the regulation of PPARα by KLF5 will be significant in the well documented downregulation of fatty acid oxidation in heart failure, possibly opening up KLF5 activators or mimetics as a therapeutic target for heart failure.
Acknowledgments
SOURCES OF FUNDING: This work is in part supported by National Institute of Health grants HL-088634, HL-110349, HL-118989, HL-129510 (to R. Tian) and T32DK007247 (to N. Roe), and by the Scientist Development Grant of the American Heart Association 12SDG12040342 (to S. W. Standage).
Footnotes
Disclosure of conflict: None.
References
- 1.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. Journal of molecular and cellular cardiology. 2008;44:968–75. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends in cardiovascular medicine. 2000;10:238–45. doi: 10.1016/s1050-1738(00)00077-3. [DOI] [PubMed] [Google Scholar]
- 3.Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ, Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-alpha activators. Circulation. 2010;121:426–35. doi: 10.1161/CIRCULATIONAHA.109.888735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nature medicine. 2011;17:1076–85. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drosatos K, Pollak NM, Pol C, Ntziachristos P, Willecke F, Valenti MC, Trent CM, Hu Y, Guo S, Aifantis I, Goldberg IJ. Cardiac Myocyte KLF5 Regulates Ppara Expression and Cardiac Function. Circulation research. 2016;118:xxx–xxx. doi: 10.1161/CIRCRESAHA.115.306383. [in this issue] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan H, Luo F, Wert SE, Zhang L, Xu Y, Ikegami M, Maeda Y, Bell SM, Whitsett JA. Kruppel-like factor 5 is required for perinatal lung morphogenesis and function. Development. 2008;135:2563–72. doi: 10.1242/dev.021964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell metabolism. 2005;1:93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, Nishimura G, Maemura K, Yamauchi T, Kubota N, Suzuki R, Kitamura T, Akira S, Kadowaki T, Nagai R. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell metabolism. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Oishi Y, Manabe I, Tobe K, Ohsugi M, Kubota T, Fujiu K, Maemura K, Kubota N, Kadowaki T, Nagai R. SUMOylation of Kruppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR-delta. Nature medicine. 2008;14:656–66. doi: 10.1038/nm1756. [DOI] [PubMed] [Google Scholar]
- 10.Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami H, Nishimatsu H, Ishikawa T, Suzuki T, Morita H, Maemura K, Sata M, Hirata Y, Komukai M, Kagechika H, Kadowaki T, Kurabayashi M, Nagai R. Kruppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nature medicine. 2002;8:856–63. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 11.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 12.Drosatos K, Drosatos-Tampakaki Z, Khan R, Homma S, Schulze PC, Zannis VI, Goldberg IJ. Inhibition of c-Jun-N-terminal kinase increases cardiac peroxisome proliferator-activated receptor alpha expression and fatty acid oxidation and prevents lipopolysaccharide-induced heart dysfunction. The Journal of biological chemistry. 2011;286:36331–9. doi: 10.1074/jbc.M111.272146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 2014;5:66–72. doi: 10.4161/viru.26907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Standage SW, Waworuntu RL, Delaney MA, Maskal SM, Bennion BG, Duffield JS, Parks WC, Liles WC, McGuire JK. Non-hematopoietic PPARα protects against cardiac injury and enhances survival in experimental polymicrobial sepsis. Critical Care Medicine. 2016 doi: 10.1097/CCM.0000000000001585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ, Nagai R. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. The Journal of clinical investigation. 2010;120:254–65. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, Kamijo Y, Gonzalez FJ, Aoyama T. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. The Journal of biological chemistry. 2000;275:22293–9. doi: 10.1074/jbc.M000248200. [DOI] [PubMed] [Google Scholar]
- 17.Prosdocimo DA, John JE, Zhang L, Efraim ES, Zhang R, Liao X, Jain MK. KLF15 and PPARalpha Cooperate to Regulate Cardiomyocyte Lipid Gene Expression and Oxidation. PPAR research. 2015;2015:201625. doi: 10.1155/2015/201625. [DOI] [PMC free article] [PubMed] [Google Scholar]