

Abstract
Background:
Pseudobulbar affect (PBA) is associated with neurological disorders or injury affecting the brain, and characterized by frequent, uncontrollable episodes of crying and/or laughing that are exaggerated or unrelated to the patient’s emotional state. Clinical trials establishing dextromethorphan and quinidine (DM/Q) as PBA treatment were conducted in patients with amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). This trial evaluated DM/Q safety in patients with PBA secondary to any neurological condition affecting the brain.
Objective:
To evaluate the safety and tolerability of DM/Q during long-term administration to patients with PBA associated with multiple neurological conditions.
Methods:
Fifty-two-week open-label study of DM/Q 30/30 mg twice daily. Safety measures included adverse events (AEs), laboratory tests, electrocardiograms (ECGs), vital signs, and physical examinations.
Clinical trial registration:
Results:
A total of 553 PBA patients with >30 different neurological conditions enrolled; 296 (53.5%) completed. The most frequently reported treatment-related AEs (TRAEs) were nausea (11.8%), dizziness (10.5%), headache (9.9%), somnolence (7.2%), fatigue (7.1%), diarrhea (6.5%), and dry mouth (5.1%). TRAEs were mostly mild/moderate, generally transient, and consistent with previous controlled trials. Serious AEs (SAEs) were reported in 126 patients (22.8%), including 47 deaths, mostly due to ALS progression and respiratory failure. No SAEs were deemed related to DM/Q treatment by investigators. ECG results suggested no clinically meaningful effect of DM/Q on myocardial repolarization. Differences in AEs across neurological disease groups appeared consistent with the known morbidity of the primary neurological conditions. Study interpretation is limited by the small size of some disease groups, the lack of a specific efficacy measure and the use of a DM/Q dose higher than the eventually approved dose.
Conclusions:
DM/Q was generally well tolerated over this 52 week trial in patients with PBA associated with a wide range of neurological conditions.
Keywords: Dextromethorphan/quinidine, Pseudobulbar affect, Safety, Tolerability
Introduction
Pseudobulbar affect (PBA) is a neurological condition that exerts a significant health burden on patients and caregivers1. PBA is associated with a wide range of neurological disorders and is characterized by frequent, sudden, uncontrollable episodes of crying and/or laughing that are greatly exaggerated or contrary to the patient’s emotional state2. Available prevalence studies suggest PBA is present in at least 5% of Parkinson’s disease (PD), 10% of multiple sclerosis (MS), stroke, traumatic brain injury (TBI), and Alzheimer’s disease (AD), 20% of progressive supranuclear palsy, and up to 50% of amyotrophic lateral sclerosis (ALS) patients3–11.
PBA is thought to arise from disruption of corticobulbar and cerebellar/pontine pathways controlling emotional expression12–14. These pathways may be disrupted by multiple neurological conditions, yet the ensuing clinical manifestations of PBA are indistinguishable, consistent with a common etiology across disorders12–14. The combination of dextromethorphan and quinidine (DM/Q) is the first pharmacotherapy approved by the US Food and Drug Administration (FDA) and European Medicines Authority (EMA) for treating PBA15,16. Dextromethorphan (DM) has many pharmacological actions, including uncompetitive N-methyl-D-aspartate (NMDA) receptor antagonism17, sigma-1 receptor agonism18, and serotonin reuptake inhibition, among others; the precise mechanism(s) accounting for PBA suppression is/are unknown19. DM is co-administered with low-dose quinidine, a potent CYP2D6 inhibitor that reduces rapid first-pass metabolism of DM. This inhibition increases DM bioavailability and half-life20,21. The efficacy, safety, and tolerability of DM/Q as PBA treatment was established in three controlled clinical trials lasting 422 or 1223,24 weeks, using fixed-dose combinations of twice daily DM/Q 30/30 mg22,23, 30/10 mg24, or 20/10 mg24 in patients with ALS22,24 or MS23,24. The present trial was designed to provide long-term safety data using the higher DM/Q 30/30 mg dose in patients with PBA, regardless of primary neurological condition.
Methods
Study design
Study 02-AVR-107 (NCT00056524) is a multicenter, 52 week, open-label safety trial. The trial began in March 2003 at 44 sites internationally (39 in the US, four in Israel, and one in Serbia and Montenegro). An extension was added to allow completing patients the option to remain on treatment past 1 year. The trial was terminated by the sponsor in June 2007, following the decision to pursue development of a lower DM/Q dose. Patients were instructed to take DM/Q 30/30 mg in the evening for 7 days and then twice daily thereafter. Patients kept a diary of dosage times and recorded any adverse events (AEs). Clinic visits occurred at screening, baseline (Day 1), and after 1, 4, 8, and 12 months of treatment or at patient discontinuation. During months when no clinic visit was scheduled, patients were contacted by telephone and asked about medication compliance and AEs.
Study population
Eligible patients were 18 to 75 years of age with a clinical diagnosis of PBA. For the purposes of this study, PBA was defined as ‘a syndrome characterized by outbursts of crying and/or laughing that occur spontaneously and inappropriately given the context in which they occur’. No specific threshold for severity of PBA symptoms was required for study entry. Patients were required to have an electrocardiogram (ECG) with no evidence of rate-corrected QT interval (QTc) prolongation (≥450 msec in men; ≥470 msec in women), heart block (isolated right bundle branch block without clinical history of heart disease was allowable), sinus bradycardia (<50 bpm), ventricular tachycardia, multifocal ventricular ectopic beats (any frequency), or unifocal ventricular ectopic beats (>5/min). Patients with ALS were required to have a vital capacity ≥50% at baseline. Patients completing prior controlled studies of twice daily DM/Q 30/30 mg to treat PBA in MS23 or ALS22 were also eligible to participate, provided they met all eligibility requirements at the time of enrollment in this study.
Exclusion criteria were myasthenia gravis; a history of ventricular tachycardia or torsades de pointes; sensitivity to quinidine or opiate drugs; major psychiatric disturbance; a history of substance abuse in the past 2 years; any major systemic disease that would interfere with interpretation of study results (e.g., malignancy, uncontrolled diabetes, dilated cardiac myopathy, ischemic or valvular heart disease); hypotension (systolic blood pressure [BP] <100 mmHg); a history of postural or any unexplained syncope; renal, hepatic, or pulmonary disease; or clinically significant deviations in standard laboratory tests. Female patients could not be pregnant or breastfeeding; those with child-bearing potential were required to use an established method of birth control.
Patients were allowed to continue existing medications, except for the following starting from 1 week before DM/Q initiation: ketoconazole, voriconazole, verapamil, diltiazem, nifedipine, sodium bicarbonate, carbonic anhydrase inhibitors, thiazide diuretics (unless urine pH ≤6.5 and on thiazide for ≥1 month at enrollment), warfarin, haloperidol, monoamine oxidase inhibitors, and tricyclic antidepressants at doses >75 mg/day (>20 mg/day for desipramine; >15 mg/day for protriptyline). Other than the study drug, medications containing dextromethorphan or quinidine were prohibited.
Safety/tolerability assessments
Safety and tolerability measures included: AEs recorded in patient diaries and during clinic visits and telephone contacts (serious adverse events [SAEs], including deaths, were required to be reported through the 30 days following final dose); vital signs (all visits); resting 12-lead ECG (screening, Day 29, and Final Visit at Week 52 or discontinuation); clinical laboratory values, including serum chemistry, hematology, and urinalysis (screening, Day 29, and Final Visit); and physical examination (screening and Final Visit).
Pharmacokinetic assessments
A blood sample was obtained on Day 29 within 12 h after dosing to determine plasma concentrations of DM, quinidine, and the DM metabolite dextrorphan (DX).
Safety-data analyses
The safety population comprised all patients receiving at least one DM/Q dose. The numbers of patients experiencing AEs were summarized by body system, relationship to study drug, and severity. SAEs and AEs that resulted in study discontinuation were also summarized descriptively. Additionally, patient demographics, disposition, concomitant medications, and AEs were analyzed by primary neurological condition. Likelihood-ratio chi-square tests were used to compare AE incidence among disease groups. Assignment of patients to subgroups for analysis by primary neurological condition was made based on similarities among nosological characteristics and without knowledge of patient data or safety outcome.
Clinical laboratory values were summarized descriptively; shifts between normal, low, or high values were analyzed using McNemar’s test. Changes in physical-examination findings (normal vs. abnormal) were assessed by McNemar’s test. Summary statistics of absolute values and percentage change from screening value were provided for systolic and diastolic BP, heart rate, respiratory rate, QT interval, QTc interval, ventricular rate, PR interval, and QRS duration. Clinically significant abnormalities were documented.
Ethics and Good Clinical Practice
The study was conducted in conformity with the standards of Good Clinical Practices and the Declaration of Helsinki. Eligible patients were informed of the study’s purposes and of anticipated AEs that recipients might experience, and ethics committee approval was obtained for each study site. Before any study procedures, a signed informed consent document was obtained.
Results
A total of 553 patients with PBA were enrolled (494 in the US, 48 in Israel, and 11 in Serbia and Montenegro). Eighty-nine of these patients entered directly from a 12 week, placebo-controlled DM/Q study to treat PBA in MS, and 56 others (9 with ALS and 47 with MS) had been previously exposed to DM and/or quinidine in clinical studies22,23. Over 30 neurological conditions were represented among the participating cohort. In addition to ALS (n = 176) and MS (n = 223), 154 participants had PBA secondary to diverse neurological conditions (Table 1). These conditions were categorized into seven disease groups as follows: ALS/motor neuron diseases (MND) (n = 199), MS (n = 223), stroke/cerebrovascular disorders (CBVD) (n = 51), TBI (n = 23), PD/movement disorders (n = 23), AD/dementia (n = 17), and ‘other PBA’ (n = 17), as shown in Table 1.
Table 1.
Categorization of patients’ primary neurological conditions into disease groups (n).
| AD/dementia (n = 17) | MS (n = 223) | Stroke/CBVD (n = 51) | ALS/MND (n = 199) | TBI (n = 23) | PD/movement disorders (n = 23) | Other PBA (n = 17) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | 15 | MS | 223 | Stroke | 46 | ALS | 176 | TBI | 21 | PD | 11 | Cerebellar ataxia | 3 |
| Frontal lobe dementia | 2 | Venous angioma | 1 | Primary lateral sclerosis | 16 | Head trauma | 2 | Parkinson syndrome | 1 | Spinocerebellar ataxia | 2 | ||
| Pontine AVM | 1 | Progressive bulbar palsy | 5 | Atypical PD | 1 | Cerebral palsy | 2 | ||||||
| Post-cerebral aneurysm surgery | 1 | Bulbar motor neuron disease or degeneration | 1 | Parkinsonian syndrome | 1 | Chronic cough | 1 | ||||||
| Brain aneurysm | 1 | Bulbar palsy | 1 | Progressive supranuclear palsy | 5 | Small-fiber neuropathy | 1 | ||||||
| Subarachnoid hemorrhage | 1 | Huntington’s disease | 1 | Leuko-encephalopathy | 1 | ||||||||
| Choreiform disorder | 1 | Post-herpes simplex virus | 1 | ||||||||||
| Movement disorder | 1 | Subdural hematoma | 1 | ||||||||||
| Corticobasilar degeneration | 1 | Viral meningio-encephalitis | 1 | ||||||||||
| Hydrocephalus | 1 | ||||||||||||
| Post-encephalitis | 1 | ||||||||||||
| Foster-Kennedy syndrome | 1 | ||||||||||||
| None known | 1 | ||||||||||||
AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; AVM: arteriovenous malformation; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.
Patient disposition
Patient disposition is shown in Table 2 and duration of study participation is shown in Figure 1. Of the 553 patients treated with DM/Q, 382 patients (69.1%) completed at least 6 months of treatment, 300 (54.2%) completed 1 year, and 296 (53.5%) completed the protocol-specified visits. One hundred forty-eight (26.8%) patients dropped out of the trial for either an AE or medication refusal due to an AE; 109 (19.7%) dropped out for other reasons. Of the completers, 262 (88.5%) elected to continue DM/Q in the optional extension. At trial termination, 20 patients (3.6%) remained in the treatment phase and 168 (64.1% of those entering the extension) remained in the extension. This report provides results from the 52 week treatment phase.
Table 2.
Patient disposition by primary neurological conditiona.
| Patient disposition, n | AD/dementia | MS | Stroke/CBVD | TBI | ALS/MND | PD/movement disorders | Other PBA | Total |
|---|---|---|---|---|---|---|---|---|
| Enrolled | n = 17 | n = 223 | n = 51 | n = 23 | n = 199 | n = 23 | n = 17 | N = 553 |
| Treated ≥6 months | 10 | 176 | 30 | 15 | 126 | 12 | 13 | 382 |
| Treated ≥12 monthsa | 5 | 149 | 27 | 10 | 86 | 11 | 12 | 300 |
| Completed treatment phase | 4 | 149 | 27 | 9 | 85 | 10 | 12 | 296 |
| Remaining at study termination | 2 | 8 | 4 | 2 | 4 | 0 | 0 | 20 |
| Did not complete due to: | 11 | 66 | 20 | 12 | 110 | 13 | 5 | 257 |
| AE | 4 | 27 | 7 | 1 | 62 | 5 | 1 | 107 |
| Medication refusal due to AE | 2 | 10 | 4 | 2 | 18 | 4 | 1 | 41 |
| Medication refusal for other than AE | 0 | 7 | 1 | 2 | 3 | 0 | 1 | 14 |
| Withdrew consent | 3 | 15 | 3 | 3 | 9 | 1 | 0 | 34 |
| Lost to follow-up | 0 | 4 | 4 | 2 | 7 | 0 | 1 | 18 |
| Protocol violation | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 3 |
| Other | 4 | 9 | 5 | 4 | 14 | 3 | 1 | 40 |
| Entered extension phase | 4 | 132 | 22 | 8 | 77 | 8 | 11 | 262 |
AD: Alzheimer’s disease; AE: adverse event; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.
aIncludes all patients who completed the treatment phase or received at least 365 days of dosing.
Figure 1.
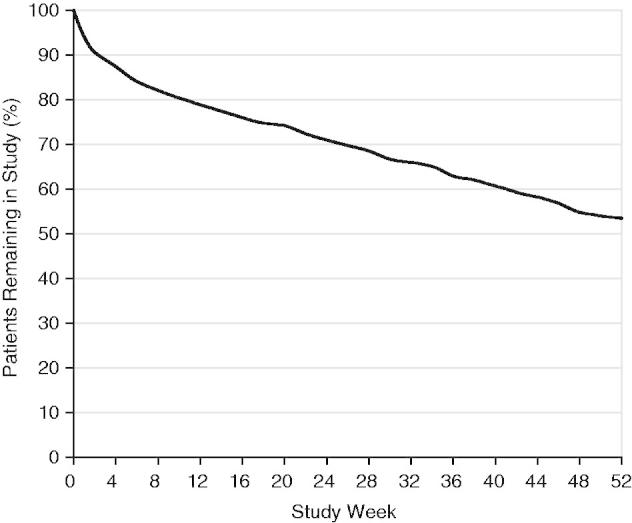
Proportion of patients remaining in study, by study week.
Patient demographics
Patient demographics by primary disease are provided in Table 3. Patients reported a median of 7.0 (range 0–420; mean 16.7 ± 32.6) PBA episodes per week at baseline. Some patients transitioned directly from a previous DM/Q study and therefore may have had low or no PBA episodes at baseline. The median patient age was 52.0 years (range 18–86), 15.9% were ≥65 years old, 58.2% were female, and 90.8% were Caucasian.
Table 3.
Patient demographics by primary neurological condition.
| Category | AD/dementia (n = 17) | MS (n = 223) | Stroke/CBVD (n = 51) | TBI (n = 23) | ALS/MND (n = 199) | PD/movement disorders (n = 23) | Other PBA (n = 17) |
|---|---|---|---|---|---|---|---|
| Age (years) | |||||||
| Mean | 63.5 | 45.7 | 54.8 | 45.6 | 55.6 | 64.9 | 49.1 |
| SD | 11.20 | 10.23 | 10.93 | 13.81 | 11.06 | 8.00 | 13.71 |
| Median | 66.0 | 47.0 | 56.0 | 44.0 | 57.0 | 66.0 | 52.0 |
| Min, max | 38, 79 | 18, 71 | 30, 80 | 18, 74 | 18, 80 | 50, 86 | 22, 67 |
| Gender, n (%) | |||||||
| Male | 10 (58.8) | 49 (22.0) | 24 (47.1) | 10 (43.5) | 116 (58.3) | 15 (65.2) | 7 (41.2) |
| Female | 7 (41.2) | 174 (78.0) | 27 (52.9) | 13 (56.5) | 83 (41.7) | 8 (34.8) | 10 (58.8) |
| Ethnicity, n (%) | |||||||
| Caucasian | 16 (94.1) | 208 (93.3) | 43 (84.3) | 18 (78.3) | 179 (89.9) | 22 (95.7) | 16 (94.1) |
| Black | 1 (5.9) | 5 (2.2) | 5 (9.8) | 1 (4.3) | 5 (2.5) | 0 (0) | 0 (0) |
| Asian | 0 (0) | 1 (0.4) | 1 (2.0) | 0 (0) | 1 (0.5) | 0 (0) | 0 (0) |
| Hispanic | 0 (0) | 8 (3.6) | 2 (3.9) | 3 (13.0) | 9 (4.5) | 1 (4.3) | 1 (5.9) |
| Other | 0 (0) | 1 (0.4) | 0 (0) | 1 (4.3) | 5 (2.5) | 0 (0) | 0 (0) |
AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; SD: standard deviation; TBI: traumatic brain injury.
Concurrent medications
Patients were taking a median of seven additional medications at baseline (range 0–30) including those for their primary neurological condition and other conditions. The number of baseline medications was similar for study completers and those who discontinued for AEs.
The most frequently reported concurrent chronic medications (taken ≥3 months during treatment) were nonsteroidal anti-inflammatory drugs and other analgesics, antidepressants, lipid-modifying agents, antithrombotics, inhibitors of gastric acid production, vitamins, anticonvulsants, and benzodiazepines (Table 4). Antipsychotics were used chronically by 2.2% of patients.
Table 4.
Common concurrent medicationsa by primary neurological condition.
| Description, % | AD/dementia (n = 17) | MS (n = 223) | Stroke/CBVD (n = 51) | TBI (n = 23) | ALS/MND (n = 199) | PD/movement disorders (n = 23) | Other PBA (n = 17) | Total (N = 553) |
|---|---|---|---|---|---|---|---|---|
| NSAIDs | 23.5 | 36.3 | 19.6 | 30.4 | 40.2 | 17.4 | 5.9 | 33.8 |
| Antidepressants | 23.5 | 23.3 | 31.4 | 30.4 | 29.1 | 34.8 | 23.5 | 26.9 |
| Tricyclic antidepressants | 0 | 4.9 | 2.0 | 13.0 | 13.1 | 4.3 | 0 | 7.6 |
| SSRIs | 5.9 | 15.2 | 21.6 | 17.4 | 10.6 | 13.0 | 5.9 | 13.6 |
| Otherb | 17.6 | 8.1 | 7.8 | 8.7 | 10.1 | 17.4 | 17.6 | 9.8 |
| Lipid modifiers | 35.3 | 22.4 | 35.3 | 21.7 | 20.6 | 13.0 | 23.5 | 23.0 |
| Antithrombotics | 35.3 | 12.1 | 52.9 | 17.4 | 24.6 | 21.7 | 5.9 | 21.5 |
| Analgesics (e.g., acetaminophen) | 35.3 | 25.6 | 19.6 | 21.7 | 14.1 | 13.0 | 5.9 | 19.9 |
| Drugs to inhibit gastric acid production | 23.5 | 15.7 | 23.5 | 21.7 | 22.6 | 17.4 | 23.5 | 19.7 |
| Vitamin E | 11.8 | 7.6 | 3.9 | 4.3 | 37.7 | 8.7 | 29.4 | 18.8 |
| Antiepileptics | 17.6 | 23.8 | 25.5 | 43.5 | 9.0 | 8.7 | 29.4 | 18.8 |
| Multivitamins, plain | 5.9 | 22.4 | 7.8 | 8.7 | 20.1 | 0 | 11.8 | 17.9 |
| Anxiolytics | 23.5 | 16.6 | 11.8 | 13.0 | 20.1 | 21.7 | 11.8 | 17.5 |
| Vitamin C | 11.8 | 12.6 | 0 | 4.3 | 30.7 | 8.7 | 0 | 17.0 |
| Antihistamines for systemic use | 17.6 | 14.8 | 7.8 | 26.1 | 18.1 | 4.3 | 17.6 | 15.6 |
| Vitamin B12 and folic acid | 11.8 | 9.9 | 7.8 | 17.4 | 20.1 | 4.3 | 11.8 | 13.6 |
| Calcium | 5.9 | 20.2 | 7.8 | 8.7 | 7.5 | 17.4 | 5.9 | 13.0 |
| Hypnotics and sedatives | 17.6 | 8.5 | 3.9 | 4.3 | 20.6 | 17.4 | 0 | 12.7 |
| Opioids | 11.8 | 11.7 | 5.9 | 8.7 | 15.1 | 4.3 | 11.8 | 11.9 |
| Laxatives | 11.8 | 9.4 | 5.9 | 4.3 | 17.1 | 0 | 11.8 | 11.4 |
| Misc. herbal | 0 | 13.0 | 2.0 | 0 | 15.1 | 0 | 0 | 10.8 |
| Beta-blocking agents | 11.8 | 6.7 | 13.7 | 4.3 | 13.1 | 17.4 | 5.9 | 10.1 |
| Thyroid preparations | 17.6 | 9.4 | 9.8 | 17.4 | 7.5 | 13.0 | 5.9 | 9.4 |
| ACE inhibitors, plain | 17.6 | 5.8 | 19.6 | 13.0 | 9.0 | 4.3 | 11.8 | 9.0 |
ACE: angiotensin-converting enzyme; AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; NSAIDs: nonsteroidal anti-inflammatory drugs; PBA: pseudobulbar affect; PD: Parkinson’s disease; SSRI: selective serotonin reuptake inhibitor; TBI: traumatic brain injury.
aUsed for ≥3 months and taken during the study; drug use represents minimum use in each category and may not include some combination products such as antihistamine cold products, combinations of vitamins and herbals, etc.
bOther includes: venlafaxine, 3.6%; bupropion, 3.3%; trazodone, 2.5%; mirtazepine, 1.3%; duloxetine, mianserin, nefazodone, reboxetine, and hydroxytryptophan, 0.2% (1 patient) each.
Common disease-specific therapies included: interferon beta, glatiramer, baclofen, and stimulants in patients with MS; donepezil, and memantine in patients with AD; riluzole in patients with ALS; dopaminergic drugs in patients with PD; antithrombotics and cardiovascular medications in patients with stroke; and antiepileptics in patients with TBI (Table 5).
Table 5.
Common concurrent disease-specific medicationsa by primary neurological condition.
| AD/dementia (n = 17) | % |
| Anticholinesterase (e.g., donepezil) | 47.1 |
| Memantine | 29.4 |
| MS (n = 223) | % |
| Interferon beta | 45.3 |
| Glatiramer | 27.4 |
| Centrally acting muscle relaxants (e.g., baclofen, tizanidine, orphenadrine) | 33.2 |
| Urinary antispasmodics (e.g., oxybutynin, tolterodine) | 25.6 |
| Psychostimulants (e.g., amphetamine derivatives, modafinil, pemoline) | 18.8 |
| Glucocorticoids | 5.8 |
| Immunosuppressants (e.g., methotrexate, mycophenolate, azathioprine) | 5.8 |
| Mitoxantrone | 4.0 |
| 4-Aminopyridine | 2.2 |
| Stroke/CBVD (n = 51) | % |
| Aspirin | 43.1 |
| Clopidogrel | 19.6 |
| Calcium channel blockers (e.g., dihydropyridine) | 11.8 |
| TBI (n = 23) | % |
| Topiramate | 13.0 |
| Carbamazepine/oxcarbazepine | 13.0 |
| Thyroid preparations | 17.4 |
| Psychostimulants (e.g., amphetamine derivatives, modafinil, pemoline) | 13.0 |
| Antimigraine (e.g., triptans, selective 5HT1 agonists) | 13.0 |
| Glucocorticoids | 8.7 |
| ALS/MND (n = 199) | % |
| Riluzole | 47.7 |
| Centrally acting muscle relaxants (e.g., baclofen, tizanidine, orphenadrine) | 31.7 |
| Coenzyme Q10 | 31.2 |
| Tetracycline antibiotics (e.g., minocycline, doxycycline) | 17.6 |
| Creatine | 15.6 |
| Beta-2-adrenergic agonists, inhalants | 14.6 |
| Beta carotene/vitamin A | 13.1 |
| Urinary antispasmodics (e.g., oxybutynin, tolterodine) | 11.1 |
| Quinine | 10.6 |
| Expectorants and mucolytics (e.g., guaifenesin, n-acetylcysteine) | 9.0 |
| Belladonna alkaloids (e.g., atropine, hyocyamine, scopolamine) | 9.0 |
| Glycopyrrolate | 7.5 |
| PD/movement disorders (n = 23) | % |
| Dopamine derivatives (± carbidopa, benserazide, or entacapone) | 65.2 |
| Dopamine agonists | 30.4 |
| Amantadine | 17.4 |
| Selegiline | 8.7 |
| Trihexyphenidyl | 0.5 |
| Other PBA (n = 17) | % |
| Phenytoin | 11.8 |
| Tiagabine | 11.8 |
5HT1: type 1 5-hydroxytryptamine (serotonin) receptor; AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.
aUsed for ≥3 months and taken during the study.
Adverse events
Over 90% of patients (n = 508; 91.8%) reported at least one AE during the 52 week trial. The most frequently reported AEs (incidence ≥15%) were nausea, headache, dizziness, fall, and diarrhea. Most AEs were mild to moderate in severity (64.8% of patients with AEs). Table 6 shows the incidence of commonly reported AEs across disease groups.
Table 6.
Incidence of adverse events reported by ≥5% and at least two patients in any primary-neurological-condition category (safety population).
| Preferred term, % (n) | AD/dementia (n = 17) | MS (n = 223) | Stroke/ CBVD (n = 51) | TBI (n = 23) | ALS/MND (n = 199) | PD/movement disorders (n = 23) | Other PBA (n = 17) | p Valuea | Total (N = 553) |
|---|---|---|---|---|---|---|---|---|---|
| Any AE | 70.6 (12) | 89.7 (200) | 90.2 (46) | 82.6 (19) | 98.0 (195) | 91.3 (21) | 88.2 (15) | 0.0004 | 91.9 (508) |
| Nausea | 11.8 (2) | 23.8 (53) | 19.6 (10) | 13.0 (3) | 31.7 (63) | 8.7 (2) | 23.5 (4) | 0.0544 | 24.8 (137) |
| Headache | 5.9 (1) | 25.1 (56) | 15.7 (8) | 26.1 (6) | 22.1 (44) | 26.1 (6) | 29.4 (5) | 0.4613 | 22.8 (126) |
| Dizziness | 5.9 (1) | 21.5 (48) | 19.6 (10) | 21.7 (5) | 17.1 (34) | 30.4 (7) | 17.6 (3) | 0.5201 | 19.5 (108) |
| Fall | 17.6 (3) | 13.5 (30) | 7.8 (4) | 13.0 (3) | 22.6 (45) | 21.7 (5) | 5.9 (1) | 0.0674 | 16.5 (91) |
| Diarrhea | 23.5 (4) | 15.7 (35) | 11.8 (6) | 13.0 (3) | 19.1 (38) | 0 (0) | 23.5 (4) | 0.2426 | 16.3 (90) |
| Fatigue | 17.6 (3) | 16.6 (37) | 7.8 (4) | 39.1 (9) | 11.1 (22) | 13.0 (3) | 17.6 (3) | 0.0141 | 14.6 (81) |
| Weakness | 0 (0) | 14.8 (33) | 3.9 (2) | 4.3 (1) | 18.6 (37) | 4.3 (1) | 11.8 (2) | 0.0245 | 13.7 (76) |
| Nasopharyngitis | 0 (0) | 16.1 (36) | 9.8 (5) | 13.0 (3) | 9.5 (19) | 4.3 (1) | 11.8 (2) | 0.1944 | 11.9 (66) |
| Somnolence | 5.9 (1) | 4.9 (11) | 15.7 (8) | 4.3 (1) | 15.6 (31) | 26.1 (6) | 11.8 (2) | 0.0019 | 10.8 (60) |
| Arthralgia | 0 (0) | 14.3 (32) | 5.9 (3) | 0 (0) | 10.1 (20) | 13.0 (3) | 5.9 (1) | 0.1334 | 10.7 (59) |
| Cough | 5.9 (1) | 10.3 (23) | 3.9 (2) | 13.0 (3) | 14.6 (29) | 4.3 (1) | 0 (0) | 0.1633 | 10.7 (59) |
| Ecchymosis | 5.9 (1) | 8.1 (18) | 5.9 (3) | 4.3 (1) | 12.6 (25) | 13.0 (3) | 17.6 (3) | 0.4343 | 9.8 (54) |
| Pain in limb | 0 (0) | 13.0 (29) | 11.8 (6) | 0 (0) | 8.5 (17) | 0 (0) | 5.9 (1) | 0.1056 | 9.6 (53) |
| Constipation | 11.8 (2) | 5.4 (12) | 5.9 (3) | 0 (0) | 15.1 (30) | 4.3 (1) | 11.8 (2) | 0.0124 | 9.0 (50) |
| Insomnia | 0 (0) | 6.7 (15) | 7.8 (4) | 8.7 (2) | 13.6 (27) | 0 (0) | 11.8 (2) | 0.1004 | 9.0 (50) |
| Back pain | 5.9 (1) | 9.9 (22) | 5.9 (3) | 0 (0) | 9.0 (18) | 4.3 (1) | 5.9 (1) | 0.6684 | 8.3 (46) |
| Dysphagia | 0 (0) | 1.8 (4) | 3.9 (2) | 0 (0) | 20.1 (40) | 0 (0) | 0 (0) | <0.0001 | 8.3 (46) |
| Dry mouth | 5.9 (1) | 5.8 (13) | 9.8 (5) | 0 (0) | 8.5 (17) | 13.0 (3) | 0 (0) | 0.4119 | 7.1 (39) |
| Edema lower limb | 5.9 (1) | 3.6 (8) | 7.8 (4) | 8.7 (2) | 9.5 (19) | 8.7 (2) | 11.8 (2) | 0.3244 | 6.9 (38) |
| Respiratory failure | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 19.1 (38) | 0 (0) | 0 (0) | <0.0001 | 6.9 (38) |
| Sore throat | 5.9 (1) | 9.0 (20) | 3.9 (2) | 4.3 (1) | 7.0 (14) | 0.4887 | 6.9 (38) | ||
| Urinary tract infection | 0 (0) | 9.9 (22) | 5.9 (3) | 4.3 (1) | 4.5 (9) | 4.3 (1) | 11.8 (2) | 0.2970 | 6.9 (38) |
| Dyspnea | 0 (0) | 3.6 (8) | 3.9 (2) | 8.7 (2) | 11.6 (23) | 4.3 (1) | 5.9 (1) | 0.0399 | 6.7 (37) |
| Fatigue aggravated | 0 (0) | 9.4 (21) | 3.9 (2) | 0 (0) | 5.0 (10) | 0 (0) | 5.9 (1) | 0.1663 | 6.1 (34) |
| Laceration | 0 (0) | 5.8 (13) | 5.9 (3) | 0 (0) | 8.0 (16) | 4.3 (1) | 5.9 (1) | 0.6724 | 6.1 (34) |
| MS aggravated | 0 (0) | 14.3 (32) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | <0.0001 | 5.8 (32) |
| Pyrexia | 5.9 (1) | 7.6 (17) | 3.9 (2) | 5.0 (10) | 4.3 (1) | 5.9 (1) | 0.7549 | 5.8 (32) | |
| Vomiting | 5.9 (1) | 7.6 (17) | 2.0 (1) | 8.7 (2) | 5.0 (10) | 4.3 (1) | 0 (0) | 0.6161 | 5.8 (32) |
| Muscle spasms | 0 (0) | 6.7 (15) | 0 (0) | 8.7 (2) | 6.0 (12) | 0 (0) | 5.9 (1) | 0.3610 | 5.4 (30) |
| Nasal congestion | 0 (0) | 6.3 (14) | 3.9 (2) | 4.3 (1) | 6.0 (12) | 0 (0) | 0 (0) | 0.6634 | 5.2 (29) |
| Anxiety | 0 (0) | 3.6 (8) | 0 (0) | 4.3 (1) | 7.5 (15) | 13.0 | 5.9 (1) | 0.1138 | 5.1 (28) |
| Joint stiffness | 0 (0) | 5.4 (12) | 2.0 (1) | 0 (0) | 6.5 (13) | 0 (0) | 11.8 (2) | 0.3292 | 5.1 (28) |
AD: Alzheimer’s disease; AE: adverse event; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.
aChi-square test.
Although chi-square tests were performed for each AE, resulting p values must be regarded as exploratory due to multiple comparisons and the small size of some groups. Differences in AE incidence across disease groups appeared consistent with the expected manifestations of the primary neurological conditions. For example, respiratory failure occurred exclusively, and dysphagia and dyspnea predominantly, in ALS/MND patients; fatigue was more than twice as common in TBI patients; somnolence was most common in PD/movement disorders patients; weakness was most common in ALS/MND and MS patients; and constipation was most common in AD/dementia, ALS/MND, and ‘other PBA’ patients.
Because of the high morbidity associated with several neurological conditions, it seems particularly important to also note the frequency of AEs that investigators considered possibly related to DM/Q treatment (TRAEs) (Table 7). Most TRAEs (91%) were mild-to-moderate in severity. Only seven occurred with incidence ≥5%: nausea (11.8%), dizziness (10.5%), headache (9.9%), somnolence (7.2%), fatigue (7.1%), diarrhea (6.5%), and dry mouth (5.1%).
Table 7.
Incidence of treatment-relateda adverse events by primary neurological condition (safety population).
| Preferred term, % | AD/dementia (n = 17) | MS (n = 223) | Stroke/CBVD (n = 51) | TBI (n = 23) | ALS/MND (n = 199) | PD/movement disorders (n = 23) | Other PBA (n = 17) | Total (N = 553) |
|---|---|---|---|---|---|---|---|---|
| Any TRAE | 23.5 | 39.9 | 51.0 | 47.8 | 59.3 | 60.9 | 52.9 | 49.0 |
| Nausea | 0 | 8.1 | 7.8 | 4.3 | 19.1 | 4.3 | 17.6 | 11.8 |
| Dizziness (excluding vertigo) | 0 | 10.3 | 7.8 | 13.0 | 10.1 | 21.7 | 17.6 | 10.5 |
| Headache NOS | 0 | 9.4 | 5.9 | 8.7 | 11.6 | 21.7 | 5.9 | 9.9 |
| Somnolence | 0 | 2.7 | 5.9 | 0 | 13.1 | 13.0 | 11.8 | 7.2 |
| Fatigue | 5.9 | 5.8 | 2.0 | 26.1 | 7.5 | 4.3 | 11.8 | 7.1 |
| Diarrhea NOS | 11.8 | 4.0 | 3.9 | 4.3 | 9.0 | 0 | 23.5 | 6.5 |
| Dry mouth | 0 | 3.1 | 5.9 | 0 | 8.0 | 8.7 | 0 | 5.1 |
Additional TRAEs that occurred in at least 5% of patients and in two or more patients in any disease category were stroke/CBVD: insomnia (not elsewhere classified), n = 3 (5.9%); TBI: lethargy, n = 3 (13.0%), abnormal dreams, n = 2 (8.7%); ALS/MND: constipation, n = 11 (5.5%); PD/movement disorders: confusion, n = 2 (8.7%); other PBA: joint stiffness, n = 2 (11.8%); flatulence, n = 2 (11.8%); dyspepsia, n = 2 (11.8%).
AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; NOS: not otherwise specified; PBA: pseudobulbar affect; PD: Parkinson’s disease; TRAE: treatment-related adverse event.
aTRAEs that occurred in at least 5% of total patients; treatment-related categories included possible, probable, highly probable, and missing.
To further assess the time course of these seven TRAEs, a post hoc analysis was conducted evaluating time to onset, duration, recurrence, and percentage of total study days with TRAE present (persistence) (Online Resource 1). Common TRAEs generally occurred early in therapy and were transient; both median time to onset and duration ranged from 4 to 8 days for all but fatigue (median duration 16 days). TRAEs did not recur in most patients; the highest incidence of recurrence was headache (42% recurrence after initial episode). TRAE presence as a percentage of total treatment days was low, ranging from a median of 2% for dizziness and headache to 22% for fatigue and sleepiness. Inclusion of patients who discontinued prior to AE resolution can reduce estimates of AE duration while increasing estimates of percentage of treatment days with AE present (by decreasing total treatment days). Omitting these patients from this analysis did not appreciably affect median duration but did decrease estimates of AE persistence (Online Resource 1).
Serious adverse events
The overall incidence of SAEs was 22.8%; no SAE was reported to be related to study medication. Most SAEs occurred in patients with ALS/MND, including all SAEs of respiratory failure, dysphagia, and pneumonia (Online Resource 2). Except for respiratory events in ALS and disease exacerbation in MS, there was no evidence of increased frequency of SAEs specific to any neurological disorder.
Forty-seven deaths occurred during the 52 week trial, most attributed to ALS progression. Thirty-one of the 39 ALS deaths were attributed to ALS-related respiratory failure or similar respiratory events. The remaining eight were attributed to ‘ALS progression’ (n = 3), cardiac arrest (n = 2), pneumonia (n = 1), infection (n = 1), and cardiorespiratory arrest and epistaxis (n = 1). One additional ALS/MND group patient with primary lateral sclerosis died of ‘cause unknown’. Following study completion, an investigator query about all ALS patient deaths provided an estimated median time from ALS diagnosis to death of 29 months. (Actual date of death was obtained for 117 patients; the remainder were censored at date of last contact.) The three deaths in MS patients were attributed to acute myelomonocytic leukemia, myocardial infarction (patient with resolving flu-like illness and extensive pre-existing skin breakdown; received DM/Q for 8 days and then discontinued for recurrent flu-like illness, hallucinations, and lethargy; and died 5 days later), and myocardial infarction and sepsis (patient with pulmonary embolism and sepsis). Of the remaining four deaths, two were attributed to stroke (patient with AD and prior stroke history who died on Day 217, and a patient with prior stroke history who died on Day 41); one was attributed to cardiac arrest (patient with AD and history of acute coronary syndrome and hypertension); and one patient committed suicide on Day 104 (patient with spinocerebellar ataxia and reactive depression). In all cases, death was considered unlikely related to DM/Q treatment. All deaths occurring during the complete DM/Q clinical development program were additionally reviewed by a group of consulting cardiologists and neurologists as part of the FDA review process for the DM/Q application for treatment of PBA. Their findings corroborated the investigators’ initial assessment that the deaths were not related to treatment but to progression of underlying neurological or other medical conditions.
AEs leading to discontinuation
One hundred forty-nine patients (26.9%) withdrew from the trial or refused further study medication due to AEs (Online Resource 3). This number includes one discontinuation during the extension for an AE beginning in the treatment phase. AE dropout was more frequent in ALS/MND (n = 80; 40.2% of ALS/MND patients), PD/movement disorders (n = 9; 39.1% of PD/movement disorders patients), and AD/dementia (n = 6; 35.3% of AD/dementia patients), and less frequent in stroke/CBVD (n = 11; 21.5%), MS (n = 37; 16.6%), TBI (n = 3; 13.0%), and ‘other PBA’ (n = 2; 11.8%). The most common AEs leading to discontinuation were respiratory failure in ALS/MND patients (14.1% of ALS/MND group; 5.1% of total), nausea (3.3%), dizziness (2.9%), headache (2.5%), and diarrhea (1.8%). Other than respiratory failure and nausea in the ALS/MND group and dizziness and headache in the PD/movement disorders group, discontinuation due to AEs appeared similar regardless of primary neurological disorder. Approximately half of the AE-related dropouts, (n = 74 or 13.5% of total patients) included AEs that were judged to be at least possibly related to treatment. In these patients, the median time to discontinuation was 16.5 days (mean 31.4; range 1–243).
Clinical laboratory values and vital signs
Laboratory test results remained stable during the trial. Shift tables showed no changes of clinical relevance. There were no clinically relevant changes in mean systolic and diastolic BP, heart rate, respiratory rate, or body temperature at any visit. Increased BP was recorded as an AE in 11 patients (2.0%), and increased systolic BP was recorded as an AE in one patient; however, these were considered ‘not related’ or ‘unlikely related’ to DM/Q treatment in all but one patient (‘possibly related’).
Physical examinations
No abnormal physical findings related to study-drug treatment were observed.
Electrocardiography
Mean ECG values showed no clinically relevant post-baseline changes (Online Resource 4). QTc intervals showed a small mean increase (QTcF 3.2; QTcB 4.6 msec) from baseline to final measurement. Five patients had QTc increases ≥60 msec; however, all values remained <500 msec during the 52 week treatment phase.
Pharmacokinetics
Blood samples were provided by 202 patients on Day 29. Mean (standard deviation) plasma concentrations were DM: 92.7 (45.6) ng/mL, DX: 78.0 (43.4) ng/mL, and quinidine: 0.15 (0.09) μg/mL. Mean DM concentration was similar to that reported in previous placebo-controlled trials using this dosage22,23.
Discussion
Long-term administration of DM/Q was well tolerated in this large, open-label study of patients with PBA associated with a wide range of neurological conditions. These results provide important safety data for DM/Q, the only approved medication for PBA, a common and distressing disorder experienced by patients with neurologic disease or injury. The low incidence of treatment-emergent AEs with DM/Q used at a higher dose than the approved dose, and occurring in a diverse, clinic-based, ‘real-world’ population, provides reassuring clarity for physicians on the risks of this treatment. The overall incidence of reported AEs is consistent with the trial length and chronic and often progressive nature of the underlying neurological conditions. Although the majority (90%) of patients reported at least one AE, only 49% reported AEs possibly related to DM/Q treatment, and only 13.4% discontinued for TRAEs. The most frequently reported AEs were consistent with those reported at higher incidence than controls in shorter, double-blind DM/Q 30/30 mg trials in PBA patients with ALS or MS22,23. In a 4 week study in ALS patients22, the most commonly reported AEs (incidence ≥15% and more than controls) among DM/Q recipients (n = 70) were nausea, dizziness, fatigue, diarrhea, and headache. In a 12 week study in MS patients23, the most commonly reported AEs (incidence ≥15% and more than placebo) among the DM/Q recipients (n = 76) were dizziness, nausea, and headache.
This patient population is susceptible to falls. Falls were reported by 16.5% of patients in this study; however, only six patients (1.1%) experienced a fall that was deemed by investigators as possibly related to treatment. In double-blind clinical trials falls were reported with similar incidence in DM/Q and placebo recipients.
Several types of AEs, particularly SAEs, were attributed to primary neurological conditions, for example respiratory depression, dyspnea, and dysphagia in ALS and disease aggravation in MS. Underlying disease contribution to AEs is supported by the AE analysis by disease group, AE data from DM/Q double-blind trials, and literature-reported mortality patterns in ALS patients22–26. Overall, the 39/176 ALS patient deaths constitute a mortality rate of 22.2%, which is lower than the death rate reported in prospective trials. In large-scale ALS-treatment trials, 1 year mortality rates ranged from approximately 26% to 35% in treatment arms and as high as 46% in placebo arms27,28. Furthermore, epidemiological studies generally suggest median survival after ALS diagnosis is approximately 15 to 19 months29–34, although at least one study has reported median survival post-diagnosis of 30 months35. The median survival after ALS diagnosis in the current study was estimated at 29 months (n = 176); however, inferences regarding potential effect of DM/Q on survival are not possible without controlled studies.
The DM/Q 30/30 mg twice daily dosage administered in this study is greater than the DM/Q 20/10 mg twice daily dosage approved by the FDA and EMA for PBA treatment and also greater than the 30/10 mg twice daily dosage approved by the EMA. Given that the incidence of some TRAEs, such as dizziness and nausea, appear dose-related in controlled trials22–24, it is likely that the AE incidence reported in this long-term study would be greater than observed with the lower approved doses. Indeed, among patients treated with DM/Q 20/10 mg for 12 weeks during the pivotal trial (n = 107) the percentages reporting headache, dizziness, and nausea were 14.0%, 10.3%, and 7.5%24 versus 22.8%, 19.5%, and 24.8%, respectively, in the present study.
Quinidine is a potent and reliable inhibitor of CYP2D6, and the dose in the approved DM/Q formulation (10 mg twice daily) is sufficient to increase DM exposure approximately 20-fold24. Other medications metabolized by CYP2D6 include antidepressants, antipsychotics, and beta-blockers. While concentrations of these medications may be increased in the presence of quinidine, potential drug interactions can be managed with simple dose adjustments. The overall number and type of concomitant medications taken in this study appear to reflect those seen in typical clinical practice. Patients who withdrew from the study for AEs did not appear to have a higher medication burden than those who completed the trial.
While quinidine is a well established type 1a antiarrhythmic drug with potential to cause dose-related QT-interval prolongation36, ventricular arrhythmia, and torsades de pointes37–40, no clinically significant arrhythmias were reported in controlled trials of DM/Q for PBA utilizing either the 30 or 10 mg doses of quinidine22–24. No patient had QTc increase >500 msec during the treatment phase. Two patients (both with MS) had a QTc >500 msec during the elective treatment extension; one had QTcB/QTcF = 493/505 msec and was withdrawn from the trial, and the other had prolonged QTc on two occasions (highest QTcB/QTcF = 526/506 msec on Day 1183) and remained in treatment until study termination. Overall ECG results suggested no clinically meaningful effect of DM/Q on myocardial repolarization or any ECG variable.
In summary, no significant ECG changes were observed on DM/Q during this 1 year study. The observed TRAEs occurred early in the treatment course, were largely mild-to-moderate, predominantly transient, and typically did not result in discontinuation. The safety and tolerability profile of DM/Q in this study was consistent with that observed in previous DM/Q trials, and the outcomes for patients receiving DM/Q followed the anticipated clinical course of the underlying neurological conditions.
Key points
Pseudobulbar Affect (PBA) is an uncontrollable disorder of emotional expression, occurring in a broad range of neurological diseases or injuries affecting the brain.
Dextromethorphan/quinidine is the only medication approved by the US Food and Drug Administration and European Medicines Authority for the treatment of PBA.
This 52 week open-label study of 553 patients demonstrates the long-term safety and tolerability of dextromethorphan and quinidine in patients with PBA secondary to a wide variety of neurological conditions.
Supplementary Material
Acknowledgments
The authors thank Laura Truskowski of INC Research for statistical analysis of the data; INC Research for data collection and management; Adrian Hepner MD, previously of Avanir Pharmaceuticals Inc., for contributing to the analysis of the study and the early stages of this manuscript; Joao Siffert MD of Avanir Pharmaceuticals Inc. and Randall E. Kaye MD, previously of Avanir Pharmaceuticals Inc., for critical review of the data and manuscript; and The Curry Rockefeller Group LLC for help in preparing the manuscript for publication.
Transparency
Declaration of funding
This work was supported by Avanir Pharmaceuticals Inc.
Declaration of financial/other relationships
G.L.P. has disclosed that he has served on the speakers bureau for and received research support from Avanir Pharmaceuticals Inc. J.P.W. has disclosed that he has served on the speakers bureau for and received research support from Avanir Pharmaceuticals Inc. C.L.-H. has disclosed that she has no significant relationships with or financial interests in any commercial companies related to this study or article. S.H.A. has disclosed that he serves on a scientific advisory board for Neuraltus Pharmaceuticals Inc.; has received a speaker honorarium from Avanir Pharmaceuticals Inc.; receives research support from the National Institutes of Health and the Muscular Dystrophy Association; and has served as an expert consultant in a medicolegal case. A.E.F. and L.E.P. have disclosed that they are employees of Avanir Pharmaceuticals Inc.
The CMRO peer reviewer on this manuscript has received an honorarium from CMRO for review work, but has no relevant financial or other relationships to disclose.
References
- Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29:775–98. doi: 10.1007/s12325-012-0043-7. [DOI] [PubMed] [Google Scholar]
- Wortzel HS, Oster TJ, Anderson CA, Arciniegas DB. Pathological laughing and crying: epidemiology, pathophysiology and treatment. CNS Drugs. 2008;22:531–45. doi: 10.2165/00023210-200822070-00001. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Arciniegas DB, Bernardini GL. Diagnosis and management of pathological laughter and crying. Mayo Clin Proc. 2006;81:1482–6. doi: 10.4065/81.11.1482. [DOI] [PubMed] [Google Scholar]
- Work S, Colamonico JA, Bradley WG, Kaye RE. Pseudobulbar affect: an under-recognized and undertreated neurological disorder. Adv Ther. 2011;28:586–601. doi: 10.1007/s12325-011-0031-3. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Crumpacker D, Fellus J. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8:1–8. doi: 10.1371/journal.pone.0072232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Gonzalez MP, Becker JT. Symptoms of depression and psychosis in Alzheimer’s disease and frontotemporal dementia: exploration of underlying mechanisms. Neuropsychiatry Neuropsychol Behav Neurol. 1996;9:154–61. [Google Scholar]
- Feinstein A, Feinstein K, Gray T, O’Connor P. Prevalence and neurobehavioral correlates of pathological laughing and crying in multiple sclerosis. Arch Neurol. 1997;54:1116–21. doi: 10.1001/archneur.1997.00550210050012. [DOI] [PubMed] [Google Scholar]
- Siddiqui MS, Fernandez HH, Garvan CW. Inappropriate crying and laughing in Parkinson disease and movement disorders. World J Biol Psychiatry. 2009;10:234–40. doi: 10.1080/15622970701639445. [DOI] [PubMed] [Google Scholar]
- House A, Dennis M, Molyneux A. Emotionalism after stroke. BMJ. 1989;298:991–4. doi: 10.1136/bmj.298.6679.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno A, Jorge RE, Robinson RG. Pathological laughing and crying following traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2004;16:426–34. doi: 10.1176/jnp.16.4.426. [DOI] [PubMed] [Google Scholar]
- Gallagher JP. Pathologic laughter and crying in ALS: a search for their origin. Acta Neurol Scand. 1989;80:114–17. doi: 10.1111/j.1600-0404.1989.tb03851.x. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Anderson SW, Martin CO. Pathological laughter and crying: a link to the cerebellum. Brain. 2001;124:1708–19. doi: 10.1093/brain/124.9.1708. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Joseph J, Press DZ, Schmahmann JD. Pathological laughing and crying in patients with multiple system atrophy-cerebellar type. Mov Disord. 2007;22:798–803. doi: 10.1002/mds.21348. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Coburn KL, Shillcutt SD. Neuroanatomy of pathological laughing and crying: a report of the American Neuropsychiatric Association Committee on Research. J Neuropsychiatry Clin Neurosci. 2009;21:75–87. doi: 10.1176/jnp.2009.21.1.75. [DOI] [PubMed] [Google Scholar]
- Nuedexta (dextromethorphan hydrobromide and quinidine sulfate) capsules. Full prescribing information. Available at: https://www.nuedexta.com/sites/default/files/pdf/Prescribing_Information.pdf [Last accessed 14 March 2014]
- European Medicines Agency. Nuedexta. Summary of product characteristics. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002560/WC500145050.pdf [Last accessed 14 March 2014]
- Choi DW, Peters S, Viseskul V. Dextrorphan and levorphanol selectively block N-Methyl-D-aspartate receptor-mediated neurotoxicity on cortical neurons. J Pharmacol Exp Ther. 1987;242:713–20. [PubMed] [Google Scholar]
- Musacchio JM, Klein M, Canoll PD. Dextromethorphan and sigma ligands: common sites but diverse effects. Life Sci. 1989;45:1721–32. doi: 10.1016/0024-3205(89)90510-9. [DOI] [PubMed] [Google Scholar]
- Werling LL, Keller A, Frank JG, Nuwayhid SJ. A comparison of the binding profiles of dextromethorphan, memantine, fluoxetine and amitriptyline: treatment of involuntary emotional expression disorder. Exp Neurol. 2007;207:248–57. doi: 10.1016/j.expneurol.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Britto MR, Valderhaug KL. Dextromethorphan: enhancing its systemic availability by way of low-dose quinidine-mediated inhibition of cytochrome P4502D6. Clin Pharmacol Ther. 1992;51:647–55. doi: 10.1038/clpt.1992.77. [DOI] [PubMed] [Google Scholar]
- Pope LE, Khalil MH, Berg JE. Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J Clin Pharmacol. 2004;44:1132–42. doi: 10.1177/0091270004269521. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Thisted RA, Appel SH, AVP-923 ALS Study Group Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63:1364–70. doi: 10.1212/01.wnl.0000142042.50528.2f. [DOI] [PubMed] [Google Scholar]
- Panitch HS, Thisted RA, Smith RA, Pseudobulbar Affect in Multiple Sclerosis Study Group Randomized, controlled trial of dextromethorphan/quinidine for pseudobulbar affect in multiple sclerosis. Ann Neurol. 2006;59:780–7. doi: 10.1002/ana.20828. [DOI] [PubMed] [Google Scholar]
- Pioro EP, Brooks BR, Cummings J, Safety, Tolerability, and Efficacy Results Trial of AVP-923 in PBA Investigators Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68:693–702. doi: 10.1002/ana.22093. [DOI] [PubMed] [Google Scholar]
- Gil J, Funalot B, Verschueren A. Causes of death amongst French patients with amyotrophic lateral sclerosis: a prospective study. Eur J Neurol. 2008;15:1245–51. doi: 10.1111/j.1468-1331.2008.02307.x. [DOI] [PubMed] [Google Scholar]
- Spataro R, Lo Re M, Piccoli T. Causes and place of death in Italian patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2010;122:217–23. doi: 10.1111/j.1600-0404.2009.01290.x. [DOI] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–91. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Louwerse ES, Weverling GJ, Bossuyt PM. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol. 1995;52:559–64. doi: 10.1001/archneur.1995.00540300031009. [DOI] [PubMed] [Google Scholar]
- Rooney J, Byrne S, Heverin M. Survival analysis of Irish amyotrophic lateral sclerosis patients diagnosed from 1995–2010. PLoS One. 2013;8:e74733. doi: 10.1371/journal.pone.0074733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccolella S, Beghi E, Palagano G, SLAP Registry Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2008;79:33–7. doi: 10.1136/jnnp.2007.118018. [DOI] [PubMed] [Google Scholar]
- Murphy M, Quinn S, Young J. Increasing incidence of ALS in Canterbury, New Zealand: a 22-year study. Neurology. 2008;71:1889–95. doi: 10.1212/01.wnl.0000336653.65605.ac. [DOI] [PubMed] [Google Scholar]
- del Aquila MA, Longstreth WT, Jr, McGuire V. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813–19. doi: 10.1212/01.wnl.0000049472.47709.3b. [DOI] [PubMed] [Google Scholar]
- Chiò A, Mora G, Leone M, Piemonte and Valle d’Aosta Register for ALS (PARALS) Early symptom progression rate is related to ALS outcome: a prospective population-based study. Neurology. 2002;59:99–103. doi: 10.1212/wnl.59.1.99. [DOI] [PubMed] [Google Scholar]
- Logroscino G, Traynor BJ, Hardiman O, EURALS Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry. 2008;79:6–11. doi: 10.1136/jnnp.2006.104828. [DOI] [PubMed] [Google Scholar]
- Millul A, Beghi E, Logroscino G. Survival of patients with amyotrophic lateral sclerosis in a population-based registry. Neuroepidemiology. 2005;25:114–19. doi: 10.1159/000086353. [DOI] [PubMed] [Google Scholar]
- Quinidine Sulfate Tablets USP Revised: June 2005 Rx only. Available at: http://pi.watson.com/data_stream.asp?product_group=1306&p=pi [Last accessed 14 March 2014]
- Holford NH, Coates PE, Guentert TW. The effect of quinidine and its metabolites on the electrocardiogram and systolic time intervals: concentration–effect relationships. Br J Clin Pharmacol. 1981;11:187–95. doi: 10.1111/j.1365-2125.1981.tb01123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coplen SE, Antman EM, Berlin JA. Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion. A meta-analysis of randomized control trials. Circulation. 1990;82:1106–16. doi: 10.1161/01.cir.82.4.1106. [DOI] [PubMed] [Google Scholar]
- Morganroth J, Goin JE. Quinidine-related mortality in the short-to-medium-term treatment of ventricular arrhythmias. A meta-analysis. Circulation. 1991;84:1977–83. doi: 10.1161/01.cir.84.5.1977. [DOI] [PubMed] [Google Scholar]
- Grace AA, Camm AJ. Quinidine. N Engl J Med. 1998;338:35–45. doi: 10.1056/NEJM199801013380107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.